Agammaglobulinemia
Agammaglobulinemia also known as Bruton’s agammaglobulinemia, X-linked agammaglobulinemia, XLA disease, BTK agammaglobulinemia, Bruton tyrosine kinase agammaglobulinemia, congenital agammaglobulinemia or hypogammaglobulinemia, is a group of rare inherited immune deficiencies characterized by a low concentration of antibodies in the blood due to the lack of particular lymphocytes in the blood and lymph 1. Antibodies are proteins (immunoglobulins, (IgM), (IgG) etc) that are critical and key components of the immune system. Antibodies are essential if the immune system is to do its job of fighting off bacteria, viruses, and other foreign substances that threaten the body. The specialized precursor cells that produce gammaglobulins (immunoglobulins), fail to develop or function properly leading to the deficiency in the number of mature lymphocyte cells called B cells. People affected by X-linked agammaglobulinemia generally begin developing frequent and recurrent bacterial infections from about 6 months of age 2. Commonly diagnosed infections include lung infections (pneumonia and bronchitis), middle ear infections, conjunctivitis, sinus infections, various skin infections, and infections that are associated with chronic diarrhea 3.
Agammaglobulinemia is the most common of the primary immunodeficiencies, accounting for approximately 50% of cases 4.
Three major types of agammaglobulinemia can be described:
- X-linked agammaglobulinemia (XLA) also called Bruton’s agammaglobulinemia,
- the much rarer X-linked agammaglobulinemia with growth hormone deficiency (about 10 cases reported), and
- Autosomal recessive agammaglobulinemia (ARA).
All of these disorders are characterized by a weakened immune system that must be strengthened by the administration of gammaglobulin in order to fight off infections.
Agammaglobulinemia or X-linked agammaglobulinemia (XLA disease) is a rare disorder that mainly affects males. The reported incidence and prevalence of X-linked agammaglobulinemia vary considerably. Some sources report that X-linked agammaglobulinemia occurs at a rate of 1 in 190,000 live births with a frequency of 1 per 100,000 newborn males and an estimated prevalence of 1 to 9 per 1,000,000 5. There is no known ethnic predisposition, but the reported incidence is highest in individuals of the White race 6.
X-linked agammaglobulinemia is caused by a gene defect or mutations in the BTK gene that blocks the growth of normal, mature immune cells called B lymphocytes and is inherited in an X-linked recessive manner 7. As a result, the body makes very little (if any) immunoglobulins (antibodies). Immunoglobulins (antibodies) play a major role in the immune response, which protects against illness and infection.
Antibodies also called gammaglobulins or immunoglobulins, are an integral part of your body’s defense mechanism against certain types of microorganisms or germs, like bacteria or viruses. Antibodies are important in the recovery from infections and protect against getting certain infections more than once. There are antibodies specifically designed to combine with each and every microbe—much like a lock and key.
The basic defect in X-linked agammaglobulinemia is an inability of the patient to produce antibodies (immunoglobulins). Antibodies (immunoglobulins) are produced by specialized cells in the body, called plasma cells. Plasma cells develop in an orderly sequence of steps beginning with stem cells located in the bone marrow (see Figures 1 and 2 below). The stem cells give rise to immature lymphocytes, called pro-B-lymphocytes. Pro-B-lymphocytes next develop into pre-B-cells, which then give rise to B-lymphocytes. Each B-lymphocyte bears on its cell surface a small amount of the immunoglobulin that it is able to produce. This cell surface immunoglobulin can bind foreign substances, (an antigen). When the B-lymphocyte comes into contact with its specific antigen, like pneumococcus or tetanus, it is triggered to mature into a plasma cell which is specialized in making and secreting large amounts of that specific antibody. Each B-cell makes a slightly different antibody, or immunoglobulin, to allow the body to respond to millions of different foreign substances.
Most patients with X-linked agammaglobulinemia have normal numbers of B-lymphocyte precursors, but very few of these go on to become B-lymphocytes. This is the underlying defect in X-linked agammaglobulinemia, a failure of B-lymphocyte precursors to mature into B-cells.
When a germ, such as bacteria, lands on a mucus membrane or enters the body, antibody molecules that recognize it stick to its surface. Antibody bound to the surface of a microorganism can have one or more effects that are beneficial. For example, some germs must attach to body cells before they can cause an infection and antibody prevents the germs from “sticking” to the cells.
Antibody on the surface of some microbes will also activate other body defenses (such as a group of blood proteins called serum complement) which can directly kill the bacteria or viruses. Finally, antibody coated bacteria are much easier for white blood cells (phagocytes) to ingest and kill than are bacteria which are not coated with antibody. All of these actions prevent germs from invading body tissues where they may cause serious infections.
People with agammaglobulinemia develop infections again and again. Common infections include ones that are due to bacteria such as Haemophilus influenzae, pneumococci (Streptococcus pneumoniae), and staphylococci. Common sites of infection include:
- Gastrointestinal tract
- Joints
- Lungs
- Skin
- Upper respiratory tract
X-linked agammaglobulinemia infants are born healthy, with no outward signs of impending illness, and do not develop recurrent infections until 6-8 months of age when maternal antibodies are no longer active.
Treatment aims to boost the immune system, which may be accomplished by administering immunoglobulins given through a vein (IVIG) or subcutaneously (SCIG).
Treatment also involves taking steps to reduce the number and severity of infections. Preventing bacterial infections is very important for people with X-linked agammaglobulinemia. Antibiotics are often needed to treat bacterial infections 4. Sudden infections in individuals with X-linked agammaglobulinemia are usually treated with antibiotics that are taken for at least twice as long as taken in healthy individuals.
A bone marrow transplant may be considered 8, 6, 9.
Figure 1. Plasma cells
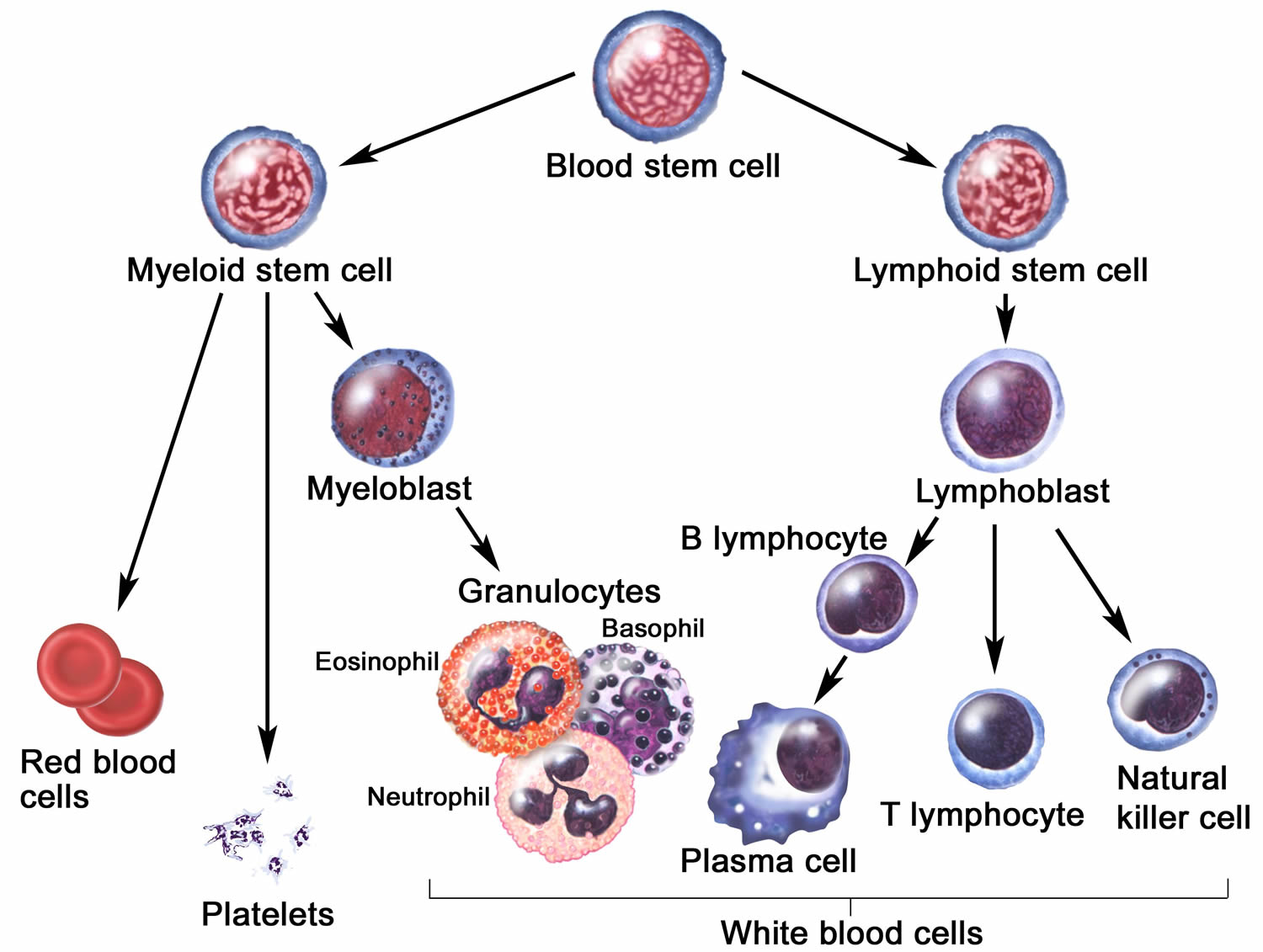
Figure 2. Cells of the immune system
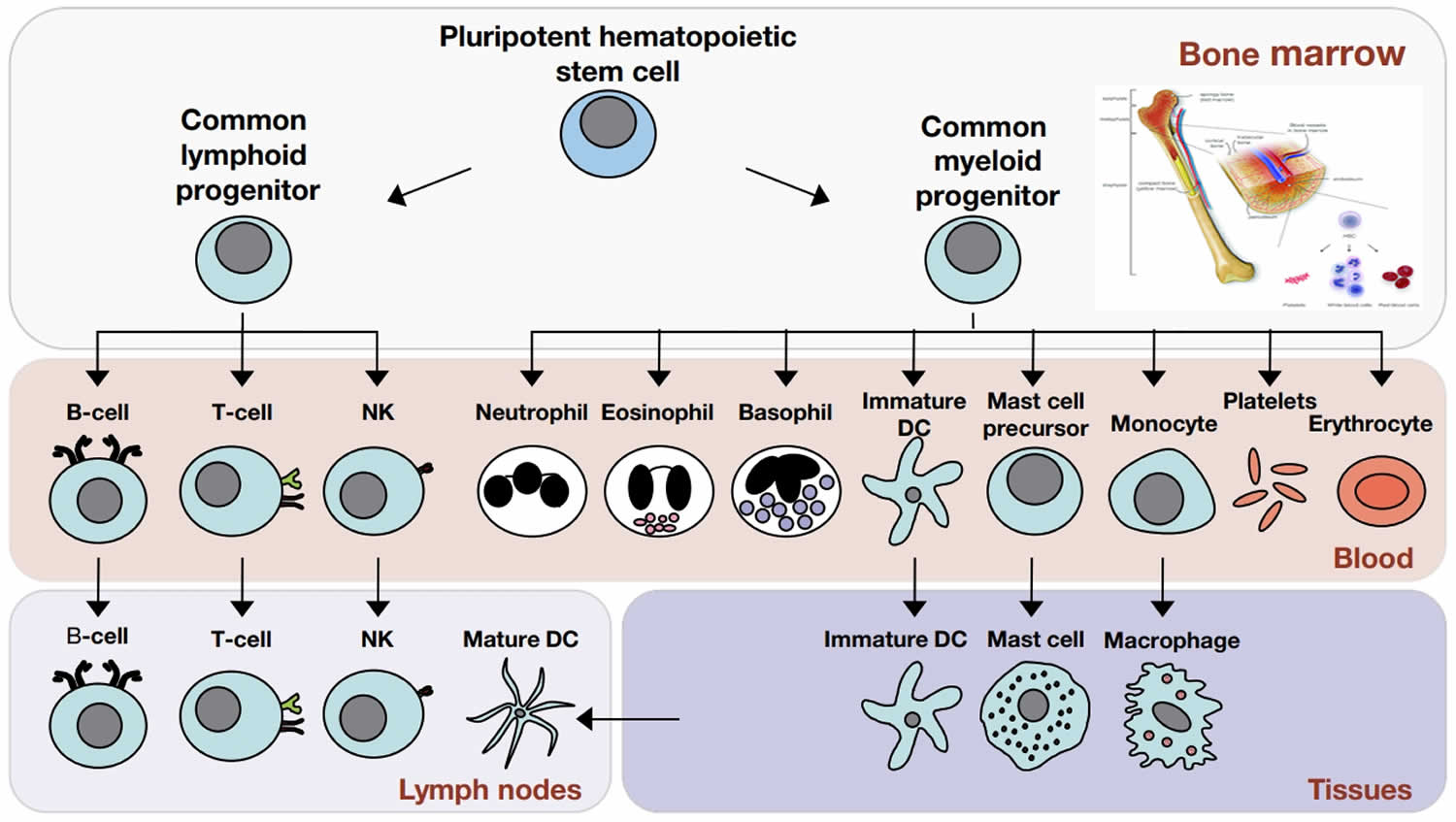
Footnote: The cells of the immune system originate in the bone marrow from pluripotent hematopoietic stem cells. Pluripotent hematopoietic stem cells give rise to a common lymphoid progenitor, which gives rise to all of the major lymphoid cell types (T‐cells, B‐cells, and Natural killer [NK] cells) or a common myeloid progenitor, which gives rise to all of the major myeloid cell types (neutrophils, eosinophils, basophils, dendritic cells (DCs), mast cells, and monocytes/macrophages) as well as the erythrocytes and megakaryocytes (which generate platelets).
X-linked agammaglobulinemia
X-linked agammaglobulinemia also called Bruton’s agammaglobulinemia, is an inherited immunodeficiency disorder that affects the immune system and occurs almost exclusively in males 10. People with X-linked agammaglobulinemia have very few mature B cells (B lymphocytes), which are specialized white blood cells that help protect the body against infection. B cells can mature into the cells that produce special proteins called antibodies or immunoglobulins. Antibodies attach to specific foreign particles and germs, marking them for destruction. Individuals with X-linked agammaglobulinemia are more susceptible to infections because their body makes very few antibodies 11. Infections can manifest in an infant as soon as the protective effect of maternal immunoglobulins wanes at around six months of age.
Children with X-linked agammaglobulinemia are usually healthy for the first 1 or 2 months of life because they are protected by antibodies acquired before birth from their mother. After this time, the maternal antibodies are cleared from the body, and the affected child begins to develop recurrent infections. In children with X-linked agammaglobulinemia, infections generally take longer to get better and then they come back again, even with antibiotic medications. The most common bacterial infections that occur in people with X-linked agammaglobulinemia are lung infections (pneumonia and bronchitis), ear infections (otitis), pink eye (conjunctivitis), and sinus infections (sinusitis). Infections that cause chronic diarrhea are also common. Recurrent infections can lead to organ damage. People with X-linked agammaglobulinemia can develop severe, life-threatening bacterial infections; however, affected individuals are not particularly vulnerable to infections caused by viruses. With treatment to replace antibodies, infections can usually be prevented, improving the quality of life for people with X-linked agammaglobulinemia.
X-linked agammaglobulinemia is a very rare disorder, primarily affecting males. Females may be carriers but have no clinical manifestations. Its prevalence in the United States is 1 in 379,000 live births and 1 in 190,000 male births.
X-linked agammaglobulinemia causes
Mutations in the Bruton tyrosine kinase (BTK) gene which is present on the long arm of the X-chromosome cause Bruton’s agammaglobulinemia or X-linked agammaglobulinemia 10. Bruton’s tyrosine kinase (BTK) gene provides instructions for making the BTK protein, which is important for the development of B cells and normal functioning of the immune system. Most mutations in the BTK gene prevent the production of any BTK protein. The absence of functional BTK protein blocks B cell development and leads to a lack of antibodies. Without antibodies, the immune system cannot properly respond to foreign invaders and prevent infection.
X-linked agammaglobulinemia inheritance pattern
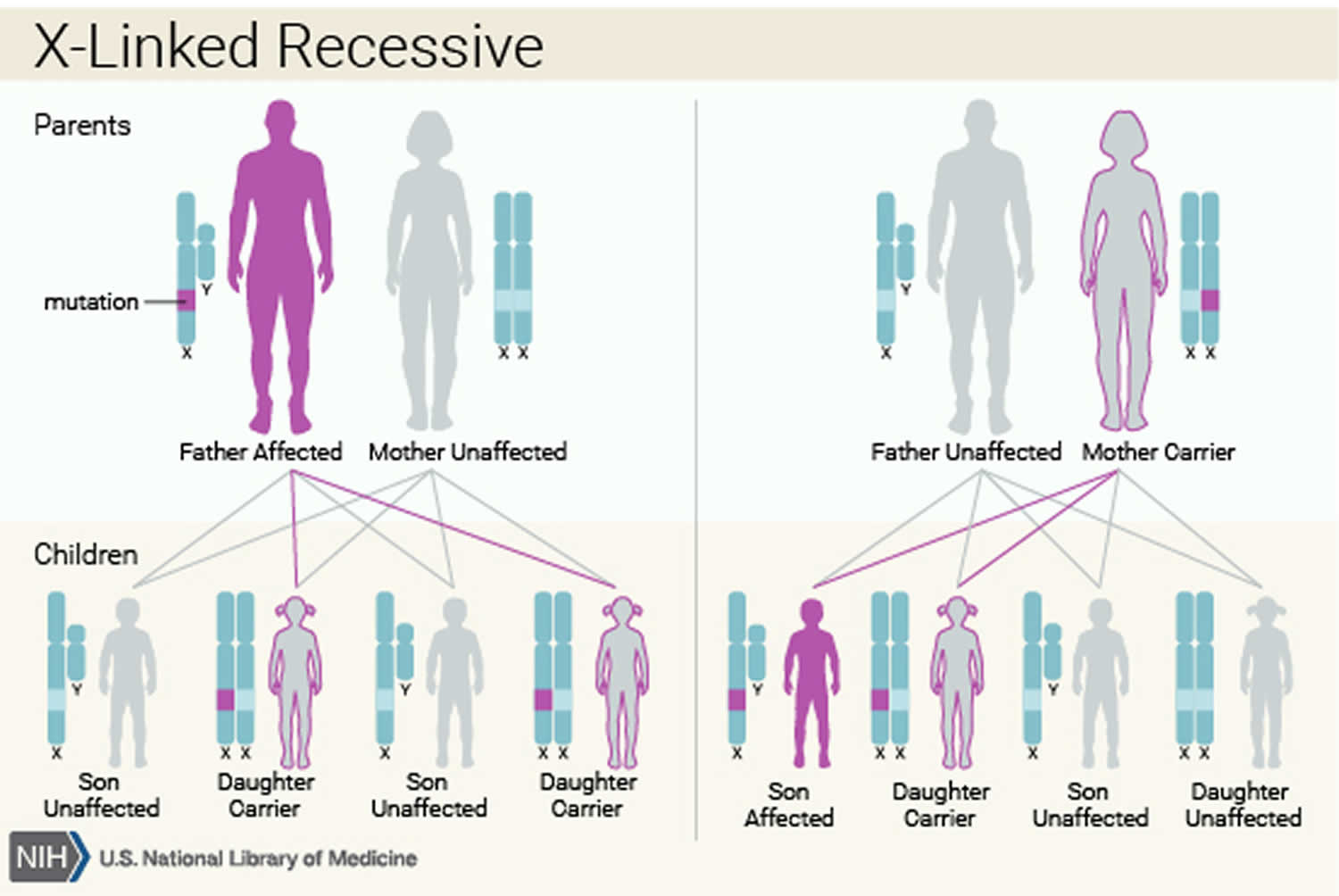
Bruton’s agammaglobulinemia is inherited in an X-linked recessive pattern. The Bruton tyrosine kinase (BTK) gene associated with Bruton’s agammaglobulinemia is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
About half of affected individuals do not have a family history of X-linked agammaglobulinemia. In most of these cases, the affected person’s mother is a carrier of one altered BTK gene. Carriers do not have the immune system abnormalities associated with X-linked agammaglobulinemia, but they can pass the altered gene to their children. In approximately 10% of the cases, the mother is not a carrier and the affected individual has a new mutation in the BTK gene. The resulting condition is known as autosomal recessive agammaglobulinemia (ARA) and describes the clinical phenotype seen in females with congenital agammaglobulinemia, which is comparable to X-linked agammaglobulinemia in males 12. The molecular defects responsible for autosomal recessive agammaglobulinemia (ARA) include mutations to the following genes: mu heavy chain (UGHN); gamma 5 (IGLL1); Igalpha (CD79A); Igbeta (CD79B); and BLNK 12. The wild-type proteins encoded by these genes have been shown to operate in collaboration with Bruton tyrosine kinase (BTK), to promote the transition from pro-B-cells to pre-B-cells in the bone marrow, during B cell maturation 13.
Figure 3. Bruton’s agammaglobulinemia X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
X-linked agammaglobulinemia symptoms
X-linked agammaglobulinemia symptoms include frequent episodes of:
- Bronchitis (airway infection)
- Chronic diarrhea
- Conjunctivitis (eye infection)
- Otitis media (middle ear infection)
- Pneumonia (lung infection)
- Sinusitis (sinus infection)
- Skin infections
- Upper respiratory tract infections
Infections typically appear in the first 4 years of life. Affected infants are usually healthy for the first few months of life until they begin to develop recurrent bacterial infections. The most common bacterial infections are ear infections, pneumonia, pink eye, sinus infections, and infections that cause chronic diarrhea. These bacterial infections can be severe and life-threatening. Most affected individuals are not vulnerable to infections caused by viruses. Infections can usually be prevented with proper treatment 7.
Other symptoms include:
- Bronchiectasis (a disease in which the small air sacs in the lungs become damaged and enlarged)
- Asthma without a known cause
The classical clinical presentation is of a young boy, who is aged between 3 months to early adulthood, with recurrent bacterial infections. During the third trimester of pregnancy, maternal IgG transfers to the fetus protecting the neonate from early infections. However, this effect wanes by six months of age, which is when the first signs of disease become apparent. The average age at diagnosis for patients with a family history of X-linked agammaglobulinemia is 2.6 years of age, while those without a positive family history are diagnosed only at 5.4 years of age on average.
However, it is important to note that infections are relatively common and knowing when to suspect X-linked agammaglobulinemia is key. A proper family history for frequent hospitalizations or deaths in boys at early ages can hint towards X-linked agammaglobulinemia well before symptoms of infections manifest. Even without a positive family history, signs hinting towards an evaluation for a primary immunodeficiency include recurrent infections, atypical infections, and unusually severe infections requiring hospitalization in a young male.
On physical examination, the absence of tonsils, chronic cough, chronic rhinitis, post-nasal drip, and clubbing can be seen. Growth charts may show evidence of failure to thrive especially in older children.
X-linked agammaglobulinemia diagnosis
The diagnosis of X-linked agammaglobulinemia comprises clinical suspicion by history, especially family history, and physical examination followed by laboratory and genetic tests 14.
Although a history of recurrent infections beginning after 6 months of age is very characteristic of X-linked agammaglobulinemia, a physical exam is also important. Lymphoid tissues are typically hypoplastic in X-linked agammaglobulinemia patients. The tonsils may be difficult to visualize, and the cervical/inguinal lymph nodes may not be palpable 12. The otoscopic exam may be used to detect signs of chronic damage, i.e., purulent otitis media, perforation of the tympanic membrane, or nasal discharge 12. Chest auscultation is performed to check for prolongation of expiration or inspiration, cough, any increase in respiratory effort, or stridor. Audible rhonchi, crackles, wheezing, and/or inspiratory squeaks suggest lung pathology and warrant further testing (e.g., lung function tests, CT scan, or biopsy) to rule out bronchiectasis. Similarly, the presence of abdominal distention justifies the need to perform an abdominal ultrasound to exclude hepatosplenomegaly 15.
Initial laboratory tests to be performed include a complete blood count with differential, quantitative serum immunoglobulin levels (IgG, IgA, and IgM), and serum specific antibody titers in response to immunization such as against tetanus or diphtheria. Serum levels of all immunoglobulins are low or nearly undetectable, and there is an absent antibody response to vaccinations. If these findings are suggestive of X-linked agammaglobulinemia, the next step is a lymphocyte phenotyping using flow cytometry which would document normal T-cell numbers but reduced to absent B-cell numbers.
Laboratory findings
- Marked reduction in all classes of serum immunoglobulins 16
- The serum IgG concentration is typically <200 mg/dL (2 g/L). Most but not all individuals with X-linked agammaglobulinemia do have some measurable serum IgG, usually between 100 and 200 mg/dL, and ~10% of individuals have serum concentration of IgG >200 mg/dL.
- The serum concentrations of IgM and IgA are typically <20 mg/dL. Particular attention should be given to serum IgM concentration. Although decreased serum concentration of IgG and IgA can be seen in children with a constitutional delay in immunoglobulin production, low serum IgM concentration is almost always associated with immunodeficiency.
- Markedly reduced numbers of B lymphocytes (CD 19+ cells) in the peripheral circulation (<1%) 17.
- Antibody titers to vaccine antigens. Individuals with X-linked agammaglobulinemia fail to make antibodies to vaccine antigens like tetanus, Haemophilus influenzae or Streptococcus pneumoniae.
- Severe neutropenia in ~10%-25% of individuals at the time of diagnosis, usually in association with pseudomonas or staphylococcal sepsis 18
These results point towards a probable diagnosis of X-linked agammaglobulinemia; however, to confirm the diagnosis, genetic testing to look for a mutation in BTK gene can be performed. A confirmed family history of X-linked agammaglobulinemia can serve as a surrogate for genetic testing.
X-linked agammaglobulinemia treatment
The first and foremost goal in patients with X-linked agammaglobulinemia should be avoidance of infections. This includes measures to prevent infections by frequent handwashing, maintaining good respiratory hygiene, and, if possible, drinking only treated water. Sudden infections in individuals with X-linked agammaglobulinemia are usually treated with antibiotics that are taken for at least twice as long as taken in healthy individuals.
The emergence of immunoglobulin replacement therapy has caused a paradigm shift in the management of patients with X-linked agammaglobulinemia. Observational studies have shown that intravenous immunoglobulin (IVIG) therapy has reduced the rate of infections and hospitalizations resulting in reduced morbidity and mortality. Some studies have shown a reduced incidence of bacterial infections from 0.4 to 0.06 per patient per year.
In the past, most people received immunoglobulin by intravenous (IV) infusion every two to four weeks. However, in the last few years, an increasing number of people have been receiving immunoglobulin by weekly subcutaneous injections (SCIG). The choice of whether to receive immunoglobulin intravenously or by subcutaneous injection may just depend on what is most convenient for the doctor and/or you. Sometimes, people with X-linked agammaglobulinemia have a reaction to gammaglobulin, which may include headaches, chills, backache, or nausea. These reactions are more likely to occur when they have a viral infection or when the brand of gammaglobulin has been changed.
However, there are a few drawbacks to the use of IVIG (intravenous immunoglobulin). Firstly, even though IVIG protects against most of the common pathogens, a few uncommon ones to which the donor pool has not been exposed are not protected against. Secondly, out of all types of immunoglobulins, only IgG is replaced while IgA and IgM are not and have their own unique functions. Finally, passive immunity through IVIG does not replace the rise in immunoglobulins seen in a healthy individual after exposure to foreign antigens 19.
In addition to IVIG, treatment with antibiotics for active infections should be done. In these individuals, recurrent pneumonia and other respiratory tract infections can lead to chronic lung problems such as bronchiectasis, chronic sinusitis, and chronic bronchitis. Thus, regular monitoring for these conditions using appropriate tests such as imaging studies is recommended as even subclinical infections can predispose individuals to develop them. Some treatment centers use chronic prophylactic antibiotics (continuous use of antibiotics) to prevent bacterial infections 20. Aggressive use of antibiotics lower the chance of chronic sinusitis and lung disease, which are common complications in individuals with X-linked agammaglobulinemia. Early diagnosis and treatment of bowel infections may decrease the risk of inflammatory bowel disease (IBD). Furthermore, children with X-linked agammaglobulinemia should not be given live viral vaccines. For example, they should be given inactivated polio vaccine (IPV) rather than the oral polio vaccine. The siblings of children with X-linked agammaglobulinemia should also be given inactivated polio vaccine (IPV) rather than oral polio vaccine in order to avoid infecting their affected sibling with live virus 20.
Lastly, despite the fact that immunoglobulin replacement is a safe and effective treatment strategy for these patients, Hematopoietic stem cell transplantation (HSCT) is an alternative. The risks of allogeneic hematopoietic stem cell transplantation such as rejection, graft-versus-host-disease make the treatment option less safe. The tedious procedure of hematopoietic stem cell transplantation and difficult procurement of a suitable donor are additional factors which make it a less popular treatment option. However, in some patients, especially in developing countries, the costs of regular IVIG or its inconvenience, and the unavailability of IVIG can lead to opting for this treatment modality.
What can I do if intravenous infusion of gammaglobulin (IVIG) is not available where I live?
Although gammaglobulin is the treatment of choice for X-linked agammaglobulinemia, there may be alternatives to intravenous gammaglobulin. In the last few years, an increasing number of people have been receiving gammaglobulin treatment by weekly subcutaneous injections 20. This is an injection (like a shot) given in the fatty layer of tissue just under the skin. Injection may also have the advantage of providing IgG levels that are relatively constant, compared with the highs and lows of levels that may occur with IV therapy. Another advantage is that injections may be given at home. Disadvantages of having injections may include discomfort where the injections are given, and the potential lack of medical supervision by doctors (because coming to the hospital for infusions may no longer be necessary). Fresh frozen plasma (FFP), used in blood transfusions, has been used in the past but carries the major risk of potential transmission of infectious diseases 21.
If you do not have access to gammaglobulin or other treatments where you live, it is recommended that you contact the various support groups for X-linked agammaglobulinemia. You can see the contact information for these support groups below. These groups may be able to help you either locate a physician or treatment center that is accessible, or provide you with information about how to obtain gammaglobulin or other treatments for X-linked agammaglobulinemia.
Organizations Supporting X-linked agammaglobulinemia
- Immune Deficiencies Foundation Australia
- PO Box 969
- Penrith NSW 2751
- Australia
- Telephone: 800-100-198
- Website: https://www.idfa.org.au
- International Patient Organization for Primary Immunodeficiencies (IPOPI)
- Rock Bottom, Trerieve
- Downderry
- PL11 3LY
- United Kingdom
- Telephone: 44-01503-250-668/961
Website: https://ipopi.org
- Jeffrey Modell Foundation (JMF)
- 780 Third Ave
- New York, NY 10017
- Fax: 212-764-4180
Website: http://www.info4pi.org - JMF is a global patient organization devoted to early and precise diagnosis, meaningful treatments, and ultimately, cures – through clinical and basic research, physician education, patient support, advocacy, public awareness and newborn screening.
Organizations Providing General Support
- Immune Deficiency Foundation
- 110 West Road, Suite 300
- Towson, MD 21204
- Toll-free: 1-800-296-4433
- Fax: +1-410-321-9165
- Website: https://primaryimmune.org/
Agammaglobulinemia causes
X-linked agammaglobulinemia (B-lymphocyte defect) is inherited as an X-linked recessive genetic trait. The abnormal gene, named BTK, has been mapped to gene locus Xq21.3-q22. A different mutation in the BTK gene causes X-linked agammaglobulinemia with growth hormone deficiency. The genetic cause of ARAG is much more complex involving other genes that have been mapped to loci on different chromosomes: 22q11.21, 14q32.33, and 9q34.13. The genes at three sites are known as IGLL1, IGHM, and LCRR8 respectively.
Chromosomes are located in the nucleus of human cells and carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes numbered from 1 through 22 are called autosomes and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 21q11.21” refers to band 11.21 on the long arm of chromosome 21. Similarly 14q32.33 refers to band 32.33 on the long arm of chromosome 14, and 9q34.13 refers to band 34.13 on the long arm of chromosome 9. The site described as Xq21.3-q22 refers to a region on the long arm of the X chromosome between bands 21.3 and 22. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother.
X-linked genetic disorders are conditions caused by an abnormal gene on the X chromosome and occur mostly in males. Females that have a disease gene present on one of their X chromosomes are carriers for that disorder. Carrier females usually do not display symptoms because females have two X chromosomes and one is inactivated so that the genes on that chromosome are nonfunctioning. It is usually the X chromosome with the abnormal gene that is inactivated. Males have one X chromosome that is inherited from their mother and if a male inherits an X chromosome that contains a disease gene he will develop the disease. Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son.
Males with X-linked disorders pass the disease gene to all of their daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male offspring.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Autosomal recessive agammaglobulinemia (ARA)
Autosomal recessive agammaglobulinemia (ARA) has been reported to be due to genes that affect B cell development. The following genes (and their official gene symbol) have been reported to cause ARA:
- mu heavy chain (IGHM)
- Lambda 5 (IGLL1)
- Ig Alpha (CD79A)
- Ig Beta (CD79B)
- BLNK (BLNK)
- PIK3R1 and PIK3CD
- SLC39A7
All of these genes code for proteins that work with BTK to support the maturation of pro-B-cells into pre-B-cells. Patients with mutations in any of these genes have clinical and laboratory findings that are very similar to those seen in patients with mutations in BTK (X-linked agammaglobulinemia). Autosomal recessive agammaglobulinemia (ARA) tend to be more severe and present earlier than cases of X-linked agammaglobulinemia. Some patients with early B cell developmental defects have yet to have a genetic cause identified, although with advances in diagnostic and genetic testing, this is improving 16.
Mu Heavy Chain Deficiency
Several cases of mu heavy chain deficiency have been reported, resulting in a clinical phenotype similar to patients with BTK mutation. The patients with mu heavy chain deletion, however, tend to present at younger ages (mean age: 11 months versus 35 months in BTK mutation) and with more severe infections. Specifically, these patients present more frequently with enteroviral infections and sepsis secondary to Pseudomonas 22. Mu heavy chain deletion is responsible as the cause for approximately 5% of agammaglobulinemia 13. Thirty percent of the mutations of the mu heavy chain gene have been found to affect the mu heavy constant region. Thirty percent of patients with mu heavy chain mutation are also found to have neutropenia 22. The majority of mutations result from large gene deletions, while a minority are caused by point mutations. While mutations in BTK may cause a partial interruption in B cell development that can lead to a “leaky” phenotype, mutations in the mu heavy chain gene results in a complete block, which inhibits B-cell maturation from the pro- to pre- B cell stage 13. There has been one case of a patient with both gene deletion and missense mutation resulting in a compound heterozygous alteration of the mu heavy chain gene. The patient presented with severe infection at 7 months of age (peritonitis, pseudomonas bacteremia, E. coli urinary tract infection, neutropenia, and hypogammaglobulinemia) 13.
Lambda 5 Deficiency
The IGLL1 gene encodes the lamba-5 and the VpreB chains, which are both components of the pre-B cell receptor complex. Lambda 5 and the VpreB chain are together known as the surrogate light chain. Few cases of IGLL1 gene mutations have been reported. One published case consisted of a 2-month-old female who presented with bilateral upper lobe pneumonia. Immunodeficiency evaluation showed absent B cells with undetectable IgA and IgM and an IgG level of 329 mg/dL. Chromosomal microarray identified a homozygous missense mutation in exon 3 of the IGLL1 gene 23. Another patient presented with recurrent otitis media at 2 months old and immunodeficiency evaluation revealed undetectable CD19 + B cells as well as hypogammaglobulinemia. This male patient was found to have mutations in exon 1 and exon 5 of lambda5/14.1 gene. Analysis of peripheral lymphocytes in this patient demonstrated 0.06% expressed CD19 with low intensity expression of surface IgM and CD38 dim, which is seen in normal B cells. Bone marrow analysis was consistent with an arrest in pro-B to pre-B cell differentiation with 85% of the few CD19 + cells expressing CD34 24.
Ig Alpha Deficiency
Ig alpha, along with Ig beta, is a transmembrane protein heterodimer responsible for signal transduction in the B cell receptor complex 25. Ig alpha and Ig beta form a disulfide bond complex and function to assist the mu heavy chain to the surface of the cell 26. A 2-year-old female patient from Turkey with agammaglobulinemia has been reported with a homozygous splice defect in exon 3 of the Ig alpha gene. Analysis of peripheral lymphocytes showed < 0.01% CD19 + B cells and bone marrow showed more than 75% of CD19 + expressed CD34, which indicated a block in differentiation from pro-B to pre-B cell 25. There have been 5 patients with mutations in Ig alpha reported, and all mutations have been reported to involve a region upstream of the transmembrane domain 27. In another case, a 6-month-old female, who presented with human herpes virus 8 (HHV8) and John Cunningham (JC) viral encephalitis, with immune evaluation pertinent for hypogammaglobulinemia and absent peripheral B cells, was found to have a novel homozygous mutation in CD79a resulting in a premature stop codon 27. A recent publication describes a 16-year-old male with agammaglobulinemia secondary to mutation in CD79a, diagnosed at 2 years of age, who developed Campylobacter jejuni spondylodiscitis, although he was on immunoglobulin replacement therapy and prophylactic antibiotics. This patient received immunoglobulin therapy with IgM supplementation, which was found to aid in complement activation and increase bacterial killing 28.
Ig Beta Deficiency
The first patient reported with Ig beta deficiency was a 15-year-old female with a long-standing history of recurrent bronchitis, which began at 5 months of age, and two episodes of pneumonia. She was diagnosed with hypogammaglobulinemia at 15 months of age 26. Although peripheral CD19 + B cells were very low, they were not completely absent (0.08%). This patient was identified as having a homozygous mutation of codon 137 in the Ig beta gene, neighboring the amino acid residue required for disulfide bonding between the Ig alpha and Ig beta proteins 26. The mutation in this patient resulted in a “leaky” phenotype, as mutant Ig beta in vitro was found to form some disulfide bonds with Ig alpha and deliver the mu heavy chain to the cell surface. This was demonstrated by the finding of a small amount of peripheral B cells in the circulation being IgM dim 26.
A 20-year-old Italian male with history of pneumonia, Salmonella enteritis, and chronic sinusitis was found to have a mutation in exon 3 of the Ig beta gene resulting in complete lack of Ig beta expression. Immunologic phenotype consisted of severe hypogammaglobulinemia, for which he had been on replacement since the age of 8 months and absent peripheral B cells 29.
In another report, a new, homozygous, null mutation in codon 13 of the Ig beta gene was found in a 15-month-old female patient with a history of recurrent upper respiratory infections, fever, ecthyma, and severe neutropenia (10 cells/mm³). Immune evaluation was positive for absence of peripheral CD19 + cells and hypogammaglobulinemia (IgG < 35 mg/dL). Parents were consanguineous and both harbored the same heterozygous mutation 30.
BLNK Deficiency
BLNK, a scaffolding protein that is phosphorylated after B cell receptor cross-linking, plays a role in B cell development. In one published report, a patient with recurrent otitis beginning at 8 months of age, two episodes of pneumonia, undetectable serum immunoglobulins and < 1% peripheral B cells, were found to have BLNK deficiency. The patient demonstrated absence of pre-B cells indicating an important role for BLNK in the pro- to pre-B cell transition [29]. It is also suspected that the older brother of this patient, who passed away from neutropenia and pseudomonal sepsis, likely had BLNK deficiency 31. There have been 5 patients with BLNK deficiency described as of this paper’s writing, although some have been cited as non-published observations 32. In one published report, a patient with agammaglobulinemia, no circulating B cells, and homozygous BLNK mutation was found to have a block in the pre-BI to pre-BII stage of B cell maturation 32.
PIK3R1 and PIK3CD Mutation
The PI3K family of kinases is involved in signal transduction, ultimately resulting in production of PIP3 and subsequent membrane recruitment and activation of AKT 33. The PI3K kinases play several other roles in the cell including signaling for growth, metabolism, survival, and more. There have been several cases of malignancies and immune dysregulation and few cases of immunodeficiency reported with overactivation or underactivation of the PI3K pathway 34. Specifically, underactivation of this pathway leads to a primarily humoral defect with near absent B cells, hypogammaglobulinemia, recurrent sinopulmonary infections, colitis, and/or autoimmunity, while T cell numbers are normal.
Class 1A PI3Ks play an important role in immunology. The PIK3R1 gene encodes p85α, p55α, and p50α, which are classified as regulatory subunits. One regulatory subunit will form a heterodimer with a catalytic subunit p110a, p110b, or PI3K delta. The PI3K delta enzyme complex is composed of p85 alpha and p110 delta, and p110 delta is encoded by the PIK3CD gene 34.
There have been 3 cases of agammaglobulinemia reported with PIK3R1 gene mutation all reported with absent B cells, elevated T cells, and agammaglobulinemia 33. The first patient described, diagnosed at 19 years of age, presented at 3.5 months old with neutropenia, interstitial pneumonia, and gastroenteritis. Later in life, she developed erythema nodosum, inflammatory colitis, and recurrent bacteremia secondary to campylobacter. Initial immune evaluation revealed hypogammaglobulinemia and absent B cells (< 1% CD19 + B cells) in the periphery and bone marrow. Analysis of bone marrow showed < 0.1% of pro-B cells, despite normal numbers of early B cell precursors, indicating near absence of cells arising from B cell lineage. The patient was found to have absent p85alpha due to a premature stop codon in the PIK3R1 gene 35. In murine studies, lack of p85 alpha has a phenotype consisting of decreased B cells, increased IL12 production from dendritic cells, and hyper-responsiveness to insulin 35.
In another published report of premature stop codon in the PIK3R gene, a 10-month-old female displayed mucosal bleeding secondary to thrombocytopenia, neutropenia, pan-hypogammaglobulinemia, and absence of B cells. Her clinical phenotype consisted of recurrent upper respiratory infections, oral thrush, and prolonged diarrhea on two occasions. After a male sibling was born, he was clinically diagnosed as having autosomal recessive agammaglobulinemia and found to have absent B cells on flow cytometry and neutropenia. Genetic testing was only obtained in the female patient 33.
There are an additional 6 cases of homozygous PIK3CD gene mutation resulting in LOF of PIK3 delta enzyme complex. These patients also displayed hypogammaglobulinemia or agammaglobulinemia as well as decreased B cells 35.
SLC39A7 Mutation
In a study by Anzilotti et. al 36, 6 individuals were identified as having hypomorphic mutations inSLC39A7, a gene encoding the protein ZIP7, which is responsible for transportation of zinc from the endoplasmic reticulum to the cytoplasm. These patients had absent B cells, agammaglobulinemia, and early infections. A subset of patients had blistering dermatoses, failure to thrive, and thrombocytopenia. These two patients with severe phenotype were cured after hematopoietic stem cell transplant. Analysis of B cell development found an arrest from the late pre-B stage to the immature B stage. These abnormal immature cells had intact expression of the genes for Rag and IL7R, but not BAFFR and CD20, which are usually expressed at the immature B cell stage 36.
X-linked agammaglobulinemia with growth hormone deficiency
Only about 10 persons in 5 or 6 families have been diagnosed with X-linked agammaglobulinemia with growth hormone deficiency. The boys in these families have reduced or undetectable numbers of B-lymphocytes. Clinicians and geneticists speculate that a second mutation in the BTK gene, very close to the mutation in this gene that causes X-linked agammaglobulinemia, is responsible for the combination of agammaglobulinemia and very short stature.
Agammaglobulinemia symptoms
The major symptoms of agammaglobulinemia are serial bacterial infections resulting from failures in specific immune responses because of defects in B-lymphocytes. These lymphocytes govern the production of antibodies. Males with X-linked primary agammaglobulinemia usually begin to show signs of such infections only late in the first year of life, after the IgG antibodies from the mother have been depleted.
Infections by almost any of the enterovirus family and the poliomyelitis virus can result in unusually severe illness in children with agammaglobulinemia. Echovirus infection can cause a group of symptoms that closely resembles dermatomyositis. These symptoms may include muscle weakness, often in the hip and shoulder areas, and difficulty swallowing. Areas of patchy, reddish skin may appear around the eyes, knuckles and elbows and occasionally on the knees and ankles. (For more information on this disorder, choose “dermatomyositis” as your search term in the Rare Disease Database.)
Infections caused by mycoplasma bacteria can lead to severe arthritis including joint swelling and pain, in children with primary agammaglobulinemia. Hemophilus influenzae is the most common mucous- producing infection (pyogenic) that occurs in people with X-linked agammaglobulinemia. Children may also have repeated infections with pneumococci, streptococci, and staphylococci bacteria, and infrequently pseudomonas infections.
Males with X-linked form of agammaglobulinemia have very low levels of IgA, IgG, and IgM antibodies circulating in their blood. Specialized white blood cells (neutrophils) are impaired in their ability to destroy bacteria, viruses, or other invading organisms (microbes). This occurs because neutrophils require antibodies from the immune system to begin to destroy invading bacteria (opsonization). The levels of circulating neutrophils in children with agammaglobulinemia may be persistently low, or may wax and wane (cyclic, transient neutropenia) in people with these disorders. The number of B-lymphocytes in children with X-linked agammaglobulinemia is less than one one-hundredth of the normal number.
Agammaglobulinemia complications
X-linked agammaglobulinemia patients are at risk for complications of the disease itself as well as secondary to treatment.
Complications associated with X-linked agammaglobulinemia usually arise from infections, especially those that have become recurrent. Susceptible individuals can become chronically ill and suffer organ damage. For example, repeated episodes of acute pneumonia may culminate in chronic lung disease and lead to bronchiectasis, which has the potential to reduce life expectancy. Bronchiectasis and chronic lung disease is a known complication of X-linked agammaglobulinemia which may progress to require lung transplantation. One series of 6 patients with X-linked agammaglobulinemia and end-stage bronchiectasis received lung transplantation in a single center. The results showed 4 out of the 6 patients developed repeated pulmonary sepsis and chronic lung allograft dysfunction in the long term 37. This demonstrates the great risks associated with lung transplant in these patients.
The likelihood that chronic infections will evolve into serious, life-threatening conditions increases with the length of delay in diagnosis. The later treatment begins, the more difficult it is to eradicate the causative organisms and prevent the systemic spread of infection to joints and vital organs 38.
Complications associated with chronic infections are, by far, the most common problem confronted by patients with X-linked agammaglobulinemia. Other, less common complications include the increased risk of developing malignancy, inflammatory conditions, or autoimmune disease. Literature suggests an increased risk of lymphoma, adenocarcinoma of gastrointestinal origin especially stomach and colon.
Complications associated with treatment are mainly those which arise from immunoglobulin replacement therapy. The replacement of immunoglobulin is a lifelong requirement for individuals with X-linked agammaglobulinemia. Regular immunoglobulin replacement therapy is known to increase life expectancy, lower the rate and severity of infections, decrease the number of hospitalizations, and reduce the need for antibiotics 12. Unfortunately, it also correlates with side effects and must, therefore, have careful monitoring. Clinicians can overcome complications associated with immunoglobulin replacement therapy by changing (1) the time interval between immunoglobulin infusions, (2) the route of administration, (3) the rate of administration, or (4) the product used for replacement 39.
Immunoglobulins used in therapy derive from the pooled serum of thousands of healthy donors who have had screening for transmissible diseases. The donor serum is processed to retain maximal amounts of IgG and only trace amounts of IgA and IgM, thus reducing the likelihood of triggering anaphylactic reactions to IgA or developing kidney damage from complex formation induced by IgM.
The length of time between infusions varies according to the route of administration. The two most common routes of administering immunoglobulins are intravenous (IVIG) and subcutaneous (SCIG). The interval between IVIG infusions can be as long as a month, while the maximum interval between SCIG infusions is usually no more than a week 40.
The rate of delivery and hence, the final immunoglobulin concentration obtained is another important factor. The higher the rate of delivery, the greater the likelihood of complications. In situations where higher immunoglobulin levels are needed to combat a recalcitrant infection, the clinician must balance the decision to increase the infusion rate against the side effect profile.
Differences in the exact composition of the product used for immunoglobulin replacement also contributes to tolerability. The purity and constituents (i.e., additives used to reduce the aggregate formation and enhance delivery) vary according to the manufacturer, and the reactions elicited vary in different individuals.
Reactions to immunoglobulin infusion therapy can be immediate or latent. The most common immediate side effect is a headache. Headache occurs within minutes and is managed using over-the-counter medications, although it can often be avoided altogether by simply slowing the rate of infusion.
Other common infusion reactions include nausea, malaise, fever/chills, chest tightness, and migraines. Infusion reactions can also categorize according to the system affected. Cardiovascular reactions include tachycardia, palpitations, flushing, and hypotension; neurologic reactions include anxiety, nervousness, irritability, tremor, fainting and seizures; respiratory system reactions include cough, chest tightness, dyspnea, wheezing, and bronchospasm; dermatologic reactions include erythema, urticaria, and eczema; musculoskeletal reactions include low backache, arthralgia, and myalgia; and gastrointestinal reactions include abdominal pain, distention, and liver dysfunction.
More serious complications can also arise. These are much less common and occur after a period of latency. Examples include aseptic meningitis, anaphylactic reactions, Stevens-Johnson syndrome, erythema multiforme, acute renal failure, acute respiratory distress syndrome, transfusion-associated lung injury, deep vein thrombosis, pulmonary edema, pulmonary embolism, cardiac arrest, shock, coma 41.
Agammaglobulinemia diagnosis
Agammaglobulinemia is confirmed by blood tests that measure levels of immunoglobulins.
Tests include:
- Flow cytometry to measure circulating B lymphocytes
- Immunoelectrophoresis – serum
- Quantitative immunoglobulins – IgG, IgA, IgM (usually measured by nephelometry)
Agammaglobulinemia treatment
The administration of intravenous gammaglobulin replacement therapy is a standard treatment for agammaglobulinemia. Intravenous gammaglobulin or subcutaneous immunoglobulin replacement therapy is used to treat agammaglobulinemias and common variable immunodeficiency. In the USA, immunoglobulin products are composed of donated plasma from 10 to 20,000 donors, which is then tested for pathogens and processed to eliminate any potential viruses and prions 42. With intravenous immunoglobulin replacement, the patient is infused every 3–4 weeks. Subcutaneous immunoglobulin infusions can be offered daily to weekly or biweekly to monthly depending on the product 43. When administered intravenously, there is a higher IgG level peak on day one compared to the peaks obtained with subcutaneous (and facilitated subcutaneous) infusions. Studies have shown a rate of 3–4 infections per patient-year, while on immunoglobulin replacement therapy 42. There is no set dose required or ideal trough for prevention of infection. It has been shown that different patients do clinically well with different trough levels, and therefore, this should be individualized based on patient’s clinical response 42.
Antibiotics are prescribed for people with agammaglobulinemia when bacterial infections occur. If a patient develops an infection, they require prompt treatment with antibiotics for a longer course, in some patients double the time, than a non-affected patient 43. Some patients are treated with antibiotics as a preventive measure (prophylactically), although there are no current published guidelines. Prophylactic antibiotics may be considered, particularly, if patients continue to have infections despite immunoglobulin replacement therapy at adequate doses 44. All people who are immunodeficient should be protected as much as possible from exposure to infectious diseases. Corticosteroids or any drug that depresses the immune system (immunosuppressant drugs) should be avoided as much as possible, as well as physical activities such as rough contact sports that risk damage to the spleen.
Monitoring for disease complications includes consideration of regular chest X-rays and sinus imaging 43. Ideally, patient should receive a baseline pulmonary function test early on in diagnosis with periodic tests to follow lung function over time 43.
In people with immunodeficiency with elevated IgM, there is a tendency to bleed excessively associated with abnormally low levels of circulating platelets in the blood (thrombocytopenia). This may complicate any surgical procedure.
Genetic counseling is recommended for people with agammaglobulinemias and their families. Other treatment is symptomatic and supportive.
Stem cell transplant
Stem cell transplant has been proposed in the past as a possible cure for X-linked agammaglobulinemia 8, 6, 9; however, there are many possible risks and complications associated with allogeneic stem cell transplant, which has limited this as an option, due to current accepted treatments available 44. There have been few cases reported of X-linked agammaglobulinemia patients receiving stem cell transplant with different conditioning regimens with varying results. The first 6 cases of X-linked agammaglobulinemia patients treated with allogeneic stem cell transplant, without utilization of a pre-conditioning regimen, did not result in engraftment in any of the 6 patients 45.
One case has been reported of a 13-year-old male with X-linked agammaglobulinemia and newly diagnosed acute myeloid leukemia (AML) who underwent allogeneic stem cell transplant with myeloablative conditioning and total body irradiation as treatment for his AML. One year after transplant, the patient had BTK gene analysis, which no longer showed his prior BTK mutation, and subsequent normal BTK expression in monocytes and B cells 9.
Another X-linked agammaglobulinemia patient, at 25 years of age, developed pre-B acute lymphocytic leukemia (ALL) with dominant negative TP53 mutation. As part of his treatment for ALL, he received myeloablative conditioning therapy, followed by allogeneic stem cell transplant. At 14 months post-transplant, the patient had normal circulating B cells; normal levels of immunoglobulins A, G, and M; and appropriate antibody response to vaccination with Prevnar-13 46.
In another report, a 28 year-old-male with X-linked agammaglobulinemia and history of severe potentially fatal infections, despite IVIG replacement therapy, received allogeneic stem cell transplant with reduced intensity conditioning with successful immune reconstitution of CD19 + B cells, IgA and IgM, and increasing IgG levels after IVIG discontinuation 6.
Agammaglobulinemia prognosis
Before regular immunoglobulin replacement therapy, most X-linked agammaglobulinemia died before the age of 10 from complications of lung disease, sepsis, or meningitis. Although chronic lung disease persists as an important factor in the mortality in patients with X-linked agammaglobulinemia, life expectancy extends into adulthood 47. Affected individuals who get diagnosed early (i.e., before five years of age) who receive regular immunoglobulin replacement therapy and are prescribed antibiotics treat or prevent infections can be expected to have a normal quality of life and live beyond the age of 40 48.
Most patients with X-linked agammaglobulinemia or autosomal recessive agammaglobulinemia (ARA) do not need to be isolated or limited in their activities. Active participation in team sports should be encouraged. Infections may require some extra attention from time to time, but children with agammaglobulinemia can participate in all regular school and extracurricular activities, and when they become adults can have productive careers and families. A full active lifestyle is to be encouraged and expected.
Agammaglobulinemia life expectancy
A large multicenter study from different countries reported survival rates at 20 years of age as low as 22%, while other countries reported survival above 70% 44. The most common cause of death reported was chronic or acute lung disease in 41% of patients. Thirty-four percent of patients had more than 24 months delay in diagnosis, and 39% of centers reported lack of genetic studies as a diagnostic challenge. A common concern reported among centers was lack of access to immunoglobulin replacement products presenting a challenge to managing patients with X-linked agammaglobulinemia 44. Additionally, many different complications were reported which highlights the heterogeneity of inflammatory, infectious, and autoimmune complications, which can be seen in patients with X-linked agammaglobulinemia. Overall, the majority of centers reported a good survival rate 49.
A study of 201 patients in a US registry found the most common cause of death in this cohort to be disseminated enteroviral infection (35%), followed by chronic lung disease (25%), then hepatitis (15%) 47. Additionally, there were 2 deaths secondary to treatments including one from hepatitis C, which was acquired from IVIG contaminated with hepatitis C virus, and another death attributed to stem cell transplant 47.
A recent study of 168 X-linked agammaglobulinemia patients in the Italian Primary Immunodeficiency Network Registry (IPINet) found a survival rate of 92.7% at 43 years of age, significantly reduced compared to the survival rate of healthy controls 50. Chronic lung disease was diagnosed in 51.8% of patients, and 13.1% of these patients had evidence of chronic lung disease at the time of diagnosis. The rate of malignancy diagnosed at follow up was 3.7%. The survival rate was further reduced in patients with a diagnosis of chronic lung disease (90.5% survival rate) 50. A study of 36 X-linked agammaglobulinemia patients in India, over the course of two decades, found the mean survival to be 11.4 years of age with a 20% mortality rate 51.
References- Agammaglobulinemia. https://rarediseases.org/rare-diseases/agammaglobulinemia/
- X-linked agammaglobulinemia. https://rarediseases.info.nih.gov/diseases/1033/x-linked-agammaglobulinemia
- Agammaglobulinemia: X-Linked and Autosomal Recessive. https://primaryimmune.org/about-primary-immunodeficiencies/specific-disease-types/agammaglobulinemia-x-linked-autosomal-recessive
- Agammaglobulinemia. https://emedicine.medscape.com/article/884942-overview
- Abolhassani H, Vitali M, Lougaris V, Giliani S, Parvaneh N, Parvaneh L, Mirminachi B, Cheraghi T, Khazaei H, Mahdaviani SA, Kiaei F, Tavakolinia N, Mohammadi J, Negahdari B, Rezaei N, Hammarstrom L, Plebani A, Aghamohammadi A. Cohort of Iranian Patients with Congenital Agammaglobulinemia: Mutation Analysis and Novel Gene Defects. Expert Rev Clin Immunol. 2016;12(4):479-86. doi: 10.1586/1744666X.2016.1139451
- Ikegame, K., Imai, K., Yamashita, M., Hoshino, A., Kanegane, H., Morio, T., Kaida, K., Inoue, T., Soma, T., Tamaki, H., Okada, M., & Ogawa, H. (2016). Allogeneic stem cell transplantation for X-linked agammaglobulinemia using reduced intensity conditioning as a model of the reconstitution of humoral immunity. Journal of hematology & oncology, 9, 9. https://doi.org/10.1186/s13045-016-0240-y
- X-linked agammaglobulinemia. https://medlineplus.gov/genetics/condition/x-linked-agammaglobulinemia
- Shillitoe B, Gennery A. X-Linked Agammaglobulinaemia: Outcomes in the modern era. Clin Immunol. 2017 Oct;183:54-62. doi: 10.1016/j.clim.2017.07.008
- Abu-Arja, R. F., Chernin, L. R., Abusin, G., Auletta, J., Cabral, L., Egler, R., Ochs, H. D., Torgerson, T. R., Lopez-Guisa, J., Hostoffer, R. W., Tcheurekdjian, H., & Cooke, K. R. (2015). Successful hematopoietic cell transplantation in a patient with X-linked agammaglobulinemia and acute myeloid leukemia. Pediatric blood & cancer, 62(9), 1674–1676. https://doi.org/10.1002/pbc.25554
- Taneja A, Chhabra A. Bruton Agammaglobulinemia. [Updated 2019 Mar 13]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK448170
- Sanford E, Farnaes L, Batalov S, Bainbridge M, Laubach S, Worthen HM, Tokita M, Kingsmore SF, Bradley J. Concomitant diagnosis of immune deficiency and Pseudomonas sepsis in a 19 month old with ecthyma gangrenosum by host whole-genome sequencing. Cold Spring Harb Mol Case Stud. 2018 Dec;4(6).
- Lackey AE, Ahmad F. X-linked Agammaglobulinemia. [Updated 2021 Jul 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549865
- Silva P, Justicia A, Regueiro A, Fariña S, Couselo JM, Loidi L. Autosomal recessive agammaglobulinemia due to defect in μ heavy chain caused by a novel mutation in the IGHM gene. Genes Immun. 2017 Sep;18(3):197-199. doi: 10.1038/gene.2017.14
- Argyropoulos KV, Palomba ML. First-Generation and Second-Generation Bruton Tyrosine Kinase Inhibitors in Waldenström Macroglobulinemia. Hematol. Oncol. Clin. North Am. 2018 Oct;32(5):853-864.
- Suri D, Rawat A, Singh S. X-linked Agammaglobulinemia. Indian J Pediatr. 2016 Apr;83(4):331-7. doi: 10.1007/s12098-015-2024-8
- Conley ME, Broides A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T, Shurtleff SA. Genetic analysis of patients with defects in early B-cell development. Immunol Rev. 2005 Feb;203:216-34. doi: 10.1111/j.0105-2896.2005.00233.x
- Nonoyama S, Tsukada S, Yamadori T, Miyawaki T, Jin YZ, Watanabe C, Morio T, Yata J, Ochs HD. Functional analysis of peripheral blood B cells in patients with X-linked agammaglobulinemia. J Immunol. 1998 Oct 15;161(8):3925-9.
- Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr. 2002 Oct;141(4):566-71. doi: 10.1067/mpd.2002.127711
- Tillman BF, Pauff JM, Satyanarayana G, Talbott M, Warner JL. Systematic review of infectious events with the Bruton tyrosine kinase inhibitor ibrutinib in the treatment of hematologic malignancies. Eur. J. Haematol. 2018 Apr;100(4):325-334.
- Smith CIE, Berglöf A. X-Linked Agammaglobulinemia. 2001 Apr 5 [Updated 2016 Aug 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1453
- Pediatric Bruton Agammaglobulinemia. https://emedicine.medscape.com/article/885625-overview
- Smith T, Cunningham-Rundles C (2019) Primary B-cell immunodeficiencies. Hum Immunol 80(6):351–362. 10.1016/j.humimm.2018.10.015
- Gemayel KT, Litman GW, Sriaroon P. Autosomal recessive agammaglobulinemia associated with an IGLL1 gene missense mutation. Ann Allergy Asthma Immunol. 2016 Oct;117(4):439-441. doi: 10.1016/j.anai.2016.07.038
- Minegishi, Y., Coustan-Smith, E., Wang, Y. H., Cooper, M. D., Campana, D., & Conley, M. E. (1998). Mutations in the human lambda5/14.1 gene result in B cell deficiency and agammaglobulinemia. The Journal of experimental medicine, 187(1), 71–77. https://doi.org/10.1084/jem.187.1.71
- Minegishi, Y., Coustan-Smith, E., Rapalus, L., Ersoy, F., Campana, D., & Conley, M. E. (1999). Mutations in Igalpha (CD79a) result in a complete block in B-cell development. The Journal of clinical investigation, 104(8), 1115–1121. https://doi.org/10.1172/JCI7696
- Dobbs AK, Yang T, Farmer D, Kager L, Parolini O, Conley ME. Cutting edge: a hypomorphic mutation in Igbeta (CD79b) in a patient with immunodeficiency and a leaky defect in B cell development. J Immunol. 2007 Aug 15;179(4):2055-9. doi: 10.4049/jimmunol.179.4.2055
- Khalili A, Plebani A, Vitali M, Abolhassani H, Lougaris V, Mirminachi B, Rezaei N, Aghamohammadi A. Autosomal recessive agammaglobulinemia: a novel non-sense mutation in CD79a. J Clin Immunol. 2014 Feb;34(2):138-41. doi: 10.1007/s10875-014-9989-3
- Langereis, J. D., Henriet, S. S., Kuipers, S., Weemaes, C., van der Burg, M., de Jonge, M. I., & van der Flier, M. (2018). IgM Augments Complement Bactericidal Activity with Serum from a Patient with a Novel CD79a Mutation. Journal of clinical immunology, 38(2), 185–192. https://doi.org/10.1007/s10875-017-0474-7
- Ferrari, S., Lougaris, V., Caraffi, S., Zuntini, R., Yang, J., Soresina, A., Meini, A., Cazzola, G., Rossi, C., Reth, M., & Plebani, A. (2007). Mutations of the Igbeta gene cause agammaglobulinemia in man. The Journal of experimental medicine, 204(9), 2047–2051. https://doi.org/10.1084/jem.20070264
- Lougaris V, Vitali M, Baronio M, Moratto D, Tampella G, Biasini A, Badolato R, Plebani A. Autosomal recessive agammaglobulinemia: the third case of Igβ deficiency due to a novel non-sense mutation. J Clin Immunol. 2014 May;34(4):425-7. doi: 10.1007/s10875-014-0033-4
- Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, Coustan-Smith E, Howard V, Campana D. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199-227. doi: 10.1146/annurev.immunol.021908.132649
- Lagresle-Peyrou C, Millili M, Luce S, Boned A, Sadek H, Rouiller J, Frange P, Cros G, Cavazzana M, André-Schmutz I, Schiff C. The BLNK adaptor protein has a nonredundant role in human B-cell differentiation. J Allergy Clin Immunol. 2014 Jul;134(1):145-54. doi: 10.1016/j.jaci.2013.12.1083
- Tang P, Upton JEM, Barton-Forbes MA, Salvadori MI, Clynick MP, Price AK, Goobie SL. Autosomal Recessive Agammaglobulinemia Due to a Homozygous Mutation in PIK3R1. J Clin Immunol. 2018 Jan;38(1):88-95. doi: 10.1007/s10875-017-0462-y
- Nunes-Santos CJ, Uzel G, Rosenzweig SD. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol. 2019 May;143(5):1676-1687. doi: 10.1016/j.jaci.2019.03.017
- Conley, M. E., Dobbs, A. K., Quintana, A. M., Bosompem, A., Wang, Y. D., Coustan-Smith, E., Smith, A. M., Perez, E. E., & Murray, P. J. (2012). Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. The Journal of experimental medicine, 209(3), 463–470. https://doi.org/10.1084/jem.20112533
- Anzilotti, C., Swan, D. J., Boisson, B., Deobagkar-Lele, M., Oliveira, C., Chabosseau, P., Engelhardt, K. R., Xu, X., Chen, R., Alvarez, L., Berlinguer-Palmini, R., Bull, K. R., Cawthorne, E., Cribbs, A. P., Crockford, T. L., Dang, T. S., Fearn, A., Fenech, E. J., de Jong, S. J., Lagerholm, B. C., … Hambleton, S. (2019). An essential role for the Zn2+ transporter ZIP7 in B cell development. Nature immunology, 20(3), 350–361. https://doi.org/10.1038/s41590-018-0295-8
- Barnes S, Kotecha S, Douglass JA, Paul E, Hore-Lacy F, Stirling R, Snell GI, Westall GP. Evolving practice: X-linked agammaglobulinemia and lung transplantation. Am J Transplant. 2015 Apr;15(4):1110-3. doi: 10.1111/ajt.13084. Epub 2015 Mar 3. Erratum in: Am J Transplant. 2015 Aug;15(8):2278. Hore-Lacey, F [corrected to Hore-Lacy, F].
- Bazregari, S., Azizi, G., Tavakol, M., Asgardoon, M. H., Kiaee, F., Tavakolinia, N., Valizadeh, A., Abolhassani, H., & Aghamohammadi, A. (2017). Evaluation of infectious and non-infectious complications in patients with primary immunodeficiency. Central-European journal of immunology, 42(4), 336–341. https://doi.org/10.5114/ceji.2017.72825
- Barahona Afonso, A. F., & João, C. M. (2016). The Production Processes and Biological Effects of Intravenous Immunoglobulin. Biomolecules, 6(1), 15. https://doi.org/10.3390/biom6010015
- Perez EE, Orange JS, Bonilla F, Chinen J, Chinn IK, Dorsey M, El-Gamal Y, Harville TO, Hossny E, Mazer B, Nelson R, Secord E, Jordan SC, Stiehm ER, Vo AA, Ballow M. Update on the use of immunoglobulin in human disease: A review of evidence. J Allergy Clin Immunol. 2017 Mar;139(3S):S1-S46. doi: 10.1016/j.jaci.2016.09.023
- Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Transfus Med Rev. 2003 Oct;17(4):241-51. doi: 10.1016/s0887-7963(03)00038-5
- Wasserman RL. Personalized Therapy: Immunoglobulin Replacement for Antibody Deficiency. Immunol Allergy Clin North Am. 2019 Feb;39(1):95-111. doi: 10.1016/j.iac.2018.08.001
- Smith CIE, Berglöf A. X-Linked Agammaglobulinemia (2001) [Updated 2016 Aug 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2021
- Cardenas-Morales, M., & Hernandez-Trujillo, V. P. (2021). Agammaglobulinemia: from X-linked to Autosomal Forms of Disease. Clinical reviews in allergy & immunology, 1–14. Advance online publication. https://doi.org/10.1007/s12016-021-08870-5
- Howard V, Myers LA, Williams DA, Wheeler G, Turner EV, Cunningham JM, Conley ME. Stem cell transplants for patients with X-linked agammaglobulinemia. Clin Immunol. 2003 May;107(2):98-102. doi: 10.1016/s1521-6616(03)00045-7
- van Zelm, M. C., Pumar, M., Shuttleworth, P., Aui, P. M., Smart, J. M., Grigg, A., & Bosco, J. J. (2019). Functional Antibody Responses Following Allogeneic Stem Cell Transplantation for TP53 Mutant pre-B-ALL in a Patient With X-Linked Agammaglobulinemia. Frontiers in immunology, 10, 895. https://doi.org/10.3389/fimmu.2019.00895
- Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, Conley ME, Cunningham-Rundles C, Ochs HD. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006 Jul;85(4):193-202. doi: 10.1097/01.md.0000229482.27398.ad
- Winkelstein, J. A., Conley, M. E., James, C., Howard, V., & Boyle, J. (2008). Adults with X-linked agammaglobulinemia: impact of disease on daily lives, quality of life, educational and socioeconomic status, knowledge of inheritance, and reproductive attitudes. Medicine, 87(5), 253–258. https://doi.org/10.1097/MD.0b013e318187ed81
- El-Sayed, Z. A., Abramova, I., Aldave, J. C., Al-Herz, W., Bezrodnik, L., Boukari, R., Bousfiha, A. A., Cancrini, C., Condino-Neto, A., Dbaibo, G., Derfalvi, B., Dogu, F., Edgar, J., Eley, B., El-Owaidy, R. H., Espinosa-Padilla, S. E., Galal, N., Haerynck, F., Hanna-Wakim, R., Hossny, E., … Sullivan, K. E. (2019). X-linked agammaglobulinemia (XLA):Phenotype, diagnosis, and therapeutic challenges around the world. The World Allergy Organization journal, 12(3), 100018. https://doi.org/10.1016/j.waojou.2019.100018
- Lougaris V, Soresina A, Baronio M, Montin D, Martino S, Signa S, Volpi S, Zecca M, Marinoni M, Baselli LA, Dellepiane RM, Carrabba M, Fabio G, Putti MC, Cinetto F, Lunardi C, Gazzurelli L, Benvenuto A, Bertolini P, Conti F, Consolini R, Ricci S, Azzari C, Leonardi L, Duse M, Pulvirenti F, Milito C, Quinti I, Cancrini C, Finocchi A, Moschese V, Cirillo E, Crescenzi L, Spadaro G, Marasco C, Vacca A, Cardinale F, Martire B, Trizzino A, Licciardello M, Cossu F, Di Matteo G, Badolato R, Ferrari S, Giliani S, Pession A, Ugazio A, Pignata C, Plebani A. Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality. J Allergy Clin Immunol. 2020 Aug;146(2):429-437. doi: 10.1016/j.jaci.2020.03.001
- Singh S, Rawat A, Suri D, Gupta A, Garg R, Saikia B, Minz RW, Sehgal S, Chan KW, Lau YL, Kamae C, Honma K, Nakagawa N, Imai K, Nonoyama S, Oshima K, Mitsuiki N, Ohara O. X-linked agammaglobulinemia: Twenty years of single-center experience from North West India. Ann Allergy Asthma Immunol. 2016 Oct;117(4):405-411. doi: 10.1016/j.anai.2016.07.044





{kind=link}