What is amyloidosis
Amyloidosis is the name for a group of rare disease that occurs when amyloid proteins are deposited in tissues and organs throughout the body – by extracellular and/or intracellular deposition 1. The amyloid proteins (insoluble abnormal amyloid fibrils) then alter the normal function of tissues. Amyloid proteins are abnormal proteins that the body cannot break down and recycle, as it does with normal proteins. Normally, proteins are broken down at about the same rate as they are produced, but these unusually stable amyloid deposits are deposited more rapidly than they can be broken down. When amyloid proteins clump together, they form amyloid deposits. The buildup of these deposits damages a person’s organs and tissues. Without treatment, this can lead to organ failure. Amyloidosis can affect different organs and tissues in different people and can affect more than one organ at the same time.
Amyloidosis most frequently affects:
- the kidneys,
- heart,
- nervous system,
- liver and spleen,
- digestive tract,
- bones and joints- especially the wrists,
- skin,
- blood vessels and clotting system,
- eyes.
Amyloid deposits can form in almost any tissue of your body. Therefore the symptoms, signs and severity of amyloidosis can vary greatly and are often vague and not specific. Many patients undergo extensive testing and see several doctors before the diagnosis is made.
Only 10% of amyloidosis deposits consist of components such as glycosaminoglycans (GAGs), apolipoprotein-E (apoE), and serum amyloid P-component (SAP), while nearly 90% of the deposits consist of amyloid fibrils that are formed by the aggregation of misfolded proteins. These proteins either arise from proteins expressed by cells at the deposition site (localized), or they precipitate systemically after production at a local site (systemic) 2. In humans, about 23 different unrelated proteins are known to form amyloid fibrils in vivo 3.
Many mechanisms of protein function contribute to amyloidogenesis, including “nonphysiologic proteolysis, defective or absent physiologic proteolysis, mutations involving changes in thermodynamic or kinetic properties, and pathways that are yet to be defined” 3.
Recent years have seen important strides forwards in treatment and the outlook for patients diagnosed with amyloidosis today is far better than it was even a few years ago. In some types of amyloidosis currently available treatments may even lead to complete cure, while in others, there may be extended symptom free survival. Twenty years ago the life expectancy of patients diagnosed with amyloidosis was usually only a few months or years, whereas now it is often 10 years or more.
Currently available treatments differ considerably for the different types of amyloidosis, but they all focus on reducing the availability of the proteins which form amyloid fibrils (precursor proteins). In many cases patients benefit considerably from these treatments, with regression of amyloid deposits once the precursor supply is stopped and subsequent substantial health improvements.
Amyloidosis Classification
Amyloid is now classified chemically. The amyloidoses are referred to with a capital A (for amyloid) followed by an abbreviation for the fibril protein. For example, in most cases formerly called primary amyloidosis and in myeloma-associated amyloidosis, the fibril protein is an immunoglobulin light chain or light chain fragment (abbreviated L); thus, patients with these amyloidoses are now said to have light chain amyloidosis (AL). Names such as AL describe the protein (light chain), but not necessarily the clinical phenotype 4.
Similarly, in most cases previously termed senile cardiac amyloidosis and in many cases previously termed familial amyloid polyneuropathy (FAP), the fibrils consist of the transport protein transthyretin (TTR); these diseases are now collectively termed ATTR.
Proteins that form amyloid fibrils differ in size, function, amino acid sequence, and native structure but become insoluble aggregates that are similar in structure and properties. Protein misfolding results in the formation of fibrils that show a common beta-sheet pattern on x-ray diffraction.
In theory, misfolded amyloid proteins can be attributed to infectious sources (prions), de novo gene mutations, errors in transcription, errors in translation, errors in post-translational modification, or protein transport. For example, in ATTR, 100 different points of single mutations, double mutations, or deletions in the TTR gene and several different phenotypes of FAP have been documented 5. Twenty-three different fibril proteins are described in human amyloidosis, with variable clinical features.
The major types of human amyloid are outlined and discussed individually in the table below. Great importance is now placed on appropriate and timely typing and classification of each type of amyloid-related disease, to focus therapy and plan appropriate monitoring 6. The common clinical amyloid entities are AL, AA, ATTR, and Aβ2M types 6.
Table 1. Human Amyloidoses Classification
| Type | Fibril Protein | Main Clinical Settings |
|---|---|---|
| Systemic | Immunoglobulin light chains | Plasma cell disorders |
| Transthyretin | Familial amyloidosis, senile cardiac amyloidosis | |
| A amyloidosis | Inflammation-associated amyloidosis, familial Mediterranean fever | |
| Beta2 -microglobulin | Dialysis-associated amyloidosis | |
| Immunoglobulin heavy chains | Systemic amyloidosis | |
| Hereditary | Fibrinogen alpha chain | Familial systemic amyloidosis |
| Apolipoprotein AI | Familial systemic amyloidosis | |
| Apolipoprotein AII | Familial systemic amyloidosis | |
| Lysozyme | Familial systemic amyloidosis | |
| Central nervous system | Beta protein precursor | Alzheimer syndrome, Down syndrome, hereditary cerebral hemorrhage with amyloidosis (Dutch) |
| Prion protein | Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker disease, fatal familial insomnia, kuru | |
| Cystatin C | Hereditary cerebral hemorrhage with amyloidosis (Icelandic) | |
| ABri precursor protein | Familial dementia (British) | |
| ADan precursor protein | Familial dementia (Danish) | |
| Ocular | Gelsolin | Familial amyloidosis (Finnish) |
| Lactoferrin | Familial corneal amyloidosis | |
| Keratoepithelin | Familial corneal dystrophies | |
| Localized | Calcitonin | Medullary thyroid carcinoma |
| Amylin* | Insulinoma, type 2 diabetes | |
| Atrial natriuretic factor amyloidosis | Isolated atrial amyloidosis | |
| Prolactin | Pituitary amyloid | |
| Keratin | Cutaneous amyloidosis | |
| Medin | Aortic amyloidosis in elderly people |
Systemic Amyloidoses
A Amyloidosis (AA)
AA Amyloidosis was previously known as secondary amyloidosis. AA Amyloidosis occurs in patients who have suffered from prolonged, chronic inflammatory or infectious diseases for many years (and occasionally with neoplasms). Worldwide, AA is the most common systemic amyloidosis, although the frequency has been shown to vary significantly in different ethnic groups 8. Most patients with AA amyloidosis have amyloid deposits in their kidneys which cause problems with kidney function, most commonly protein in the urine. There are always amyloid deposits in the spleen, which may be enlarged. There may also be amyloid deposits in other parts of the body, which usually do not cause symptoms.
In more than 90% of cases, proteinuria (protein in the urine) is the first sign. Patients may also have:
- Nephrotic syndrome:
- large amounts of protein in the urine (>3.5 g/day)
- low albumin in the blood
- peripheral edema- swollen ankles
- End stage kidney failure: the kidneys stop functioning altogether and the patient needs either dialysis or a kidney transplant in order to survive.
- Rarely:
- hematuria (blood in the urine)
- renal tubular defects
- nephrogenic diabetes insipidus
- renal calcification (calcium deposits in the kidneys)
- AA amyloid deposits leading to illness may also occur, rarely, in the:
- thyroid gland (goiter)
- liver (enlarged liver)
- There may be AA amyloid deposits in the heart and the gut but these usually do not cause any symptoms.
The amyloid fibrils making up the amyloid deposits are formed from a protein called amyloid A protein which is derived from a normal blood protein of unknown function, called serum amyloid A protein (SAA). SAA is known as the “precursor” protein in AA amyloidosis. Serum amyloid A protein (SAA) is produced in the liver and circulates in the serum bound to high-density lipoprotein (HDL) and is found in very small quantities (less than 5 mg per liter) in the blood of all healthy people. But when there is almost any kind of inflammation, infection or injury in the body, serum amyloid A protein (SAA) production increases greatly, along with the production of another normal trace protein called C-reactive protein (CRP). Production of some other blood proteins also increases, although to a much smaller extent, as part of this normal reaction to disease, called the acute phase response 2. The level of SAA increases more dramatically than that of any other protein and may reach 2,000 mg per liter.
If there is recovery from whatever disease or injury caused the acute phase response, then levels of serum amyloid A protein (SAA) drop rapidly. However, if the disease persists, SAA levels remain high. In up to 10% of patients with sustained high levels of serum amyloid A protein (SAA) persisting for several years, the SAA forms amyloid A fibrils in the tissues and these amyloid deposits cause AA amyloidosis.
Some of the conditions associated with A Amyloidosis (AA) include the following:
- Rheumatoid arthritis (RA) 9
- Alzheimer disease 10
- Multiple myeloma 11
- Juvenile idiopathic arthritis 12
- Ankylosing spondylitis 13
- Psoriasis and psoriatic arthritis 14
- Still disease 15
- Behçet syndrome 16
- Familial Mediterranean fever 17
- Crohn disease 18
- Leprosy 19
- Osteomyelitis 20
- Tuberculosis 21
- Chronic bronchiectasis 22
- Chronic pyelonephritis (kidney infection) in paraplegic patients 23
- Chronic infections in intravenous drug users 23
- Castleman disease 24
- Hodgkin’s disease and non-Hodgkin lymphoma 25
- Renal cell carcinoma 26
- Gastrointestinal, lung, or urogenital carcinoma 27
- Basal cell carcinoma (BCC)
- Hairy cell leukaemia
- Cryopyrin-associated periodic syndromes 28
- Whipple’s disease
- A small number of patients with AA amyloidosis do not have detectable evidence of a prolonged inflammatory disease. Some of these are carriers of inherited fever syndrome genes. Inherited fever syndromes are very rare in the general population, though some are relatively common in certain ethnic groups. People with inherited fever syndromes usually suffer from repeated episodes of fever and general unwellness, sometimes with other symptoms such as rashes, stomachaches and painful swollen joints. Each time that such an episode occurs, the patient recovers, usually within a few days, without any clear cause being identified.
Some patients may be exposed to chronic inflammation and high serum amyloid A protein (SAA) levels for decades yet may never develop AA amyloidosis. Others develop AA amyloidosis after just a few years of chronic inflammation. For example, children with Familial Mediterranean Fever may develop rapidly progressive kidney failure due to AA amyloidosis, even before the Familial Mediterranean Fever is diagnosed and treatment is established. The reason for this is unclear. There is some evidence that genetic factors related to variations within the gene for SAA may play a part in determining the likelihood that high levels of SAA will form amyloid deposits. It is not possible at present to predict which patients with chronic inflammation will develop AA amyloidosis if the inflammation is not treated successfully and which will not.
However, it is clear that successful treatment of inflammatory conditions, with suppression of SAA levels does prevent the development of AA amyloidosis.
Therapy has traditionally been aimed at the underlying inflammatory condition to reduce the production of the precursor amyloid protein, SAA. Disease-modifying antirheumatic drugs (DMARDs) such as colchicine, a microtubule inhibitor and weak immunosuppressant, can prevent secondary renal failure due to amyloid deposition specifically in familial Mediterranean fever.
Newer therapies have become more targeted to avoid the cytotoxicity of older agents (eg, chlorambucil, cyclophosphamide). The SAA amyloid seen in Cryopyrin-associated periodic syndromes was reduced with a biologic interleukin (IL)–1β trap called rilonacept.
Tumor necrosis factor–alpha (TNF-alpha) is also thought to be involved in amyloid deposition 29. Aggressive use of newer biologic therapies for RA, such as etanercept (a TNF-alpha blocker), and tocilizumab 30 have been used to decrease the concentration of SAA, serum creatinine, creatinine clearance, and proteinuria in renal AA associated with rheumatoid arthritis 31. One study compared etanercept with tocilizumab and showed that IL-6 inhibition may be a more effective therapy 32.
Additionally, SAA isoforms have been studied using high-resolution 2-dimensional gel electrophoresis and peptide mapping by reverse-phase chromatography, electrospray ionization tandem mass spectrometry, and genetic analysis down to the post-translational modification level. SAA is coded by 4 genes: SAA1, AAA2, SAA3, and SAA4. The SAA1 gene contributes to most of the deposits and contains a single nucleotide polymorphism that defines at least 3 haplotypes. The saa1.3 allele was found to be a risk factor and a poor prognostic indicator in Japanese RA patients. Genetic analysis has proved useful in selecting patients for biologic therapy and for predicting outcome 33. Early treatment is essential to prevent the long-term complications of AA amyloidosis 34.
AL Amyloidosis (Light chain amyloidosis or AL)
AL amyloidosis (light chain amyloidosis) was previously known as primary amyloidosis or myeloma-associated amyloidosis and is currently the most common type of amyloidosis in developed countries. AL amyloidosis is commoner in men than in women and although most patients with AL amyloidosis are aged over 45, it occasionally occurs at younger ages. If left untreated AL amyloidosis (light chain amyloidosis) is progressive and may lead to death within a year.
Patients with AL amyloidosis (light chain amyloidosis) have an underlying disorder in which there is overproduction of amyloidogenic proteins called light chains (clonal immunoglobulin light chain or light chain fragment). The “L” in the name AL amyloidosis stands for “light chain.” Abnormal free light chains can be measured in the blood in about 95% of patients with AL amyloidosis. Light chains are parts of antibodies, also known as immunoglobulins (abbreviated as Ig). Immunoglobulins (or antibody) are among the most abundant protein components in the blood, constituting about 20% of the total protein in plasma by weight. Immunoglobulins (Ig) are produced by a type of immune system cell called plasma cells (differentiated B-lymphocytes or B cells). Plasma cells develop from B lymphocytes (B cells), a type of white blood cell that is made in the bone marrow. Normally, when bacteria or viruses enter the body, some of the B cells will change into plasma cells. The plasma cells make antibodies (immunoglobulins) to fight bacteria and viruses, to stop infection and disease.
Normal B cells and plasma cells stop multiplying and producing antibodies once the body has recovered from an infection, but in the B cell/plasma cell disorders the cells do not shut down as and when they should, but instead continue to churn out antibodies or antibody fragments (light chains) into the bloodstream. These abnormal proteins do not serve any defence function against disease, but in many cases they also do no harm.
In AL amyloidosis a line, or clone, of identical B cells or plasma cells behaves abnormally, continuously producing large quantities of just one part, or fragment, of a particular antibody called a monoclonal immunoglobulin light chain. AL amyloidosis develops when the abnormal proteins produced (usually light chains or fragments of light chains) are amyloidogenic. This means that they aggregate (clump) together in the tissues in the form of long thread-like fibers that the body cannot easily destroy and clear away. These fibrous aggregates are the amyloid fibrils which form amyloid deposits and as they accumulate in the tissues of the body they disrupt the structure and function of whichever organs are involved.
The abnormal plasma cells are usually, but not always, located in the bone marrow. Most patients with AL amyloidosis have relatively low numbers of abnormal plasma cells in the bone marrow but this can be variable. 15% of patients with AL amyloidosis can have many abnormal plasma cells in the bone marrow which may be directly damaging to bones or affect the function of the bone marrow.
Light chain amyloidosis is a monoclonal plasma cell disorder closely related to multiple myeloma, as some patients fulfill diagnostic criteria for multiple myeloma. Typical organs involved include the heart, kidney, peripheral nervous system, gastrointestinal tract, respiratory tract, and nearly any other organ.
The symptoms of AL amyloidosis depend on which tissues and organs are affected.
- Kidney failure
- Most people with AL amyloidosis have a build-up of amyloid proteins in their kidneys, and are at risk of kidney failure.
- Symptoms of kidney failure include:
- swelling, often in the legs, caused by fluid retention (edema)
- tiredness
- weakness
- loss of appetite
- Heart failure
- Deposits of amyloid in the heart can cause the muscles to become stiffer, making it more difficult to pump blood around the body. This may result in heart failure, which can cause:
- shortness of breath
- edema
- an abnormal heartbeat (arrhythmia)
- Deposits of amyloid in the heart can cause the muscles to become stiffer, making it more difficult to pump blood around the body. This may result in heart failure, which can cause:
- Other symptoms
- Amyloid proteins can also build up in other areas, like the liver, spleen, nerves or digestive system. Symptoms can include:
- feeling lightheaded or fainting, particularly after standing or sitting up
- numbness or a tingling feeling in the hands and feet (peripheral neuropathy)
- nausea, diarrhea or constipation
- numbness, tingling and pain in the wrist, hand and fingers (carpal tunnel syndrome)
- easy bruising
- Amyloid proteins can also build up in other areas, like the liver, spleen, nerves or digestive system. Symptoms can include:
AL amyloidosis does not affect the brain, so it does not cause any problems with memory or thinking, for example.
AL amyloidosis treatment is directed towards the abnormal plasma cells (usually in the bone marrow), producing abnormal light chains that form amyloid deposits. AL amyloidosis treatment usually mirrors the management of multiple myeloma (ie, chemotherapy). Selected patients have received benefit from high-dose melphalan and autologous stem-cell transplantation, with reports of prolonged survival in some studies.
The most current guidelines recommend high-dose steroids for isolated organ involvement, but transplantation should be considered early 35. Any transplantation should also be followed by high-dose intravenous melphalan supported with stem-cell transplantation to try to prevent future amyloid deposition in the transplanted organ 35.
Other agents used in light chain amyloidosis have included bortezomib, rituximab, immunomodulatory agents, and standard-dose alkylating agents (eg, melphalan, cyclophosphamide), thalidomide, and lenalidomide 36. Bortezomib is a proteasome inhibitor that is well tolerated in multiple myeloma 37. Patients younger than 65 years may be candidates for stem cell transplantation with melphalan and dexamethasone or thalidomide-cyclophosphamide-dexamethasone regimens 35.
The response is then classified as either partial, complete, or no response. If a partial or complete response is achieved, patients are closely monitored 35. Imaging and some biomarkers like N-terminal pro-brain natriuretic peptide (NT-proBNP), B-type natriuretic peptide (BNP), troponin, and free light-chain concentration can be useful in gauging clinical response, especially in cardiac amyloid disease 38. If no response is seen, the patient becomes a candidate for novel agents that usually include alkylating agents combined with novel agents such as lenalidomide 39.
Heavy chain amyloidosis (AH amyloidosis)
In a few cases, immunoglobulin chain amyloidosis fibrils contain only heavy-chain sequences rather than light-chain sequences, and the disease is termed AH amyloidosis rather than AL amyloidosis. Electron microscopy may be helpful in the detection of small deposits and in the differentiation of amyloid from other types of renal fibrillar deposits 40.
Transthyretin amyloidosis (ATTR amyloidosis)
ATTR amyloidosis is a form of systemic amyloidosis caused by amyloid deposits made up of a protein called transthyretin (TTR). ATTR amyloidosis means A for amyloid and the TTR is short for the protein “transthyretin”. Transthyretin (TTR) is a protein mainly produced in the liver with a smaller portion also coming from the choroid plexus of the brain and functions as a transporter of thyroxine (T4) and retinol (vitamin A)-binding protein 41. All the transthyretin (TTR) in the blood is produced by the liver. TTR in the brain and the eye is made separately by a structure called the choroid plexus, which is located within the brain and produces the cerebrospinal fluid that bathes the brain and spinal cord. In the past, because ATTR often involves nerve or cardiac involvement, some terms were used when the chemical variations were less defined. Examples of these outdated terms include FAP (Familial Amyloid Polyneuropathy) and FAC (Familial Amyloid Cardiomyopathy). Today, the different forms of ATTR are termed according to the “chemically based” name of the transthyretin protein variation. An example of this would be ATTRV30M (for ATTR Val30Met or p.Val50Met according to the Human Genome Variation Society recommendation), which is the most common ATTR variation. The most common TTR variants in the United States are:
- Val30Met (also the most found worldwide). What was once termed FAP (Familial amyloid polyneuropathy) is most commonly caused by Val30Met.
- Thr60Ala
- Leu58His
- Ser77Tyr
- Val122Ile — predominantly seen in the African-American population; associated with cardiomyopathy (heart conditions). What was once termed FAC (Familial amyloid cardiomyopathy) is commonly caused by Val122lle.
Transthyretin amyloidosis (ATTR amyloidosis) can be either hereditary or acquired (non-hereditary):
- Hereditary ATTR amyloidosis is caused by a mutation in the gene for TTR, inherited from one parent. The disease therefore runs in families, though the timing, development and severity of the disease can vary greatly. Hereditary ATTR amyloidosis is the most commonly recognized form of hereditary systemic amyloidosis but it is nevertheless a very rare disease. More than 150 amyloidogenic variants (mutations) of TTR have been observed and different mutations may cause different disease manifestations. Despite being extremely rare in most parts of the world, hereditary ATTR amyloidosis is common in some very localised parts of Portugal, Sweden and Japan. It may also be common, but under-diagnosed in several other regions including Spain, France, Brazil, Argentina, Cyprus, Bulgaria and Ireland.
- In acquired (non-hereditary) ATTR amyloidosis, the amyloid is formed by the normal, so‑called ‘wild-type’ protein. This disease is not hereditary. It is known as wild-type transthyretin amyloidosis or wild-type ATTR (ATTRwt) amyloidosis (formerly called senile systemic amyloidosis or cardiac TTR amyloidosis). Wild-type ATTR (ATTRwt) amyloidosis is a slowly progressive disease affecting elderly people, mostly men. The symptoms usually start after age 65. There is no mutation in the TTR gene so the condition is not hereditary (it does not run in families). Normal, ‘wild-type’ TTR protein misfolds to form the amyloid deposits, with obvious major clinical effect in the heart although there may also be carpal tunnel syndrome in some people, and occasionally peripheral neuropathy. It has long been known that wild-type TTR commonly forms microscopic amyloid deposits in elderly people. These types of amyloid deposits are found at autopsy in 1 in 4 people over age 80. It was believed until recently that they usually caused no symptoms and clinical disease caused by this type of amyloid was very rarely diagnosed. Some patients with small wild type ATTR amyloidosis deposits in the heart and minimal symptoms may not require any treatment. No-one knows just how common symptomatic wild type ATTR amyloidosis really is but discovery of new imaging techniques, has shown it to be much more common than previously recognised. It is believed to be underdiagnosed at present. This diagnosis may become more common in future as the population ages and diagnostic methods continue to improve.
The clinical presentation and effects of transthyretin amyloidosis (ATTR amyloidosis) vary widely depending on which organs are mostly affected.
- Symptoms of hereditary ATTR amyloidosis may include:
- peripheral neuropathy: limb weakness and pain, loss of sensation
- autonomic neuropathy: disturbances of bowel, bladder and blood pressure and sexual dysfunction
- heart failure – symptoms result from stiffening of the heart due to amyloid deposits (restrictive cardiomyopathy). They may include:
- shortness of breath (dyspnea), sometimes just after mild exertion
- palpitations and abnormal heart rhythms, most frequently atrial fibrillation or atrial flutter
- leg swelling (edema)
- weight loss
- nausea
- fatigue
- dizziness and collapse (syncope or fainting), which may occur after exertion, or after eating
- disrupted sleep
- angina (chest pain)
- disease due to amyloid deposits in the:
- eye
- kidneys
- thyroid gland
- adrenal glands
- blood vessels
- Symptoms may appear as early as age 20, or as late as age 80. There is often little correlation between the underlying mutation and the clinical disease features. Within families the pattern is usually quite consistent for:
- age of onset
- rate of disease progression
- involvement of different body systems
- In some families all affected members have just neuropathy, while in other families all affected members have both neuropathy and cardiac disease. In a few cases certain mutations have been associated with either particularly severe disease or with relatively limited disease.
- Patients carrying a mutation in their genes do not always develop disease. Some cases have been reported where people over age 60 have no disease despite having two copies of the TTR gene mutation which causes production of the Val30Met TTR protein variant.
- Symptoms of wild-type ATTR amyloidosis result from stiffening of the heart due to amyloid deposits (restrictive cardiomyopathy). These symptoms may include:
- shortness of breath, sometimes just after mild exertion
- palpitations and abnormal heart rhythms, most frequently atrial fibrillation or atrial flutter
- leg swelling (edema)
- weight loss
- nausea
- fatigue
- dizziness and collapse (syncope or fainting), which may occur after exertion, or after eating
- disrupted sleep
- angina (chest pain)
- Almost 50% of patients with wild-type ATTR (ATTRwt) amyloidosis experience carpal tunnel syndrome – tingling and pain in the wrists, pins and needles in the hands. Carpal tunnel syndrome often appears 3-5 years before the symptoms of heart disease.
- It not clear at present just how commonly the deposits actually do lead to symptomatic disease. Recent developments with diagnostic tests such as cardiac magnetic resonance (CMR) have greatly improved the detection of amyloid in the heart during life. This has led to the belief that wild-type ATTR (ATTRwt) amyloidosis may be far more common than was previously thought. Cardiologists are gradually becoming more aware that wild-type amyloidosis may cause otherwise unexplained heart failure and are referring these patients more frequently to the National Amyloidosis Center.
Treatment of all types of amyloidosis is currently based on the following principles:
- Reducing the supply of amyloid forming precursor proteins.
- Genetic-based therapies:
- Small interfering RNA.
- Antisense oligonucleotides.
- Genetic-based therapies:
- Supporting the function of organs containing amyloid.
Reducing variant TTR supply
For most ATTR variations, the liver is the main source of amyloid production. However, the liver itself is not affected by the disease in most cases and the amyloid burden causes damage in other parts of the body. A liver transplant is very helpful in reducing (or stopping) the amyloid deposits. It can stabilize or improve neurological symptoms as well as gastrointestinal problems (which can correct poor nutrition and overall health). However, the statistics vary as to who can benefit from these transplants, with the more common ATTR Val30Met having the highest success rate. Liver transplantation should be performed in Val30Met patients as early as possible as it removes the main source of mutant transport protein transthyretin and dramatically reduces the progression of neuropathy (up to 70%) and can double the median survival. The outcome of liver transplantation is largely dependent on the mutation that exists in the patient. In some cases, amyloid deposition does completely stop after transplantation, so research is ongoing in this area. For those patients with cardiac symptoms, studies have shown that heart problems may continue after a liver transplant. In some situations, a combined heart and liver transplant will help a patient with an ATTR variant that produces advanced cardiac problems.
Several medications have been approved by the Food and Drug Administration (FDA) for treating patients with ATTR amyloidosis. Other medications continue to be investigated. In 2018, two drugs were approved by the FDA for ATTR polyneuropathy of hereditary transthyretin-mediated (hATTR) amyloidosis in adults. The first was Patisiran (Onpattro) lipid complex injection, a first of its kind RNA interference therapeutic. This drug aims to silencing the gene expression. The second drug approved in 2018 is Inotersen (Tegsedi) which reduces the production of TTR protein through a once a week subcutaneous injection. In 2019, Tafamidis (Vyndamax and Vyndaqel) were approved by the FDA for ATTR cardiomyopathy. These drugs are for oral administration taken once daily. In clinical trials, new therapies include aiming to treat the root cause of the disease, destabilized and folded TTR, using monoclonal antibodies to specifically target and clear misfolded (toxic) form a the TTR amyloid protein, and using a drug designed to reduce the production of transthyretin (TTR protein) in all types of TTR amyloidosis. Another RNA interference therapeutic is also currently in clinical trial. Advances in other treatments are likely, with new studies and clinical trials currently in view. It is possible that ATTR can cause serious health complications, so it should not be taken lightly. However, do not assume that disability or severe health issues are stamped on your future. There are treatments available and research continues.
ATTR silencers
These medications act on the liver to decrease the production of TTR. Two ATTR silencers approved by the FDA to treat patients with the hereditary type of ATTR who also have neuropathy.
- Patisiran (Onpattro®) is an infusion that is given every three weeks.
- Inotersen (Tegsedi®) is an injection given once a week and requires weekly lab work.
ATTR stabilizers
These medications stabilize the TTR protein, which in turn prevents it from breaking apart and forming amyloid fibrils.
- Tafamidis (Vyndamax®, Vyndaqel®) is approved by the FDA for patients with hereditary or wild-type ATTR that has affected their heart.
- AG10 is a medication currently being tested in a clinical trial.
- Diflunsial (Dolobid®) is a nonsteroidal anti-inflammatory drug (NSAID) that has been shown to also stabilize the TTR protein. However, this medication has not been fully studied in patients with ATTR that has affected the heart and also may not be tolerated due to side effects.
Fibril disruptors
These medications may help break up and clear ATTR amyloid fibrils. Doxycycline (antibiotic) and green tea extract (over-the-counter supplement)have only been tested in small studies and there is limited evidence that these medications would be helpful in treating amyloidosis.
An antibody that removes TTR amylod fibrils, called PRX-004, is being tested in clinical trials.
Supportive treatment
Supportive treatment is helpful for various symptoms, including peripheral neuropathy, autonomic neuropathy, and cardiac and kidney problems, and can change the quality of life for many people. There are several medications that can be prescribed to treat peripheral neuropathy, which can cause tingling or burning in some parts of the body. These medications can help with pain relief and nerve damage. If a patient has autonomic neuropathy, symptoms can vary, with common problems affecting blood pressure, heart rate, digestion, and perspiration, depending on the location of the damage to the nerves. Other gastrointestinal dysfunctions may require treatment for symptoms that include poor nutritional health, diarrhea or constipation, and nausea or vomiting. Doctors can prescribe medications to help with these symptoms to lessen the pain and the symptom itself.
Management of heart problems, heart failure, and kidney dialysis (when needed) make a significant improvement on a patient’s quality of life. Reversing any damage to the organs and other parts of the body is difficult to achieve. If treatment begins during the early onset of clinical symptoms, the overall success rate is higher, so early detection is essential.
Heart disease treatment
ATTR amyloid deposits in the heart cause the heart to stiffen which can lead to symptoms of heart failure. Patients can benefit from supportive treatment measures for heart failure. However many standard medications used for heart failure are not helpful for patients with cardiac amyloidosis. Careful attention to fluid balance is important.
Fluid balance
The most important principle of treatment for cardiac amyloidosis is strict fluid balance control. Specialist heart failure nurse involvement may help patients to achieve this. Many patients with ATTR cardiac amyloidosis should limit their fluid intake. This advice is extremely important, but is often overlooked.
When there is cardiac amyloidosis, the heart may be too stiff to pump the blood efficiently around the body. This can lead to fluid build- up, causing leg swelling (oedema) and breathlessness due to fluid in the lungs. This problem is exacerbated if the patient drinks too much fluid.
Fluid excess can be avoided by careful attention to the 3 Ds:
- Diet
- Fluid intake should be steady and should usually not exceed 1.5 liters per day.
- Salt intake should be limited. This includes attention not just to salt deliberately added to the food during cooking or at the table but also to ready prepared foods with high salt content such as processed foods, crisps, bacon, canned meats, sausages, canned soups and smoked fish. Apart from that, a balanced, healthy diet is always advisable. It can be very helpful to meet with a dietician for precise and personalised dietary advice.
- Diuretics (water tablets)
- Doctors will often prescribe diuretics (water tablets) which increase the amount of urine produced and help the body to lose excess salt and water in the urine. This can help to reduce ankle swelling and breathlessness. Diuretics prescribed may include furosemide and spironolactone. Taking these drugs is not a substitute for avoidance of excessive dietary salt and water. Patients should follow their doctor’s advice carefully regarding the dose of diuretic and the time of day when the tablet should be taken.
- Daily weights
- Some patients benefit from recording their weight regularly, usually daily or weekly. It is important that weight should be measured consistently – using the same scales, at the same time of day. This is usually best done first thing in the morning after passing urine, just wearing underclothes. Several litres of fluid can accumulate in the body without it being very noticeable. An increase in weight can be an early sign of fluid overload. The doctor or nurse can then recommend appropriate measures such as increased diuretic dose, before the patient even feels unwell because of the fluid overload.
Heart transplantation
For hereditary ATTR amyloidosis, combined heart and liver transplant has been performed in a few dozen cases around the world. This operation is only an option for a very small minority of patients, and it carries significant risks.
Treatment of peripheral neuropathy symptoms
Medications that may help to alleviate neuropathic pain include gabapentin, pregabalin and duloxetine. Medical staff can give advice regarding appropriate foot care and footwear. This is important in order to prevent painless ulcers at pressure points and to protect areas of the foot that lack sensation.
Treatment of autonomic neuropathy symptoms
If there is orthostatic hypotension (drops in blood pressure and faintness on standing up from sitting or lying positions), elastic stockings may be recommended. Patients may benefit from instruction in how to change position carefully from lying to sitting, sitting to standing and standing to walking. Drug treatment with midodrine or fludrocortisone may also be helpful to maintain blood pressure and allow higher diuretic doses. Care should be taken to avoid dehydration if there is vomiting and diarrhoea. Intravenous fluids and anti‑nausea drugs may be necessary, but it is important to avoid fluid overload if there is heart disease. There are drugs that can help to control diarrhoea and constipation, and others that can help to combat erectile dysfunction.
Dealing with postural hypotension:
- After sitting down for more than 1/2 hour always stand for about 30 seconds before moving off.
- If feeling light headed or you have ringing in your ears when walking, stand still for a few moments until it passes. Sometimes you can walk through it after some practice!
- If it gets really bad always ask for help or sit down.
- When you get a cold or virus the postural hypotension can get worse. It may help by taking the standard doses of acetaminophen (paracetamol).
- When getting up from bed in the morning, always sit on the side for a few moments before walking off.
- Never run and always pace yourself.
- Always allow more time than you think you need.
Beta-2-microglobulin amyloidosis (Abeta2M)
Beta-2 microglobulin amyloidosis also called B2M amyloidosis, AB2M, Abeta2m and DRA (for Dialysis-Related Amyloidosis), is amyloidosis associated with the B2M (beta-2 microglobulin) protein. Beta-2 microglobulin amyloidosis occurs with end stage renal (kidney) failure after a patient has been on hemodialysis for many years, but it can also occur in patients who use continuous ambulatory peritoneal dialysis (CAPD) and, rarely, patients with renal failure who are not on dialysis. After a patient has been on dialysis for more than 15 to 20 years, research data suggests an incidence of 95% or higher. In Europe, studies show the rate at 100% after 13 years. Most experts agree that beta-2 microglobulin amyloidosis rarely occurs before 5 years of dialysis. The beta-2 microglobulin (B2M) protein is normally found on the surface of many white blood cells and is present in small amounts in the blood, cerebral spinal fluid, and urine. With certain conditions and diseases, if the body produces an overabundance of these cells, then the body reacts by raising the level of the B2M (beta-2 microglobulin) protein 42. There may also be an increase of B2M protein if the body destroys these cells due to a disease.
Healthy kidneys can clear out substances and toxins and remove them from the body. As kidney failure begins, the normal B2M protein increases. After many years of dialysis and with the ‘overexpression’ of this normal protein triggering misfolded amyloid proteins, the kidneys are unable to clear them out of the body, which then elevates the AB2M amyloid in the blood. As a result, the accumulation and deposit of AB2M amyloid protein is often found in bones, joints and tendons.
The level of B2M does not diagnose a specific disease, but may help determine how advanced a disease is. The increased level of B2M in the body can occur with several diseases such as AB2M amyloidosis, multiple myeloma, lymphomas, or leukemia, among many others. In patients with renal failure, the beta-2 microglobulin (B2M) protein accumulates in the serum, leading to secondary osteoarticular destruction and dialysis-related amyloidosis (DRA) 43. Beta-2 microglobulin amyloidosis commonly is associated with deposits in the carpal ligaments, synovium, and bone, resulting in carpal tunnel syndrome, destructive arthropathy, bone cysts, and fractures. Other organs involved include the heart, gastrointestinal tract, liver, lungs, prostate, adrenals, and tongue.
Some studies suggest that carpal tunnel syndrome is the most common symptom. It is caused by pressure on the “median nerve” from amyloid deposits, resulting in tingling, numbness and pain in the fingers and wrist. Other common symptoms may include joint pain and/or stiffness (arthralgia) and soft tissue masses, causing tenderness and pain. In addition, bone cysts may occur (making hollow cavities in the bone) which in turn may lead to fractures.
Serious complications may arise, especially if the patient has been on dialysis for a very long time –longer than 20 to 30 years. For example, if ligaments and discs are severely damaged by the AB2M amyloid deposition, paraplegia is possible. In some cases, patients may develop cardiac (heart) symptoms, such as arrhythmia (irregular heartbeat), which can range from mild to severe.
Congestive heart failure is rare, but may occur, due to extreme amyloid deposition in the heart. In addition, gastrointestinal (GI) bleeding has been found in some patients, but this is considered a rare occurrence.
Like all the amyloidosis diseases, the diagnosis of amyloidosis is based on the evidence of amyloid deposits and a defined tissue or organ involvement.
A lab test is performed that can analyze blood, urine, or fluid for increased levels of the B2M protein. Determining the level of B2M provides the doctor with more information about the health of the patient and the lab test is very helpful as a marker for detecting kidney damage, for distinguishing between different types of kidney disorders, or for tumors that occur with some blood cell cancers.
It is recommended that the patient have typing done in a reference laboratory at an experienced amyloidosis center, and the typing should agree with the clinical features and symptoms of the patient’s disease. Unfortunately, costs of typing are not always covered by insurance, but it is very important to have correct typing before any treatment is initiated. A biopsy sample is taken from a suspected affected area, like the synovium (the smooth lining of a joint) or from a cystic bone lesion. Then this biopsy sample is sent to a lab for Congo-red staining. The lab will stain the biopsy sample and, if it turns an apple green color under a ‘polarizing’ microscope, then amyloidosis is confirmed. The lab will also define the type of the specific amyloid protein. Using an “immunostaining” technique, the sample is tested with the antibodies of different amyloid proteins. If this test shows the correct reaction to the B2M protein, then the diagnosis is AB2M amyloidosis.
In addition, there are various imaging tests that can support the diagnosis. A doctor may use x-rays, CT (computed tomography) scans, ultrasonography (ultrasound), and MRIs (magnetic resonance imaging) on the various joints, tendons, and bones that are affected by AB2M amyloidosis.
In some cases, joint erosion or cystic bone lesions or fractures may be observed before the AB2M diagnosis, resulting in the need to test for this amyloidosis disease.
There is no treatment known for beta-2-microglobulin amyloidosis itself. Progress has been observed when some patients use dialysis membranes that are known to have a higher clearance of beta-2-microglobulin microglobulin (B2M). Studies indicate that this may result in a delay in the onset of AB2M amyloidosis and the associated symptoms for these patients. Traut et al 44 reported that patients using polyamide high-flux membranes had lower beta-2 microglobulin (B2M) concentrations compared with those patients on low-flux dialyzers. They postulated that the difference was mediated by an increase in beta-2 microglobulin (B2M) mRNA, lower concentrations of beta-2 microglobulin (B2M) released from the blood cells, and or better beta-2 microglobulin (B2M) clearance in patients treated with high-flux dialyzers 44. Yamamoto et al 45 have investigated Lixelle adsorbent columns that remove serum beta-2 microglobulin (B2M) safely in dialysis patients and significantly improved quality of life, strength, C-reactive protein levels, and B2M concentration.
Kidneys rarely recover or improve when end stage renal failure has set in. For some patients a kidney transplant is an option, depending on the patient’s overall health, and can slow or stop the advancement of AB2M amyloidosis.
Other medical treatment consists of treating the symptoms and, if possible, surgically removing the amyloid deposits. For some symptoms, doctors prescribe corticosteroids and NSAIDs (non-steroidal anti-inflammatory drugs). Physical and occupational therapy may be helpful, in addition to wrist splints, cervical collars, and other braces and supports. If the patient develops cardiac or gastrointestinal (GI) symptoms, a cardiologist or gastroenterologist should be consulted and individual treatments prescribed for that patient’s needs.
In addition, carpal tunnel release surgery can significantly reduce the pressure on the median nerve in the wrist, and surgery can be done to aid compression on the spinal cord to improve pain and discomfort. Other surgery on joints or tendons may be helpful to reduce pain and restore some function.
Leukocyte chemotactic factor 2 (ALECT2) amyloidosis
ALECT2 amyloidosis was first discovered in 2008 in a 61 year old woman whose kidney was removed because of kidney cancer 46. Seven years earlier she had been diagnosed with amyloidosis of unknown type. Since then she had suffered from slowly progressive kidney failure. Pathological analysis of the amyloid fibrils from the kidney that had been removed identified a protein called leukocyte chemotactic factor 2 (LECT2). Leukocyte chemotactic factor 2 (LECT2) is a protein that is made in the liver, circulates in the blood, and its complete function in the body is still undetermined. Scientists believe that it may be part of cartilage reconstruction and tissue repair, autoimmune response, or it may be part of your cell growth, among other possibilities.
The underlying cause of ALECT2 amyloidosis is unknown. It is also unclear whether ALECT2 amyloid deposition is a slow process beginning early in life and remaining undetected for years or whether it is a rapid process beginning later in life. ALECT2 amyloidosis generally appears in elderly people (usually after age 60), with only 5 reported cases in people aged under 50. Current research shows that patients have been diagnosed as young as 43 years of age and up to 88 years of age. The average age of patients is around 66 years old.
While there still exists a possibility that ALECT2 has some hereditary and genetic connection, the classification of this disease is currently listed as an ‘acquired’ systemic amyloidosis disease, and not a hereditary one. This is because there is no one genetic mutation found in any patient that has been diagnosed with ALECT2. However, the scientists question if ALECT2 is a “digenic disease.” “Di” means “two” in this case, which means that it could be the result of a combination of two disorders in the genetic sequence, involving a genetic mutation that has not yet been identified. Apart from one case of 2 brothers with ALECT2 amyloidosis, none of the patients described in the literature had any family history of amyloidosis. All patients with ALECT2 amyloidosis whose LECT2 gene has been examined have a particular, common variant (polymorphism) of the LECT2 gene (called homozygosity for G nucleotide at position 172) but no causative mutations have been described in this disease.
With the overproduction of this normal protein triggering misfolded amyloid proteins, the unknown cause for this overproduction means that there is no absolute conclusion in the classification for ALECT2 at this time. Research continues in this area.
In the US, ALECT2 amyloidosis may be a relatively common cause of amyloidosis that was previously either classified as “unknown type”, or misdiagnosed as a different type of amyloidosis. In US studies it accounted for 2.5-10% of all cases of kidney amyloidosis and 25% of all cases of liver amyloidosis. Recent data from the Mayoclinic suggest that ALECT2 amyloidosis is now as common as AA amyloidosis in the US (in this study the four most common types were AL – 61.7%, ATTR – 24.5%, AA 3.7% and ALECT 3.6%) In the UK ALECT2 amyloidosis accounts for about 1% of all patients referred with biopsy-proven kidney amyloid.
About 90% of all cases described occurred in people of Hispanic origin, and it was particularly common in the Southwestern US. It also appears to be more common in people of Punjabi, Arab, North African and Native American Indian origin than in those of other ethnic origins.
ALECT2 organ involvement is mainly associated with kidney damage. Recent studies show that liver involvement with ALECT2 could also be more common than previously thought. However, even though amyloid protein has been discovered in liver specimens, most patients have not presented with symptoms from liver damage. Along with kidney damage and liver involvement, there have been reports of findings of amyloid in the spleen, colon, and adrenal glands in a number of ALECT2 patients, although symptoms were also not often evident in those areas.
Patients with ALECT2 amyloidosis affecting the kidneys usually come to medical attention because of progressive chronic kidney failure. Protein in the urine is generally a less prominent disease feature than in other types of amyloidosis affecting the kidneys. Chronic hypertension and diabetes are common in patients diagnosed with ALECT2 amyloidosis and a minority (up to 13% in one study) have kidney cancer.
Many experts agree that ALECT2 amyloidosis should be considered while looking for a diagnosis when a patient has evidence of renal (kidney) disease. A patient’s symptoms may appear as renal failure or “nephrotic syndrome.” Nephrotic syndrome is a group of symptoms that relate to kidney problems, including: protein found in the urine, high cholesterol levels, low blood protein levels, and swelling.
Although there is still little known about ALECT2 when compared to other forms of amyloidosis diseases, it is extremely important to get an accurate diagnosis of ALECT2 and not mistake it for the more common AL (or any other) amyloidosis disease that may cause kidney damage. With AL amyloidosis (light chain amyloidosis) and so many of the amyloidosis diseases, the treatments vary one from another and can be harmful to the patient if a certain treatment is prescribed for an amyloidosis disease that is not accurately diagnosed.
Many cases of liver ALECT2 amyloidosis were diagnosed incidentally in liver biopsies performed for unrelated conditions. The most common clinical abnormality associated with liver ALECT2 amyloidosis is raised alkaline phosphatase (ALP), an abnormality commonly seen in many liver diseases.
Clinically significant liver ALECT2 amyloidosis is rare in patients diagnosed with kidney ALECT2 amyloid, and clinically significant kidney ALECT2 amyloid is unusual in those diagnosed with liver ALECT2 amyloidosis. ALECT2 amyloid deposits have been described in other organs such as the spleen and prostate, but not in the heart.
There are no specific treatments for ALECT2 amyloidosis.
Patients with ALECT2 amyloidosis affecting the kidneys should receive supportive treatment, as for kidney failure caused by other types of amyloidosis. ALECT2 amyloidosis in the liver may not require treatment.
All patients with ALECT2 amyloidosis affecting their kidney function should be seen regularly by a nephrologist who is expert in amyloidosis. Important principles include:
- carefully monitoring and maintenance of fluid balance
- regular testing of kidney functions with blood and urine tests
- use of diuretics when required
Abnormal kidney function due to ALECT2 amyloidosis may affect the ability of the kidneys to produce urine. This means that the body is unable to cope well with excess fluids. Patients with fluid overload may develop swelling in the legs (edema) or difficulty breathing due to heart failure.
If kidney damage becomes severe or results in renal failure, dialysis is necessary. Outcomes after kidney transplantation appear to be good, although ALECT2 amyloidosis can slowly recur in the graft. It is still unknown if renal transplantation is a viable option for all patients with ALECT2 amyloidosis and is considered on a case-by-case basis. These cases will continue to be documented to determine the success rates over time.
Patients with ALECT2 amyloidosis affecting the kidneys have better overall survival than those with AL or AA amyloidosis affecting the kidneys, presumably because the heart is not affected by ALECT2 amyloidosis.
Cryopyrin-associated periodic syndrome (CAPS)-associated amyloidosis
Types of CAPS include the following:
- Familial cold autoinflammatory syndrome (FCAS)
- Muckle-Wells syndrome (MWS)
- Neonatal-onset multisystem inflammatory disease (NOMID)
These disorders are typically associated with heterozygous mutations in the NLRP3 (CIAS1) gene, which encodes the cryopyrin (NALP3) protein, and are inherited in an autosomal dominant manner 47. The inflammation in CAPS is driven by excessive release of interleukin (IL)–1β. [51] IL-1β release is normally regulated by an intracellular protein complex known as the inflammasome that maps to a gene sequence called NLRP3. Mutations in NLRP3 may cause an aberrant cryopyrin protein inside the inflammasome, leading to the release of too much IL-1β and subsequent multisystem inflammation.
Secondary to increased IL-1β, CAPS patients have chronically elevated levels of acute-phase reactants, especially serum amyloid A (SAA) and high sensitivity C-reactive protein (hsCRP) 48. With elevated SAA coupled with multisystem cytokine dysregulation, multisystem amyloid deposition can be severe, with the most feared complication being renal failure. By blocking the action of IL-1β or down-regulating its production, inflammation and therefore amyloid deposition can be reduced 49.
In a randomized double-blind CAPS therapy trial, the soluble decoy receptor rilonacept was shown to provide rapid and profound symptom improvement, in addition to improvement in measures of inflammation such as hsCRP and SAA levels 49. In the second part of the study, continued treatment with rilonacept maintained improvements, where disease activity worsened with discontinuation of the drug 49.
Canakinumab is a competing human immunoglobulin G (IgG) monoclonal antibody that also targets IL-1β and has shown efficacy in autoinflammatory conditions that, if untreated, are fatal by age 20 years in about 20% of individuals 28. Both agents have the potential to cause infections, but they are usually mild and treatable.
Muckle-Wells syndrome
Muckle-Wells syndrome is another autoinflammatory syndrome secondary to a mutation in the CIAS gene encoding cryopyrin, a component of the inflammasome that regulates the processing of IL-1β. Patients commonly experience sensorineural hearing loss and other neuropathies 50. The IL-1β receptor antagonist anakinra has been shown to improve the signs and symptoms in Muckle-Wells syndrome by decreasing serum (hsCRP and SAA) and cytokines such as IL-6, IL-8, IL-12, and IL-1β. In some cases, it improved sensory deafness, as well as the laboratory values for markers of inflammation Muckle-Wells syndrome 51.
Hereditary Renal Amyloidoses
Hereditary amyloidoses encompass a group of conditions that each are related to mutations in a specific protein. The most common form is transthyretin amyloidosis (usually neuropathic), but non-neuropathic amyloidoses are likely the result of abnormalities in lysozyme, fibrinogen, alpha-chain, or apolipoprotein A-I and A-II 52. Consider these diseases when a renal biopsy demonstrates amyloid deposition and when they are likely diagnoses (rather than light chain amyloidosis [AL] or A amyloidosis [AA]) because the family history suggests an autosomal dominant disease. Again, the definitive diagnosis is made using immunohistologic staining of the biopsy material with antibodies specific for the candidate amyloid precursor proteins. Clinical correlation is required to diagnose amyloid types, even if a hereditary form is detected by amyloid protein typing 53.
Apolipoprotein AI amyloidosis (apoAI) is an autosomal dominant amyloidosis caused by point mutations in the apoAI gene. Usually, this amyloidosis is a prominent renal amyloid but can also form in many locations. ApoAI (likely of normal sequence) is the fibril precursor in localized amyloid plaques in the aortae of elderly people. ApoAI can present either as a nonhereditary form with wild-type protein deposits in atherosclerotic plaques or as a hereditary form due to germline mutations in the apoA1 gene 54. Currently, more than 50 apoAI variants are known and 13 are associated with amyloidosis 54. As more gene locations are found, the clinical phenotypes are slowly being elucidated.
Fibrinogen amyloidosis (AFib) is an autosomal dominant amyloidosis caused by point mutations in the fibrinogen alpha chain gene. If DNA sequences indicate a mutant amyloid precursor protein, protein analysis of the deposits must provide the definitive evidence in laboratories with sophisticated methods 53.
Lysozyme amyloidosis (ALys) is an autosomal dominant amyloidosis caused by point mutations in the lysozyme gene.
Apolipoprotein AII amyloidosis (AapoAII) is an autosomal dominant amyloidosis caused by point mutations in the apoAII gene. The 2 kindreds described with this disorder have each carried a point mutation in the stop codon, leading to production of an abnormally long protein.
Central nervous system amyloidoses
Beta protein amyloid
The amyloid beta precursor protein (AβPP), which is a transmembrane glycoprotein, is the precursor protein in beta protein amyloid (A). Three distinct clinical settings are as follows:
- Alzheimer disease has a normal-sequence protein, except in some cases of familial Alzheimer disease, in which mutant beta protein is inherited in an autosomal dominant manner.
- Down syndrome has a normal-sequence protein that forms amyloids in most patients by the fifth decade of life.
- Hereditary cerebral hemorrhage with amyloidosis (HCHWA), Dutch type, is inherited in an autosomal dominant manner. The beta protein contains a point mutation. These patients typically present with cerebral hemorrhage followed by dementia.
The accumulation of amyloid-beta peptide (Aβ) in the brain both in the form of plaques in the cerebral cortex and in blood vessel as cerebral amyloid angiopathy (CAA) causes progressive cognitive decline. Researchers have used immunization strategies to neutralize amyloid fibrils before deposition. Experimental models and human clinical trials have shown that accumulation of Aβ plaques can be reversed by immunotherapy. Aβ immunization results in solubilization of plaque Aβ42 which, at least in part, exits the brain via the perivascular pathway, causing a transient increase in the severity of CAA. The extent to which these vascular alterations following Aβ immunization in Alzheimer disease are reflected in changes in cognitive function remains to be determined 55.
Newer research has focused on finding a way to prevent the parent molecules from fragmenting and then aggregating to form toxic oligomers. Aβ peptide is known as a factor in the pathology of Alzheimer disease. Aβ aggregation is dependent on monomer concentration, nucleus formation, fibril elongation, and fibril fragmentation. Cellular, kinetic, and radiolabeling experiments have demonstrated that secondary nucleation occurs on the surface of only specific types of Aβ fibrils. The work focused on Aβ42, which could be a target molecule for future therapies to prevent nucleation events, oligomer formation rates, and neurotoxic effects 56.
Other target fibrils with secondary amyloidogenic nucleation potential are also being investigated across large-scale comparative data sets 57.
Prion protein amyloidosis (APrP)
The precursor protein in APrP is a prion protein, which is a plasma membrane glycoprotein. The etiology is either infectious (ie, kuru) and transmissible spongiform encephalitis (TSE) or genetic (ie, Creutzfeldt-Jakob disease [CJD], Gerstmann-Sträussler-Scheinker [GSS] syndrome, fatal familial insomnia [FFI]). The infectious prion protein is a homologous protein encoded by a host chromosomal gene, which induces a conformational change in a native protease-sensitive protein, increasing the content of beta-pleated sheets. The accumulation of these beta-pleated sheets renders the protein protease-resistant and therefore amyloidogenic 58. Patients with TSE, CJD, GSS, and FFI carry autosomal dominant amyloidogenic mutations in the prion protein gene; therefore, the amyloidosis forms even in the absence of an infectious trigger.
Similar infectious animal disorders include scrapie in sheep and goats and bovine spongiform encephalitis (ie, mad cow disease).
Cystatin C amyloidosis
The precursor protein in cystatin C amyloidosis (ACys) is cystatin C, which is a cysteine protease inhibitor that contains a point mutation. This condition is clinically termed Hereditary cerebral hemorrhage with amyloidosis (HCHWA), Icelandic type.
ACys is autosomal dominant. Clinical presentation includes multiple strokes and mental status changes beginning in the second or third decade of life. Many of the patients die by age 40 years. This disease is documented in a 7-generation pedigree in northwest Iceland. The pathogenesis is one of mutant cystatin that is widely distributed in tissues, but fibrils form only in the cerebral vessels; therefore, local conditions must play a role in fibril formation.
Non-amyloid beta cerebral amyloidosis (chromosome 13 dementias)
Two syndromes (British and Danish familial dementia) that share many aspects of clinical Alzheimer disease have been identified. Findings include the presence of neurofibrillary tangles, parenchymal preamyloid and amyloid deposits, cerebral amyloid angiopathy (CAA), and amyloid-associated proteins. Both conditions have been linked to specific mutations on chromosome 13; they cause abnormally long protein products (ABri and ADan) that ultimately result in different amyloid fibrils.
Localized amyloidosis
Localized amyloidosis means that just a single organ or part of the body is affected. About 12% of all newly diagnosed amyloidosis patients at the National Amyloidosis Centre have localized amyloidosis 59. In these rare conditions, amyloid deposits are found in just one part of the body. Localized amyloidosis almost never progress to systemic amyloidosis. Localised amyloidosis is usually of AL amyloidosis (light chain amyloidosis) type and is a very different disease from systemic amyloidosis 60. Localized amyloidosis may require no treatment or may be completely curable, for example by surgical excision of the amyloid deposits. Localized amyloidosis almost never progresses to systemic amyloidosis.
In localized AL amyloidosis, local foci of abnormal plasma cells producing AL amyloid deposits limited to just one place may be present in the 59:
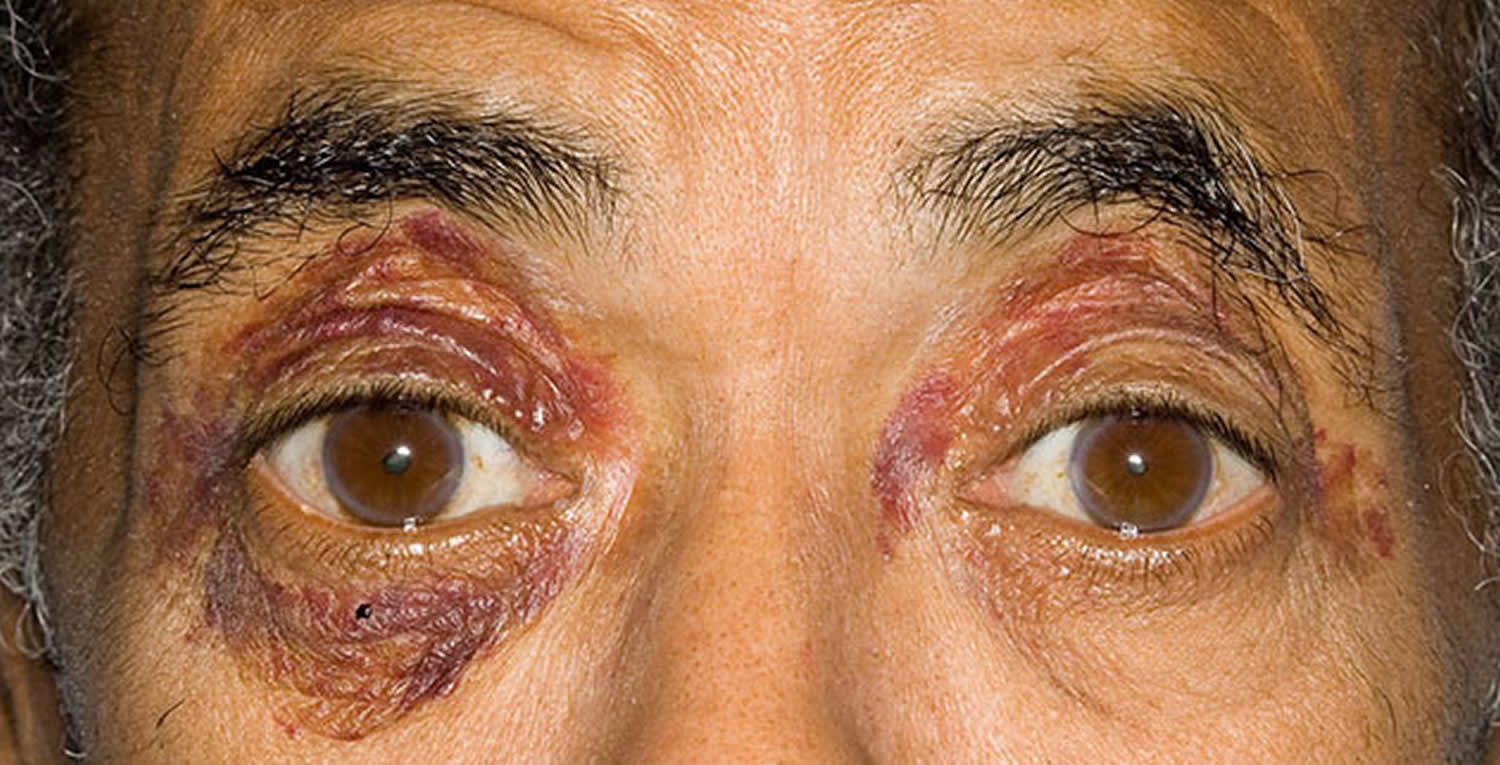
- Skin. Around 14% of patients diagnosed with localized amyloidosis at the National Amyloidosis Centre have amyloid deposits affecting the skin. Localized amyloidosis in the skin may be macular (flat lesions), papular (raised lesions under 5 mm in diameter –sometimes known as lichen amyloidosis), or nodular (raised lesions over 5 mm in diameter). The condition
may appear in adults of any age, most commonly in people aged around 60. It is equally common in men and women. Skin of the head and neck region is commonly affected and there may be several skin lesions in different locations. Lichen amyloidosis often affects the shins, thighs and forearms. In some patients, local friction from nylon brushes, towels and other rough materials may contribute to the production of macular and lichenoid lesions.- Macular cutaneous amyloidosis: amyloid fibrils are derived from a skin protein called galactin. There is an itchy rash, with scaly, brownish lesions that may be flat or raised (macules or papules). It is common in people with dark skin.
- Hereditary cutaneous amyloid: rare, with unknown fibril type, sometimes with an associated disease affecting many body systems.
- Upper airways and respiratory tract. Localized amyloid in the respiratory tract is usually of AL type. Amyloid in the larynx is more frequent in older people but occasionally affects young adults. This may cause hoarseness or a sensation of ‘fullness’ in the throat, choking and shortness of breath on exertion. This condition is usually quite benign but sometimes it may be progressive. Tracheobronchial amyloid affects the airways below the throat and above the lungs. It is very rare, and usually affects people aged over 50. There may be shortness of breath and cough. They may cough up blood and there may be recurrent pneumonia. The lung tissue itself is the most common site for localized respiratory amyloid deposits. The deposits usually appear as nodules on chest X-ray. The nodules may grow slowly and often there are few symptoms. Frequently the diagnosis is made after nodules are detected incidentally on a chest X-ray. Treatment may not be required if the local amyloid deposits cause few or no symptoms. If there are troublesome symptoms, then the local amyloid deposits may be removed, either by surgery of by laser therapy.
- Genital and urinary system. Localized amyloid deposits may be found in the bladder or genital tract. They are usually of AL type but occasionally may be of ATTR type. There may be no symptoms, or there may be irritation or burning on passing urine, or blood in the urine. When patients undergo urological investigation with cystoscopy, the amyloid deposits may at first be mistaken for tumors, before biopsy examination shows localized amyloid. These deposits can usually be treated by surgical removal (resection).
- Eye. Local amyloid deposits may affect the cornea, the conjunctiva or the orbit of the eye. When the orbit is affected, the lesions may disrupt eye movements and the structure of the orbit. Amyloid deposits in the eye may be of AL type. Other, very rare types have been identified, with precursor proteins called lactoferrin and keratoepithelin. If these deposits are symptomatic, they may be treated with local excision.
- Lymph nodes (rare). They often cause no symptoms and require no specific treatment.
- Gastrointestinal tract (rare). Localised amyloid deposits of AL type may, rarely, be found in the stomach, or the small or large intestine. Sometimes these do not cause symptoms and are detected incidentally when patients undergo routine gastroscopy (examination of the stomach with a flexible viewing instrument inserted via the mouth and esophagus) or colonoscopy (examination of the inner lining of the large intestine with a flexible viewing instrument inserted into the rectum). These localized amyloid deposits may cause symptoms such as bleeding from the gut, heartburn or constipation. Supportive care is usually recommended for these patients.
- Localized amyloid in other sites. Small amyloid deposits of non-AL type are sometimes detected in the blood vessel walls, and in benign or malignant tumors of glandular tissue. Such deposits are usually detected incidentally when these tissues are biopsied for a reason unrelated to amyloidosis. They generally do not cause any disease and do not require treatment or follow up.
Localized AL amyloidosis is not usually associated with detection of a serum monoclonal protein/paraprotein in blood tests 59.
It should be noted that serum monoclonal protein/paraprotein may sometimes be detected in patients with myeloma or in a benign condition called ‘monoclonal gammopathy of uncertain significance’ (MGUS), which is not associated with disease and does not require any treatment. Around 3% of people aged over 50 and 5% of those aged over 70 have MGUS, so some people may just coincidentally have both localized AL amyloidosis and MGUS at the same time. It is important that full investigation is performed to definitely rule out systemic AL amyloidosis in these people.
Localised amyloid is diagnosed by biopsy of the affected tissue. In this procedure, a small tissue sample is obtained, processed and examined under the microscope. When systemic amyloidosis is present, biopsy samples may be obtained either from the affected organs, or from other, easily accessible sites, such as the fat under the skin of the abdomen, or the rectum. However, as localized amyloidosis only affects a single site, the biopsy specimen must be obtained from that site itself. This may be simple if the site affected is easily accessible, such as the skin, or more complex if the site is difficult to access, for example the lungs. Specialist assistance (for example bronchoscopic biopsy of lung lesions) may then be necessary in order to obtain biopsy specimens.
When localized amyloid deposits are detected in a single site, doctors need to rule out systemic amyloidosis by assessing functions of all the vital organs (heart, liver, kidney and nerves). This assessment includes blood and urine tests to evaluate organ functions, bone marrow biopsy and further investigations to evaluate any disease symptoms. Tests such as ECG, echocardiography and cardiac MRI may be used to evaluate heart function and to assess the heart for amyloid deposits. A SAP scan is a type of nuclear medicine imaging test which uses iodine-123 (123I) and serum amyloid P component (SAP) to diagnose amyloidosis. If the SAP scan shows no amyloid deposits and if the other test results also do not show any evidence of amyloid, then localized amyloidosis can be diagnosed.
Other localized amyloidoses
Cerebral amyloid angiopathy (CAA)
Cerebral amyloid angiopathy (CAA) is the most common form of localized amyloidosis 60. Cerebral amyloid angiopathy (CAA) is a neurological condition that causes amyloid protein to build up in the walls of blood vessels in the brain 61. Cerebral amyloid angiopathy (CAA) is totally unrelated to all other forms of local and systemic amyloidosis and patients with this condition are treated by neurologists and stroke physicians, not by amyloidosis physicians.
In CAA, amyloid fibrils composed of beta-protein form Aβ amyloid deposits in the walls of the blood vessels in the brain. This type of amyloid deposit is also found within the brain substance in patients with Alzheimer’s disease. Despite this close relationship and an increased tendency for the two diseases to occur together, most patients with Alzheimer’s disease do not have cerebral amyloid angiopathy (CAA), and most with CAA do not have Alzheimer’s disease. But in CAA the amyloid in the blood vessels of the brain is known to be a common cause of strokes (bleeds in the brain, known as intracerebral hemorrhages) and problems with memory or thinking (sometimes leading to dementia).
CAA is actually a common process in aging and usually harmless 62. In some people with severe CAA, however, the protein deposits cause the blood vessel walls to crack, in which case blood can leak out and damage the brain. The damage from this process is called a bleeding (or hemorrhagic) stroke 63.
Cerebral amyloid angiopathy (CAA) affects elderly people, almost always aged over 60, and becomes more common with increasing age. It is estimated that CAA is present in the brain in 20-40% of the elderly general population (it’s occasionally diagnosed in people in their 50’s or 60’s but is much more common in people in their 70’s and 80’s) and in 50-60% of elderly people with dementia 64. Alzheimer’s disease also becomes more common as people age, so elderly people may have both Alzheimer’s and CAA. Apart from increasing age, and a gene called apolipoprotein E may slightly affect the risk for CAA, their association with the disease appears to be too weak to cause multiple cases to cluster in a family and is not useful for predicting who will or won’t get the disease. Most people with this gene do not develop CAA and many with the disease do not have the risky gene.
There are some families (primarily from Denmark, Iceland, and Belgium) that inherit CAA in a dominant fashion, but most CAA patients do not have any affected family members 65. Although there may be some increased genetic risk in relatives of CAA patients, experts believe this risk to be small and rarely see two affected people in the same family.
Neurologists and stroke physicians can diagnose CAA on the basis of clinical data combined with information from blood-sensitive brain imaging scans, looking for areas of bleeding near the surface of the brain.
There are no specific drugs available at present for CAA, but drug pressure lowering with antihypertensive drugs in patients who have had a CAA related stroke reduces the risk of future stroke. There are also a number of new drugs in various stages of development, and but there is not yet an effective treatment for CAA.
Gelsolin amyloidosis
This disorders was first reported in 1969 and found to be heritable in a Finnish family in an autosomal dominant fashion. A flurry of research uncovered the molecular dysfunction of the disease but a treatment approach has yet to be devised. Gelsolin amyloidosis has now been described in countries all over the world and is often undiagnosed or misdiagnosed 66.
Amyloid fibrils include a gelsolin fragment that contains a point mutation. Two amyloidogenic gelsolin mutations are described. “A G654A or G654T DNA mutation in the gelsolin coding area (q32–34) of chromosome 9, which changes an aspartate at position 187 in the gelsolin protein to an asparagine or tyrosine (D187N/Y) residue respectively. These mutations lead to gelsolin fragment formation and amyloidogenesis” 66.
The precursor protein in gelsolin amyloidosis (AGel) is the ubiquitous actin-modulating protein gelsolin, but the mutated form lacks a crucial calcium binding site, allowing it to unfold and expose the amino acid chain to proteolysis while processed in the Golgi and again after exocytosis, forming an 8- and 5-kd amyloidogenic fragment 67. When the gelsolin gene is transcribed, some is spliced to form cytoplasm and some is secreted from the cell, but only the secreted forms deposit as amyloid secondary to faulty post-translational modification of gelsolin 68. Mass spectrometric-based proteomic analysis and immunohistochemical staining can reliably differentiate amyloid as gelsolin type 69.
Most early symptoms are ocular; thus, the ophthalmologist is crucial in early diagnosis. Clinical characteristics include slowly progressive cranial neuropathies, distal peripheral neuropathy, and lattice corneal dystrophy. Gelsolin gene sequencing is the only confirmatory test, but tissue samples can also be stained with tagged, commercially available gelsolin antibodies to provide a solid pathology diagnosis 70. An extremely detailed review of this condition, along with the recent scientific developments, was published by Solomon et al in 2012 66.
Atrial natriuretic factor amyloidosis
The precursor protein is atrial natriuretic factor (ANF), a hormone controlling salt and water homeostasis that is synthesized by the cardiac atria. Amyloid deposits are localized to the cardiac atria. This condition is highly prevalent in elderly people and generally is of little clinical significance. ANF amyloidosis (AANF) is most common in patients with long-standing congestive heart failure, presumably because of persistent ANF production. Proper diagnosis requires extensive testing, including paraffin blocks of tissue for molecular typing by liquid chromatography-tandem mass spectrometry and observation of Congo red staining on affected atrial tissues 71. No known relation exists to the amyloidoses that involve the cardiac ventricles (ie, AL, ATTR).
Keratoepithelin amyloidosis and lactoferrin amyloidosis
Point mutations occur in a gene termed BIGH3, which encodes keratoepithelin and leads to autosomal dominant corneal dystrophies characterized by the accumulation of corneal amyloid. Some BIGH3 mutations cause amyloid deposits, and others cause nonfibrillar corneal deposits. Another protein, lactoferrin, is also reported as the major fibril protein in familial subepithelial corneal amyloidosis. The relationship between keratoepithelin and lactoferrin in familial corneal amyloidosis is not yet clear.
Calcitonin amyloid
In calcitonin amyloid (ACal), the precursor protein is calcitonin, a calcium regulatory hormone synthesized by the thyroid. Patients with medullary carcinoma of the thyroid may develop localized amyloid deposition in the tumors, consisting of normal-sequence procalcitonin (ACal). The presumed pathogenesis is increased local calcitonin production, leading to a sufficiently high local concentration of the peptide and causing polymerization and fibril formation.
Islet amyloid polypeptide amyloidosis
In islet amyloid polypeptide amyloidosis (AIAPP), the precursor protein is an islet amyloid polypeptide (IAPP), also known as amylin. IAPP is a protein secreted by the islet beta cells that are stored with insulin in the secretory granules and released in concert with insulin. Normally, IAPP modulates insulin activity in skeletal muscle, influencing energy homeostasis, satiety, blood glucose levels, adiposity, and even body weight 72. IAPP amyloid is found in insulinomas and in the pancreas of many patients with diabetes mellitus type 2 (DM2). IAPP could be toxic at the competitive or non-competitive level. IAPP analogs are now being investigated in the treatment of DM2 and obesity 72.
Prolactin amyloid
In prolactin amyloid (Apro), prolactin or prolactin fragments are found in the pituitary amyloid. This condition is often observed in elderly people and has also been reported in an amyloidoma in a patient with a prolactin-producing pituitary tumor. The spherical amyloid deposits have a characteristic MRI appearance that differs from common pituitary adenomas 73.
Keratin amyloid
Some forms of cutaneous amyloid react with antikeratin antibodies. The identity of the fibrils is not chemically confirmed in keratin amyloid (Aker). Aker is often described as primary cutaneous amyloidosis and is extremely rare 74.
Medin amyloid
Aortic medial amyloid occurs in most people older than 60 years. Medin amyloid (AMed) is derived from a proteolytic fragment of lactadherin, a glycoprotein expressed by mammary epithelium.
Nonfibrillar Components of Amyloid
All types of amyloid deposits contain not only the major fibrillar component (solubility in water, buffers of low ionic strength), but also nonfibrillar components that are soluble in conventional ionic-strength buffers. The role of the minor components in amyloid deposition is not clear. These components do not appear to be absolutely required for fibril formation, but they may enhance fibril formation or stabilize formed fibrils.
The nonfibrillar components, contained in all types of amyloid, are discussed below. However, note that other components found in some types of amyloid include complement components, proteases, and membrane constituents.
Pentagonal component
Pentagonal (P) component comprises approximately 5% of the total protein in amyloid deposits. This component is derived from the circulating serum amyloid P (SAP) component, which behaves as an acute-phase reactant. The P component is one of the pentraxin group of proteins, with homology to C-reactive protein. In experimental animals, amyloid deposition is slowed without the P component.
Radiolabeled material homes to amyloid deposits; therefore, this component can be used in amyloid scans to localize and quantify amyloidosis and to monitor therapy response. Radiolabeled P component scanning has proven clinically useful in England, where the technology was developed, but it is available in only a few centers worldwide.
Apolipoprotein E
Apolipoprotein E (apoE) is found in all types of amyloid deposits.
One allele, ApoE4, increases the risk for beta protein deposition, which is associated with Alzheimer disease. ApoE4 as a risk factor for other forms of amyloidosis is controversial.
The role of apoE in amyloid formation is not known.
Glycosaminoglycans (GAGs)
GAGs are heteropolysaccharides composed of long, unbranched polysaccharides that contain a repeating disaccharide unit. These proteoglycans are basement membrane components intimately associated with all types of tissue amyloid deposits. Amyloidotic organs contain increased amounts of GAGs, which may be tightly bound to amyloid fibrils. Heparan sulfate and dermatan sulfate are the GAGs most often associated with amyloidosis.
Heparan sulfate and dermatan sulfate have an unknown role in amyloidogenesis. Studies of A amyloidosis (AA) and light chain amyloidosis (AL) amyloid have shown marked restriction of the heterogeneity of the glycosaminoglycan chains, suggesting that particular subclasses of heparan and dermatan sulfates are involved.
Compounds that bind to heparan sulfate proteoglycans (eg, anionic sulfonates) decrease fibril deposition in murine models of AA and have been suggested as potential therapeutic agents.
Mechanisms of Amyloid Formation
Amyloid protein structures
In all forms of amyloidosis, the cell secretes the precursor protein in a soluble form that becomes insoluble at some tissue site, compromising organ function. All the amyloid precursor proteins are relatively small (ie, molecular weights 4000-25,000) and do not share any amino acid sequence homology. The secondary protein structures of most soluble precursor proteins (except for SAA and chromosomal prion protein [Prpc]) have substantial beta-pleated sheet structure, while extensive beta-sheet structure occurs in all of the deposited fibrils.
In some cases, hereditary abnormalities (primarily point mutations or polymorphisms) in the precursor proteins are present (eg, lysozyme, fibrinogen, cystatin C, gelsolin). In other cases, fibrils form from normal-sequence molecules (eg, AL, β2 M). In other cases, normal-sequence proteins can form amyloid, but mutations underlying inflammatory milieu accelerate the process (eg, TTR, beta protein precursor or CAPS).
Deposition location
In localized amyloidoses, the deposits form close to the precursor synthesis site; however, in systemic amyloidoses, the deposits may form either locally or at a distance from the precursor-producing cells. Amyloid deposits primarily are extracellular, but reports exist of fibrillar structures within macrophages and plasma cells.
Proteolysis and protein fragments
In some types of amyloidosis (eg, always in AA, often in AL and ATTR), the amyloid precursors undergo proteolysis, which may enhance folding into an amyloidogenic structural intermediate. In addition, some of the amyloidoses may have a normal proteolytic process that is disturbed, yielding a high concentration of an amyloidogenic intermediate. For example, it was shown that the mast cells of allergic responses may also participate in the development of secondary or amyloid AA in chronic inflammatory conditions. Mast cells hasten the partial degradation of the SAA protein that can produce highly amyloidogenic N-terminal fragments of SAA. However, factors that lead to different organ tropisms for the different amyloidoses are still largely unknown.
Whether the proteolysis occurs before or after tissue deposition is unclear in patients in whom beta protein fragments are observed in tissue deposits. In some types of amyloid (eg, AL, Aβ, ATTR), nonfibrillar forms of the same molecules can accumulate before fibril formation; thus, nonfibrillar deposits, in some cases, may represent intermediate deposition.
Amyloidosis symptoms
You may not experience signs and symptoms of amyloidosis until the condition is advanced. When signs and symptoms are evident, they depend on which of your organs are affected.
Signs and symptoms of amyloidosis may include:
- Swelling of your ankles and legs
- Severe fatigue and weakness
- Shortness of breath
- Numbness, tingling or pain in your hands or feet, especially pain in your wrist (carpal tunnel syndrome)
- Diarrhea, possibly with blood, or constipation
- Unintentional, significant weight loss
- An enlarged tongue
- Skin changes, such as thickening or easy bruising, and purplish patches around the eyes
- An irregular heartbeat
- Difficulty swallowing
Amyloidosis causes
In general, amyloidosis is caused by the buildup of an abnormal protein called amyloid. Amyloid is produced in your bone marrow and can be deposited in any tissue or organ. The specific cause of your condition depends on the type of amyloidosis you have.
There are several types of amyloidosis, including:
- AL amyloidosis (immunoglobulin light chain amyloidosis) is the most common type and can affect your heart, kidneys, skin, nerves and liver. Previously known as primary amyloidosis, AL amyloidosis occurs when your bone marrow produces abnormal antibodies that can’t be broken down. The antibodies are deposited in your tissues as amyloid, interfering with normal function.
- AA amyloidosis mostly affects your kidneys but occasionally your digestive tract, liver or heart. It was previously known as secondary amyloidosis. It occurs along with chronic infectious or inflammatory diseases, such as rheumatoid arthritis or inflammatory bowel disease.
- Hereditary amyloidosis (familial amyloidosis) is an inherited disorder that often affects the liver, nerves, heart and kidneys. Many different types of gene abnormalities present at birth are associated with an increased risk of amyloid disease. The type and location of an amyloid gene abnormality can affect the risk of certain complications, the age at which symptoms first appear, and the way the disease progresses over time.
- Dialysis-related amyloidosis develops when proteins in blood are deposited in joints and tendons — causing pain, stiffness and fluid in the joints, as well as carpal tunnel syndrome. This type generally affects people on long-term dialysis.
- Wild-type amyloidosis. This variety of amyloidosis occurs when the TTR protein made by the liver is normal but produces amyloid for unknown reasons. Formerly known as senile systemic amyloidosis, wild-type amyloidosis tends to affect men over age 70 and typically targets the heart. It can also cause carpal tunnel syndrome.
- Localized amyloidosis. This type of amyloidosis often has a better prognosis than the varieties that affect multiple organ systems. Typical sites for localized amyloidosis include the bladder, skin, throat or lungs. Correct diagnosis is important so that treatments that affect the entire body can be avoided.
Risk factors for amyloidosis
Anyone can develop amyloidosis. Factors that increase your risk include:
- Age. Most people diagnosed with AL amyloidosis, the most common type, are between ages 60 and 70, although earlier onset occurs.
- Sex. Nearly 70 percent of people with AL amyloidosis are men.
- Other diseases. Having a chronic infectious or inflammatory disease increases your risk of AA amyloidosis.
- Family history. Some types of amyloidosis are hereditary.
- Kidney dialysis. Dialysis can’t always remove large proteins from the blood. If you’re on dialysis, abnormal proteins can build up in your blood and eventually be deposited in tissue. This condition is less common with modern dialysis techniques.
- Race. People of African descent appear to be at higher risk of carrying a genetic mutation associated with the type of amyloidosis that can harm the heart.
Complications of amyloidosis
The potential complications of amyloidosis depend on which organs the amyloid deposits affect. Amyloidosis can seriously damage your:
- Kidneys. Amyloid can harm the kidneys’ filtering system, causing protein to leak from your blood into your urine. The kidneys’ ability to remove waste products from your body is lowered, which may eventually lead to kidney failure.
- Heart. Amyloid reduces your heart’s ability to fill with blood between heartbeats. Less blood is pumped with each beat, and you may experience shortness of breath. If amyloidosis affects your heart’s electrical system, your heart rhythm may be disturbed.
- Nervous system. You may experience pain, numbness or tingling of the fingers or numbness, lack of feeling or a burning sensation in your toes or the soles of your feet. If amyloid affects the nerves that control your bowel function, you may experience periods of alternating constipation and diarrhea.
If the condition affects nerves that control blood pressure, you may experience dizziness or near fainting when standing too quickly.
Diagnosis of Amyloidosis
Amyloidosis is often overlooked because the signs and symptoms can mimic those of more-common diseases. Diagnosis as early as possible can help prevent further organ damage. Precise diagnosis is important because treatment varies greatly, depending on your specific condition.
Your doctor is likely to start with a thorough medical history and physical exam. After that, you may have:
- Laboratory tests. Your blood and urine may be analyzed for abnormal protein that can indicate amyloidosis. Depending on your signs and symptoms, you may also have thyroid and liver function tests.
- Biopsy. A tissue sample may be taken and checked for signs of amyloidosis. The biopsy may be taken from your abdominal fat, bone marrow, or an organ such as your liver or kidney. Tissue analysis can help determine the type of amyloid deposit.
- Imaging tests. Images of the organs affected by amyloidosis can help establish the extent of your disease. Echocardiogram may be used to assess the size and functioning of your heart. Other imaging tests can evaluate the extent of amyloidosis in your liver or spleen.
- SAP scintigraphy (SAP scan). Serum amyloid P component (SAP) is a normal blood protein, present in everybody, which is always present in amyloid deposits, in all types of amyloidosis because it binds strongly to amyloid fibrils of all types. In healthy people there are very small quantities of SAP and it is only present inside the bloodstream, but not in the organs. In the bodies of patients with amyloidosis, in addition to the small quantities of SAP in the blood there are large quantities of SAP coating the amyloid deposits in the organs. In the SAP scan, all of this coating shows up clearly, as if it has been highlighted, or labelled. This signal is picked up using a detector called a gamma camera. All body parts where radioactive signal is detected must contain amyloid deposits. So a very clear picture is obtained of the location and quantity of the amyloid deposits in organs throughout the body. The SAP scan is performed either the day after or 2-3 hours after the injection of the radiolabelled SAP protein. The patient is scanned on an open device called a whole body gamma camera scanner. The scan takes place while lying flat, still and fully clothed for about 25 minutes on the scanner. A technician stays in the room throughout the procedure and a carer may also stay, if desired.
Amyloidosis is diagnosed when Congo red–binding material is demonstrated in a biopsy specimen. For example, in cardiac amyloidosis, the definitive diagnosis of the type of amyloid can be made using an endomyocardial biopsy specimen, with Congo red and immunologic staining of the tissue sample. Alternatively, when noninvasive testing suggests cardiac amyloidosis, studying a subcutaneous fat aspiration instead of endomyocardial biopsy, thereby avoiding an invasive procedure, often provides a specific diagnosis. Because different types of amyloidosis require different approaches to treatment, determining only that a patient has a diagnosis of amyloidosis is no longer adequate. A clinical situation may suggest the type of amyloidosis, but the diagnosis generally must be confirmed by immunostaining a biopsy specimen. Antibodies against the major amyloid fibril precursors are commercially available. For example, AL, ATTR, and Aβ2 M can present as carpal tunnel syndrome or gastrointestinal amyloidosis, but each has a different etiology and requires a different treatment approach.
Similarly, determining whether the amyloid is of the AL or ATTR type is often difficult in patients with cardiac amyloidosis, because the clinical picture usually is similar. Without immunostaining to identify the type of deposited protein, an incorrect diagnosis can lead to ineffective and, perhaps, harmful treatment. Be wary of drawing diagnostic conclusions from indirect tests (eg, monoclonal serum proteins) because the results of these presumptive diagnostic tests can be misleading; for example, monoclonal serum immunoglobulins are common in patients older than 70 years, but the most common form of cardiac amyloidosis is derived from TTR.
New adjuncts to traditional laboratory testing are now available. The serum free light chain test is commercially available and can aid in screening, diagnosis, prognosis, and treatment monitoring by detecting detects low concentrations of free light chains and can measure the ratio of kappa chains to lambda chains 75.
Organ biopsies
It is important to recognize that not all biopsy sites offer the same sensitivity. The best sites from which to obtain a biopsy specimen in systemic amyloidosis are the abdominal fat pad and rectal mucosa (approaching 90% sensitivity for fat pad and 73-84% for rectal mucosa) 76. While some imaging modalities can strongly suggest amyloidosis, a tissue sample showing birefringent material is still the criterion standard. This is definitely the case in terms of cardiac specific amyloid. While, endomyocardial biopsy is the best confirmatory test for local cardiac amyloid deposition, it can be very risk averse and requires a center of excellence with the full complement of immunohistochemical and molecular-based testing 77.
When the subcutaneous fat aspiration biopsy (the least invasive biopsy site) does not provide information to reach a firm diagnosis, biopsy specimens can be obtained from other organs. In addition, an advantage to performing a biopsy of an involved organ (eg, kidney, heart) is that it definitively establishes a cause-and-effect relationship between the organ dysfunction and amyloid deposition.
Other sites that are often sampled but have poor sensitivity for the diagnosis of amyloid include the salivary glands, skin, tongue, gingiva, stomach, and bone marrow. A recent study suggests that the duodenum may be the most sensitive biopsy site compared with the gingiva, esophagus, or gastric antrum in AA renal disease 78.
Treatment of Amyloidosis
There’s no cure for amyloidosis. But treatment can help manage signs and symptoms and limit further production of amyloid protein. Specific treatments depend on the type of amyloidosis and target the source of the amyloid production.
AL amyloidosis. Many of the same chemotherapy medications that treat multiple myeloma are used in AL amyloidosis to stop the growth of abnormal cells that produce amyloid.
Autologous blood stem cell transplant (ASCT) offers an additional treatment option in some cases. This procedure involves collecting your own stem cells from your blood and storing them for a short time while you have high-dose chemotherapy. The stem cells are then returned to your body via a vein.
ASCT is most appropriate for people whose disease isn’t advanced and whose heart isn’t greatly affected.
AA amyloidosis. Treatments target the underlying condition — for example, an anti-inflammatory medication to treat rheumatoid arthritis.
Hereditary amyloidosis. Liver transplantation may be an option because the protein that causes this form of amyloidosis is made in the liver.
Dialysis-related amyloidosis. Treatments include changing your mode of dialysis or having a kidney transplant. Beta-2m-adsorbing columns for dialysis – β2-m–adsorbing columns have demonstrated some benefit in dialysis-related amyloidosis. This addition to long-term dialysis may reduce inflammation and accumulation of fibrils without major adverse effects 79.
Doxycycline. A pilot study demonstrated reduction in arthralgia and increased range of motion with doxycycline treatment. The authors theorize that doxycycline inhibits β2-microglobulin fibrillogenesis and inhibits the accumulation in bone architecture. Their trial monitored long-term dialysis patients with severe amyloid arthropathy. Using a low dose of 100 mg of doxycycline daily, they were able to provide pain reduction, increased range of motion, and no adverse effects at 5 months 80.
Immunotherapy. Aβ immunotherapies have shown some promise in the treatment of Alzheimer disease. Agents with great potential include the passive monoclonal antibodies bapineuzumab 45 and solanezumab 81. However, both of these humanized monoclonal antibodies have failed to provide meaningful cognitive changes in clinical trials 82. Further studies using this strategy are ongoing.
Several other monoclonal antibodies to target Aβ deposition include “PF-04360365 (ponezumab), which targets the free C-terminus of Aβ, and specifically Aβ34–41; MABT5102A, which binds to Aβ monomers, oligomers, and fibrils with equally high affinity; GSK933776A, which, similar to bapineuzumab, targets the N-terminal sequence of Aβ; BAN2401, which targets Aβ protofibrils; and gantenerumab, which targets the N-terminus and central portion of Aβ” 83.
Gantenerumab
Gantenerumab is the first fully human monoclonal antibody that binds regions of Aβ configuration not present in the structure of the native monomeric Aβ. Therefore, gantenerumab preferentially binds to aggregated Aβ 84. Phase II and III clinical trials have shown disease-modifying potential in Alzheimer disease, with reductions in brain amyloid as measured by carbon 11 [11 C]–labeled Pittsburgh compound B PET but no meaningful neurocognitive improvements. It was very interesting that “live-cell imaging indicated that a clearance of fluorescent-labeled gantenerumab bound to amyloid deposits occurred in a dose-dependent manner within hours via active intracellular uptake by brain-activated microglia adjacent to amyloid plaques” 83. This approach may continue to yield more clinically significant results in the future.
Tafamidis
Transthyretin amyloidosis is related to abnormal conformational changes and transthyretin aggregation leading to severe neuropathy. After learning the structure and function of transthyretin, researchers developed a molecule and later a drug that could stabilized the native tetrameric state of transthyretin 85.
A phase II, open-label, single-treatment arm evaluated the pharmacodynamics, efficacy, and safety of this drug, called tafamidis, in patients with non-Val30Met transthyretin (TTR) amyloidosis. Twenty-one patients with 8 different non-Val30Met mutations received daily oral tafamidis, and many experienced improved quality of life measures, while pro-BNP and echocardiographic features improved 86. Another study showed that long-term use continued to stabilize TTR at 30 months. In a cross-over scheme, patients previously have given placebo were able to slow their neurologic decline, proving that earlier treatment is better 87. The success of tafamidis in a subset of amyloidosis is very encouraging for future molecular targets.
Tocilizumab
A subset of patients with amyloidosis (AA) in the setting of arthritis were monitored for benefits from the IL-6 receptor antibody tocilizumab. After a year of therapy with the standard regimen for rheumatoid arthritis (RA) (8 mg/kg 4 days per weeks) a significant reduction in renal dysfunction, serum AA concentration, and urinary protein secretion was observed in conjunction with reduced disease activity. These patients had been previously treated with etanercept without these benefits. This study again highlights the importance of treating the underlying condition, but certain biologic targets may have more downstream effects on amyloidogenesis.
Other
Other innovative medicines (RNA interference, antisense oligonucleotides) have been developed to block hepatic production of both mutant and wild type TTR (noxious in late-onset forms of NAH after age 50 y), and to remove amyloid deposits (monoclonal anti-SAP) 88. Clinical trials should first include patients with late-onset FAP or non-met30 TTR familial amyloid polyneuropathy who are less responsive to LT7 and patients in whom tafamidis is ineffective or inappropriate. Initial and periodic cardiac assessment is necessary, as cardiac impairment is inevitable and largely responsible for mortality. Symptomatic treatment is crucial to improve these patients’ quality of life.
Supportive care for Amyloidosis
To manage ongoing signs and symptoms of amyloidosis, your doctor also may recommend:
- Pain medication
- Fluid retention medication (diuretic) and a low-salt diet
- Blood-thinning medication
- Medication to control your heart rate.
For example, cardiac amyloidosis requires judicious use of rate and rhythm control agents to prevent fatal arrhythmias. Amyloid can infiltrate the conduction system and the coronary arteries 89. Clues to aid in early detection are the presence of cardiac arrhythmia with early heart failure evidenced by abnormal echocardiogram 90. Certain medications such as digoxin or calcium channel blockers are dangerous to the already-hindered conduction system in cardiac amyloid. In addition, higher doses of angiotensin-converting enzyme inhibitors can also foster dangerous hypotension and are not recommended 91. Beta-blockers are relatively contraindicated in cardiac amyloidosis and are associated with a higher death rate 92. Amiodarone (200 mg orally 5 days per week), however, prevents arrhythmias and should be considered first and continued indefinitely 93.
Lifestyle and home remedies for amyloidosis
These tips can help you live with amyloidosis:
- Pace yourself. If you feel short of breath, take a break. You’ll need to avoid strenuous activities, but you may be able to continue normal daily activities, such as going to work. Talk to your doctor about an appropriate level of activity for you.
- Eat a balanced diet. Good nutrition is important to provide your body with adequate energy. Follow a low-salt diet if your doctor recommends it.
Coping and support for amyloidosis
A diagnosis of amyloidosis can be extremely challenging. Here are some suggestions that may make dealing with amyloidosis easier:
- Find someone to talk with. You may feel comfortable discussing your feelings with a friend or family member, or you might prefer meeting with a formal support group.
- Set reasonable goals. Having goals helps you feel in control and can give you a sense of purpose. Choose goals you can reach.
- Amyloidosis & Kidney Disease. https://www.niddk.nih.gov/health-information/kidney-disease/amyloidosis
- Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, et al. A primer of amyloid nomenclature. Amyloid. 2007 Sep. 14(3):179-83.
- Buxbaum JN. The systemic amyloidoses. Curr Opin Rheumatol. 2004 Jan. 16(1):67-75.
- Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, et al. A primer of amyloid nomenclature. Amyloid. 2007 Sep. 14(3):179-83.
- Ando Y, Ueda M. Novel methods for detecting amyloidogenic proteins in transthyretin related amyloidosis. Front Biosci. 2008. 13:5548-58.
- Hazenberg BP. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013 May. 39(2):323-45.
- https://emedicine.medscape.com/article/335414-overview
- Buck FS, Koss MN, Sherrod AE, Wu A, Takahashi M. Ethnic distribution of amyloidosis: an autopsy study. Mod Pathol. 1989 Jul. 2(4):372-7.
- Shin JK, Jung YH, Bae MN, Baek IW, Kim KJ, Cho CS. Successful treatment of protein-losing enteropathy due to AA amyloidosis with octreotide in a patient with rheumatoid arthritis. Mod Rheumatol. 2013 Mar. 23(2):406-11.
- Tam JH, Pasternak SH. Amyloid and Alzheimer’s disease: inside and out. Can J Neurol Sci. 2012 May. 39(3):286-98.
- Basha HI, Raj E, Bachuwa G. Cardiac amyloidosis masquerading as biventricular hypertrophy in a patient with multiple myeloma. BMJ Case Rep. 2013 Jul 29. 2013
- Saha A, Chopra Y, Theis JD, Vrana JA, Sethi S. AA amyloidosis associated with systemic-onset juvenile idiopathic arthritis. Am J Kidney Dis. 2013 Oct. 62(4):834-8.
- Cammelli D. [Extra-articular manifestations of seronegative spondylarthritis]. Recenti Prog Med. 2006 May. 97(5):280-9.
- Bergis M, Dega H, Planquois V, Benichou O, Dubertret L. [Amyloidosis complicating psoriatic arthritis]. Ann Dermatol Venereol. 2003 Nov. 130(11):1039-42.
- Kishida D, Okuda Y, Onishi M, Takebayashi M, Matoba K, Jouyama K. Successful tocilizumab treatment in a patient with adult-onset Still’s disease complicated by chronic active hepatitis B and amyloid A amyloidosis. Mod Rheumatol. 2011 Apr. 21(2):215-8.
- Erten S, Perçinel S, Olmez U, Ensari A, Düzgün N. Behçet’s disease associated with diarrhea and secondary amyloidosis. Turk J Gastroenterol. 2011 Feb. 22(1):106-7.
- Lane T, Loeffler JM, Rowczenio DM, Gilbertson JA, Bybee A, Russell TL. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum. 2013 Apr. 65(4):1116-21.
- Denis MA, Cosyns JP, Persu A, Dewit O, de Galocsy C, Hoang P. Control of AA amyloidosis complicating Crohn’s disease: a clinico-pathological study. Eur J Clin Invest. 2013 Mar. 43(3):292-301.
- Silva Júnior GB, Barbosa OA, Barros Rde M, Carvalho Pdos R, Mendoza TR, Barreto DM. [Amyloidosis and end-stage renal disease associated with leprosy]. Rev Soc Bras Med Trop. 2010 Jul-Aug. 43(4):474-6.
- Neugebauer C, Graf R. [Expert opinion problems in the evaluation of osteomyelitis]. Orthopade. 2004 May. 33(5):603-11; quiz 612.
- Lekpa FK, Ndongo S, Pouye A, Tiendrebeogo JW, Ndao AC, Ka MM. [Amyloidosis in sub-Saharan Africa]. Med Sante Trop. 2012 Jul-Sep. 22(3):275-8.
- Akçay S, Akman B, Ozdemir H, Eyüboglu FO, Karacan O, Ozdemir N. Bronchiectasis-related amyloidosis as a cause of chronic renal failure. Ren Fail. 2002 Nov. 24(6):815-23.
- Introduction to AA Amyloidosis. https://www.amyloidosis.org.uk/about-amyloidosis/understanding-aa-amyloidosis/aa-amyloidosis
- Caro-Cuenca MT, Ortega-Salas R, Espinosa-Hernández M. Renal AA amyloidosis in a Castleman’s disease patient. Nefrologia. 2012. 32(5):699-700.
- Caro-Cuenca MT, Ortega-Salas R, Espinosa-Hernández M. Renal AA amyloidosis in a Castleman’s disease patient. Blood. 2013 Jul 18. 122(3):458-9.
- Nobata H, Suga N, Itoh A, Miura N, Kitagawa W, Morita H, et al. Systemic AA amyloidosis in a patient with lung metastasis from renal cell carcinoma. Amyloid. 2012 Dec. 19(4):197-200.
- Rivera R, Kaul V, DeCross A, Whitney-Miller C. Primary gastric amyloidosis presenting as an isolated gastric mass. Gastrointest Endosc. 2012 Jul. 76(1):186-7.
- Yokota S, Kikuchi M, Nozawa T, Kizawa T, Kanetaka T, Miyamae T, et al. [An approach to the patients with cryopyrin-associated periodic syndrome (CAPS) : a new biologic response modifier, canakinumab]. Nihon Rinsho Meneki Gakkai Kaishi. 2012. 35(1):23-9.
- Smith GR, Tymms KE, Falk M. Etanercept treatment of renal amyloidosis complicating rheumatoid arthritis. Intern Med J. 2004 Sep-Oct. 34(9-10):570-2.
- Miyagawa I, Nakayamada S, Saito K, Hanami K, Nawata M, Sawamukai N. Study on the safety and efficacy of tocilizumab, an anti-IL-6 receptor antibody, in patients with rheumatoid arthritis complicated with AA amyloidosis. Mod Rheumatol. 2013 Oct 21.
- Nakamura T, Higashi S, Tomoda K, Tsukano M, Baba S. Efficacy of etanercept in patients with AA amyloidosis secondary to rheumatoid arthritis. Clin Exp Rheumatol. 2007 Jul-Aug. 25(4):518-22.
- Okuda Y, Ohnishi M, Matoba K, Jouyama K, Yamada A, Sawada N, et al. Comparison of the clinical utility of tocilizumab and anti-TNF therapy in AA amyloidosis complicating rheumatic diseases. Mod Rheumatol. 2014 Jan. 24(1):137-43.
- Nakamura T. Clinical strategies for amyloid A amyloidosis secondary to rheumatoid arthritis. Mod Rheumatol. 2008. 18(2):109-18.
- Lane T, Loeffler JM, Rowczenio DM, Gilbertson JA, Bybee A, Russell TL, et al. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum. 2013 Apr. 65(4):1116-21.
- Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011 May 10. 29(14):1924-33.
- Sissoko M, Sanchorawala V, Seldin D, Sworder B, Angelino K, Broce M, et al. Clinical presentation and treatment responses in IgM-related AL amyloidosis. Amyloid. 2015. 22 (4):229-35.
- Kastritis E, Anagnostopoulos A, Roussou M, Toumanidis S, Pamboukas C, Migkou M, et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007 Oct. 92(10):1351-8.
- Wechalekar AD, Goodman HJ, Lachmann HJ, Offer M, Hawkins PN, Gillmore JD. Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood. 2007 Jan 15. 109(2):457-64.
- Dispenzieri A, Gertz MA, Hayman SR, et al. A phase II study of pomalidomide and dexamethasone in previously treated light-chain (AL) amyloidosis. J Clin Oncol. 2010. 28:579s.
- Picken MM. Immunoglobulin light and heavy chain amyloidosis AL/AH: renal pathology and differential diagnosis. Contrib Nephrol. 2007. 153:135-55.
- Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019 Jul;15(7):387-404. doi: 10.1038/s41582-019-0210-4
- Floege J, Bartsch A, Schulze M, Shaldon S, Koch KM, Smeby LC. Clearance and synthesis rates of beta 2-microglobulin in patients undergoing hemodialysis and in normal subjects. J Lab Clin Med. 1991 Aug. 118(2):153-65.
- Otsubo S, Kimata N, Okutsu I, Oshikawa K, Ueda S, Sugimoto H, et al. Characteristics of dialysis-related amyloidosis in patients on haemodialysis therapy for more than 30 years. Nephrol Dial Transplant. 2009 May. 24(5):1593-8.
- Traut M, Haufe CC, Eismann U, Deppisch RM, Stein G, Wolf G. Increased binding of beta-2-microglobulin to blood cells in dialysis patients treated with high-flux dialyzers compared with low-flux membranes contributed to reduced beta-2-microglobulin concentrations. Results of a cross-over study. Blood Purif. 2007. 25(5-6):432-40.
- Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci. 2002 Oct. 3(10):824-8.
- ALECT2 amyloidosis. https://www.amyloidosis.org.uk/about-amyloidosis/other-types-of-amyloidosis/alect2-amyloidosis
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001 Nov. 29(3):301-5.
- Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006 Aug 10. 355(6):581-92.
- Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008 Aug. 58(8):2443-52.
- Stew BT, Fishpool SJ, Owens D, Quine S. Muckle-Wells syndrome: a treatable cause of congenital sensorineural hearing loss. B-ENT. 2013. 9(2):161-3.
- Yamazaki T, Masumoto J, Agematsu K, Sawai N, Kobayashi S, Shigemura T, et al. Anakinra improves sensory deafness in a Japanese patient with Muckle-Wells syndrome, possibly by inhibiting the cryopyrin inflammasome. Arthritis Rheum. 2008 Mar. 58(3):864-8.
- Granel B, Valleix S, Serratrice J, Chérin P, Texeira A, Disdier P, et al. Lysozyme amyloidosis: report of 4 cases and a review of the literature. Medicine (Baltimore). 2006 Jan. 85(1):66-73.
- Picken MM, Linke RP. Nephrotic syndrome due to an amyloidogenic mutation in fibrinogen A alpha chain. J Am Soc Nephrol. 2009 Aug. 20(8):1681-5.
- Eriksson M, Schönland S, Yumlu S, Hegenbart U, von Hutten H, Gioeva Z, et al. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: identification of three novel mutations in the APOA1 gene. J Mol Diagn. 2009 May. 11(3):257-62.
- Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, et al. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain. 2008 Dec. 131:3299-310.
- Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L. Proliferation of amyloid-ß42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A. 2013 Jun 11. 110(24):9758-63.
- Emily M, Talvas A, Delamarche C. MetAmyl: a METa-predictor for AMYLoid proteins. PLoS One. 2013. 8(11):e79722.
- Rezaei H. Prion protein oligomerization. Curr Alzheimer Res. 2008 Dec. 5(6):572-8.
- Localised Amyloidosis. https://www.amyloidosis.org.uk/about-amyloidosis/other-types-of-amyloidosis/localised-amyloidosis
- https://docs.google.com/viewer?url=https%3A%2F%2Fwww.ucl.ac.uk%2Fdrupal%2Fsite_amyloidosis%2Fsites%2Famyloidosis%2Ffiles%2Flocalised_amyloidosis.pdf
- https://www.angiopathy.org/educational-material
- What is Cerebral Amyloid Angiopathy (CAA)? https://www.angiopathy.org/new-page-2
- What are the symptoms of CAA? https://www.angiopathy.org/q-what-are-the-symptoms-of-caa
- What are the risk factors that cause some people to get CAA? https://www.angiopathy.org/q-what-are-the-risk-factors-that-cause-some-people-to-get-caa
- If my family member has CAA, will I get it too? https://www.angiopathy.org/q-if-my-family-member-has-caa-will-i-get-it-too
- Solomon JP, Page LJ, Balch WE, Kelly JW. Gelsolin amyloidosis: genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit Rev Biochem Mol Biol. 2012 May-Jun. 47(3):282-96.
- Chen CD, Huff ME, Matteson J, Page L, Phillips R, Kelly JW. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca(2+) stabilization. EMBO J. 2001 Nov 15. 20(22):6277-87.
- Kangas H, Paunio T, Kalkkinen N, Jalanko A, Peltonen L. In vitro expression analysis shows that the secretory form of gelsolin is the sole source of amyloid in gelsolin-related amyloidosis. Hum Mol Genet. 1996 Sep. 5(9):1237-43.
- Ida CM, Yan X, Jentoft ME, Kip NS, Scheithauer BW, Morris JM. Pituicytoma with gelsolin amyloid deposition. Endocr Pathol. 2013 Sep. 24(3):149-55.
- Haverland N, Pottiez G, Wiederin J, Ciborowski P. Immunoreactivity of anti-gelsolin antibodies: implications for biomarker validation. J Transl Med. 2010 Dec 20. 8:137.
- Podduturi V, Armstrong DR, Hitchcock MA, Roberts WC, Guileyardo JM. Isolated atrial amyloidosis and the importance of molecular classification. Proc (Bayl Univ Med Cent). 2013 Oct. 26(4):387-9.
- Abedini A, Schmidt AM. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013 Apr 17. 587(8):1119-27.
- Levine SN, Ishaq S, Nanda A, Wilson JD, Gonzalez-Toledo E. Occurrence of extensive spherical amyloid deposits in a prolactin-secreting pituitary macroadenoma: a radiologic-pathologic correlation. Ann Diagn Pathol. 2013 Aug. 17(4):361-6.
- Wenson SF, Jessup CJ, Johnson MM, Cohen LM, Mahmoodi M. Primary cutaneous amyloidosis of the external ear: a clinicopathological and immunohistochemical study of 17 cases. J Cutan Pathol. 2012 Feb. 39(2):263-9.
- Rao M, Lamont JL, Chan J, Concannon TW, Comenzo R, Ratichek SJ, et al. Serum Free Light Chain Analysis for the Diagnosis, Management, and Prognosis of Plasma Cell Dyscrasias: Future Research Needs: Identification of Future Research Needs From Comparative Effectiveness Review No. 73 [Internet]. 2012 Sep.
- West S, Singleton JD. Rheumatology Secrets. 2nd ed. Philadelpha, PA: Hanley and Belfus; 2003. 519.
- Thiene G, Bruneval P, Veinot J, Leone O. Diagnostic use of the endomyocardial biopsy: a consensus statement. Virchows Arch. 2013 Jul. 463(1):1-5.
- Yilmaz M, Unsal A, Sokmen M, Harmankaya O, Alkim C, Kabukcuoglu F, et al. Duodenal biopsy for diagnosis of renal involvement in amyloidosis. Clin Nephrol. 2012 Feb. 77(2):114-8.
- Yamamoto Y, Hirawa N, Yamaguchi S, Ogawa N, Takeda H, Shibuya K, et al. Long-term efficacy and safety of the small-sized ß2-microglobulin adsorption column for dialysis-related amyloidosis. Ther Apher Dial. 2011 Oct. 15(5):466-74.
- Montagna G, Cazzulani B, Obici L, Uggetti C, Giorgetti S, Porcari R. Benefit of doxycycline treatment on articular disability caused by dialysis related amyloidosis. Amyloid. 2013 Sep. 20(3):173-8.
- Senior K. Dosing in phase II trial of Alzheimer’s vaccine suspended. Lancet Neurol. 2002 May. 1(1):3.
- Vellas B, Carrillo MC, Sampaio C, Brashear HR, Siemers E, Hampel H. Designing drug trials for Alzheimer’s disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD Task Force. Alzheimers Dement. 2013 Jul. 9(4):438-44.
- Novakovic D, Feligioni M, Scaccianoce S, Caruso A, Piccinin S, Schepisi C, et al. Profile of gantenerumab and its potential in the treatment of Alzheimer’s disease. Drug Des Devel Ther. 2013. 7:1359-64.
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, et al. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009 Aug 26. 29(34):10582-7.
- Bannykh SI, Balch WE, Kelly JW, Page LJ, Shelton GD. Formation of gelsolin amyloid fibrils in the rough endoplasmic reticulum of skeletal muscle in the gelsolin mouse model of inclusion body myositis: comparative analysis to human sporadic inclusion body myositis. Ultrastruct Pathol. 2013 Oct. 37(5):304-11.
- Merlini G, Planté-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013 Dec. 6(6):1011-20.
- Coelho T, Maia LF, da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013 Nov. 260(11):2802-14.
- Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016 Jun 25. 387 (10038):2641-54.
- Selvanayagam JB, Hawkins PN, Paul B, Myerson SG, Neubauer S. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol. 2007 Nov 27. 50(22):2101-10.
- Falk RH, Rubinow A, Cohen AS. Cardiac arrhythmias in systemic amyloidosis: correlation with echocardiographic abnormalities. J Am Coll Cardiol. 1984 Jan. 3(1):107-13.
- Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, et al. Al amyloidosis. Orphanet J Rare Dis. 2012 Aug 21. 7:54.
- Soni A, LeLorier P. Sudden death in nondilated cardiomyopathies: pathophysiology and prevention. Curr Heart Fail Rep. 2005 Sep. 2(3):118-23.
- Palladini G, Perfetti V, Obici L, Caccialanza R, Semino A, Adami F. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004 Apr 15. 103(8):2936-8.




{kind=link}