
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis also known as ALS or Lou Gehrig’s disease (a famous New York Yankees baseball player who was diagnosed with it in 1939), is a type of motor neuron disease that affect voluntary muscle-controlling nerves in your brain and spinal cord leading to muscle wasting (muscle atrophy), weakness, paralysis and ultimately death 1, 2. Voluntary muscles are those you use to move to produce movements intentionally like chewing, walking, and talking. Amyotrophic lateral sclerosis is a progressive neuromuscular condition, meaning the symptoms get worse over time and some people die within 2–5 years from onset of symptoms due to respiratory failure 3, 4, 5. However, about 10 percent of people with amyotrophic lateral sclerosis live 10 years or more. The famous physicist Stephen Hawking, pictured above, lived for more than 50 years after he was diagnosed with ALS. Maintaining an optimistic outlook can help improve quality of life for people with ALS.
At the present time, the cause of amyotrophic lateral sclerosis (ALS) is unknown. ALS is mainly sporadic in origin (sporadic ALS), but ALS is inherited in 5% to 10% of people (familial ALS) 4. Hereditary forms of the disease (familial ALS or FALS), are predominantly autosomal dominant and rarely X-linked or recessive 3. If a parent has familial ALS that is inherited an autosomal dominant, each child has a 50% chance of developing it, too. If you have this type of ALS, your care team may suggest genetic counseling. Researchers continue to study possible causes of ALS. Most theories center on a complex interaction between genetic and environmental factors.
Progression of ALS is usually linear, without remissions or exacerbations 6. Whereas the rate of progression varies between individuals, the pattern of progression is relatively predictable. The most common pattern in patients with same side (unilateral) limb onset (the predominant form), progresses to include the opposite side (contralateral) limb, then the other same side (ipsilateral) extremity (i.e., the left leg if the initial weakness was in the left arm), followed by the other contralateral extremity, before ultimately affecting the bulbar muscles (muscles that are involved in speaking, swallowing, chewing, and holding the jaw in place).
ALS has no cure and there is no effective treatment to reverse its progression, but treatments can ease symptoms and improve quality of life. The clinical management of people with ALS is complex and requires a comprehensive and multidisciplinary approach that include team of doctors trained in many areas and other health care professionals to provide your care, coordinated diagnostic tests, access to dietary and nutritional counseling, lung and breathing care, follow-up care, and the support of social workers 5, 7. This might prolong your survival and improve your quality of life. The United States Food and Drug Administration (FDA) has approved three medicines for treating ALS, riluzole (Rilutek, Exservan, Tiglutik kit), edaravone (Radicava), and sodium phenylbutyrate and taurursodiol (Relyvrio), have a modest effect on survival 8.
ALS affects persons of all races and ethnicities; however, whites, males (male: female = 1.5:1), non-Hispanics, persons aged ≥60 years, and those with a family history of amyotrophic lateral sclerosis are more likely to develop the disease 9, 10. From the National ALS Registry, approximately 32,000 Americans currently have ALS 11. The annual incidence rate is 1 to 2 cases per 100,000. Amyotrophic lateral sclerosis is most commonly diagnosed in middle age and affects more men than women. It usually presents with problems in dexterity or gait resulting from muscle weakness 1. Difficulty in speaking or swallowing is the initial symptom in the bulbar form of the disease 1. Over a period of months or years, patients with ALS develop severe, progressive muscular weakness and other symptoms caused by loss of function in both upper and lower motor neurons. Sphincter control, sensory function, intellectual abilities and skin integrity are preserved. Patients become completely disabled, often requiring ventilatory support and gastrostomy. Death usually occurs within five years of diagnosis and is attributed to respiratory failure or cachexia (a wasting syndrome that leads to loss of skeletal muscle and fat) 1.
Amyotrophic lateral sclerosis first symptoms develop gradually and can vary from person to person. They include:
- Difficulty walking or doing normal daily activities
- Weakness in your hands, legs or feet
- Hand weakness or clumsiness
- Tripping and falling
- Slow or slurred speech (called dysarthria or “thick speech”) and difficulty in projecting your voice
- Swallowing difficulties
- Muscle cramps and twitching (called fasciculations) in your arms, shoulders and tongue
- Uncontrollable crying, laughing or yawning (pseudobulbar affect)
- Difficulty holding your head up or maintaining posture
- In some cases, cognitive (thinking) and/or behavior changes
In more advanced stages, ALS causes shortness of breath and difficulty in breathing and swallowing, which is what eventally lead to a person’s death.
Approximately 20 percent of individuals with ALS also develop frontotemporal dementia (FTD) 12, 13, 14, 15. Changes in personality and behavior may make it difficult for affected individuals to interact with others in a socially appropriate manner. Communication skills worsen as the disease progresses. It is unclear how the development of ALS and frontotemporal dementia (FTD) are related. Individuals who develop both conditions are diagnosed as having ALS-frontotemporal dementia (FTD).
Amyotrophic lateral sclerosis (ALS) is difficult to diagnose early because it can mimic other neurological diseases. Furthermore, there is no single test that can definitely diagnose ALS. Your doctor will conduct a physical exam and review your full medical history. A neurologic examination will test your reflexes, muscle strength, and other responses and will be held at regular intervals to assess whether symptoms such as muscle weakness, muscle wasting, and spasticity are progressively getting worse.
Muscle and imaging tests to rule out other diseases and confirm the diagnosis include:
- Electromyography (EMG) is a recording technique that detects electrical activity of muscle fibers and can help diagnose ALS.
- A nerve conduction study (NCS) measures the electrical activity of your nerves and muscles by assessing the nerve’s ability to send a signal along the nerve or to the muscle.
- Magnetic resonance imaging (MRI) is a noninvasive procedure that uses a magnetic field and radio waves to produce detailed images of the brain and spinal cord.
- Blood and urine tests may be performed based on your symptoms, test results, and findings from the examination by a doctor. A physician may order these tests to eliminate the possibility of other diseases.
- Spinal tap (lumbar puncture). This involves removing a sample of your spinal fluid for laboratory testing using a small needle inserted between two vertebrae in your lower back.
- A muscle biopsy may be performed if your doctor believes you may have a muscle disease other than ALS. Under local anesthesia, a small sample of muscle is removed and sent to the lab for analysis.
There is no treatment to reverse damage to motor neurons or cure amyotrophic lateral sclerosis. However, treatments can make living with the disease easier. Supportive health care is best provided by multidisciplinary teams of healthcare professionals such as physicians; pharmacists; physical, occupational, speech, and respiratory therapists; nutritionists; social workers; clinical psychologists; and home care and hospice nurses. These teams can design an individualized treatment plan and provide special equipment aimed at keeping people as mobile, comfortable, and independent as possible.
Doctors may use the following medications approved by the U.S. Food and Drug Administration (FDA) to support a treatment plan for amyotrophic lateral sclerosis:
- Riluzole (Rilutek, Exservan, Tiglutik kit). Riluzole (Rilutek) is an oral medication believed to reduce damage to motor neurons by decreasing levels of glutamate, which transports messages between nerve cells and motor neurons. Taken orally, riluzole can increase life expectancy by 3 to 6 months. The thickened liquid form (Tiglutik) or the tablet (Exservan) that dissolves on the tongue may be preferred if you have swallowing difficulties. Riluzole (Rilutek) can cause side effects such as dizziness, gastrointestinal conditions and liver function changes. Your health care provider will monitor your blood counts and liver function while you’re taking the medicine. It is important to note that the elimination of riluzole will be affected by CYP1A2 inhibitors like caffeine and theophylline.
- Edaravone (Radicava). Edaravone (Radicava) is given intravenously through a vein in your arm or orally as a pill, can reduce the decline in daily functioning in people with ALS. Its effect on life span isn’t yet known. Side effects can include bruising, headache and shortness of breath. This medicine is given daily for two weeks a month.
- Sodium phenylbutyrate and taurursodiol (Relyvrio). This medicine, recently approved by the FDA, can slow the rate of decline in people with ALS. In particular, it may help people with performing daily tasks. It also may help people with ALS live longer, but more study is needed. Potential side effects of the medicine include diarrhea, belly pain, nausea and upper respiratory infection. People with disorders that affect bile acid circulation may experience diarrhea that gets worse when taking this medicine.
Other medications may be prescribed to help you manage symptoms including muscle cramps, stiffness, excess saliva and phlegm, and the unwanted episodes of crying and/or laughing, or other emotional displays. Medications may also help you with any pain, depression, sleep disturbances, and constipation.
ALS Resource Links
- ALS Association
- MDA ALS Division
- Northeast Amyotrophic Lateral Sclerosis Consortium (NEALS)
- Mayo Clinic ALS Information
- >National Institutes of Health (NIH) ALS Clinical Trials
- National Library of Medicine ALS Information
- National Institutes of Health ALS Information
- Emory ALS Center
- ALS Therapy Development Institute
- Les Turner ALS Foundation
- ALS Biorepository Brain Bank
- CReATe
- CReATe
- ALS Family Charitable Foundation
Is there a cure for ALS?
Currently there is no known cure or treatment that halts or reverses the progression of ALS.
While the search for an effective treatment and cure continues, multidisciplinary teams across the globe are assisting patients and their families to adjust to the many challenges of living with ALS. These teams of specialists use devices and therapies to help patients manage their ALS symptoms and to allow people with the disease to maintain their independence and quality of life. This multidisciplinary approach has also been shown to prolong survival of people who have ALS.
Treatments and interventions may include:
- proper body positioning
- exercise regimens, physical and occupational therapy
- devices and supports to help people walk
- braces and splints for the legs and arms
- customized wheelchairs
- home assessment to make it easier to get around in the house.
Who gets ALS?
About 60% of the people reported to have ALS in the United States are men, and 93% of patients are Caucasian. Within the United States, the prevalence of amyotrophic lateral sclerosis is estimated at 5.2 per 100,000 16. Worldwide the incidence of ALS is approximately 1.6 cases per 100,000 persons annually, with similar rates demonstrated within the United States 17, 18. Based on US population studies, a little more than 5,600 people in the US are diagnosed with ALS each year – approximately 15 new cases per day. It is estimated that as many as 32,000 Americans have ALS at any given time.
Most people develop ALS between the ages of 40 and 70, with an average age of 55 at the time of diagnosis. However, rare cases of the disease do occur in persons in their 20s and 30s.
For sporadic ALS, male-to-female incidence ratios ranging from 1.3 to 1.5 have been demonstrated. Increased incidence is also noted with increases in age, particularly after 40 years of age 19. Globally, a higher incidence is associated with White ethnicity 20, 21. The only established risk factors for ALS are age and family history; however, a growing body of evidence suggests cigarette smoking may also be a risk factor 22.
Approximately 50% of people diagnosed with ALS live at least three or more years after diagnosis. About 25% live five years or more and up to 10% live more than 10 years.
What is upper motor neuron?
Upper motor neurons originate in the gray matter of the brain and send long axons which carry the impulses for movement down a descending tract of the spinal cord. These axons synapse with lower motor neurons in the ventral horn of the spinal cord. The axons of the lower motor neurons exit the ventral root of the spinal cord to innervate the muscles or glands of the body. An upper motor neuron is actually an interneuron and not a true motor neuron: it is so named because the cell originates in the upper part of the central nervous system (brain) and regulates the activity of lower motor neurons. Only a lower motor neuron is a true motor neuron because it conveys action potentials from the central nervous system (CNS) to skeletal muscles in the periphery.
Upper motor neurons from the cerebral cortex are essential for the planning and execution of voluntary movements of the body. Other upper motor neurons originate in motor centers of the brainstem: the vestibular nuclei, reticular formation, superior colliculus, and red nucleus. Upper motor neurons from the brainstem help regulate posture, balance, muscle tone, and reflexive movements of the head and trunk.
The primary tract which carries signals for voluntary movement is known as the pyramidal tract. The pyramidal tract divides further into the corticospinal tract and the corticobulbar tract. Injury or lesions to upper motor neuron’s are common because of the vast areas covered by the motor neuron pathways. upper motor neuron lesions are designated as any damage to the motor neurons that reside above nuclei of cranial nerves or the anterior horn cells of the spinal cord. Damage to upper motor neuron’s lead to a characteristic set of clinical symptoms known as the upper motor neuron syndrome. These symptoms can include weakness, spasticity, clonus, and hyperreflexia. upper motor neuron’s lesions have a wide differential diagnosis which ranges from cerebrovascular accidents, traumatic brain injury, malignancy, infections, inflammatory disorders, neurodegenerative disorders, and metabolic disorders.
Figure 1. Upper motor neuron
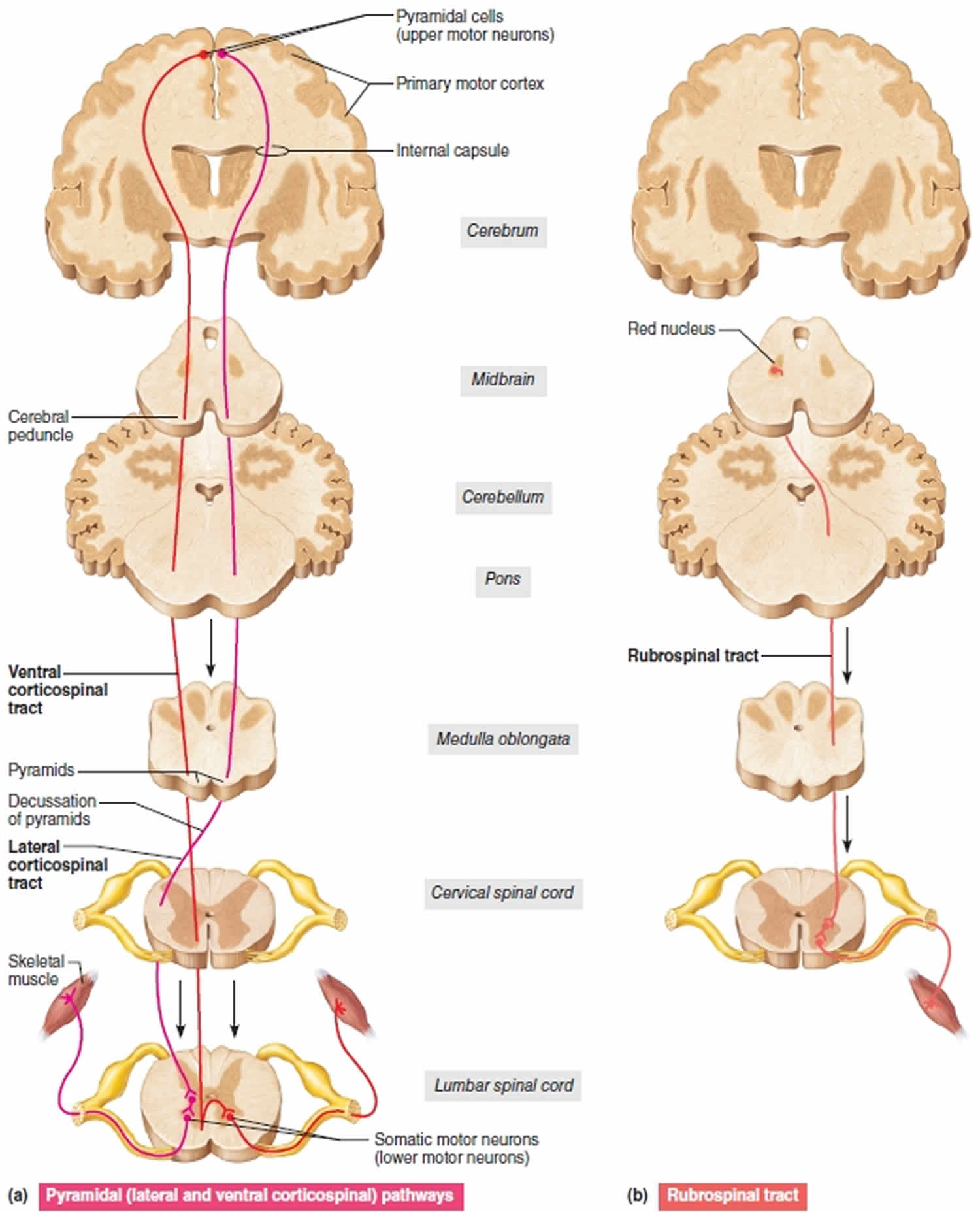
Footnote: Three upper motor neuron descending pathways by which the brain influences movement. Cross sections up to the cerebrum, which is shown in frontal section. (a) Pyramidal (lateral and ventral corticospinal pathways) originate from the cerebral cortex and control skilled, voluntary movements. (b) The rubrospinal tract, one of the indirect pathways, helps regulate muscle tone.
Figure 2. Upper and lower motor neuron
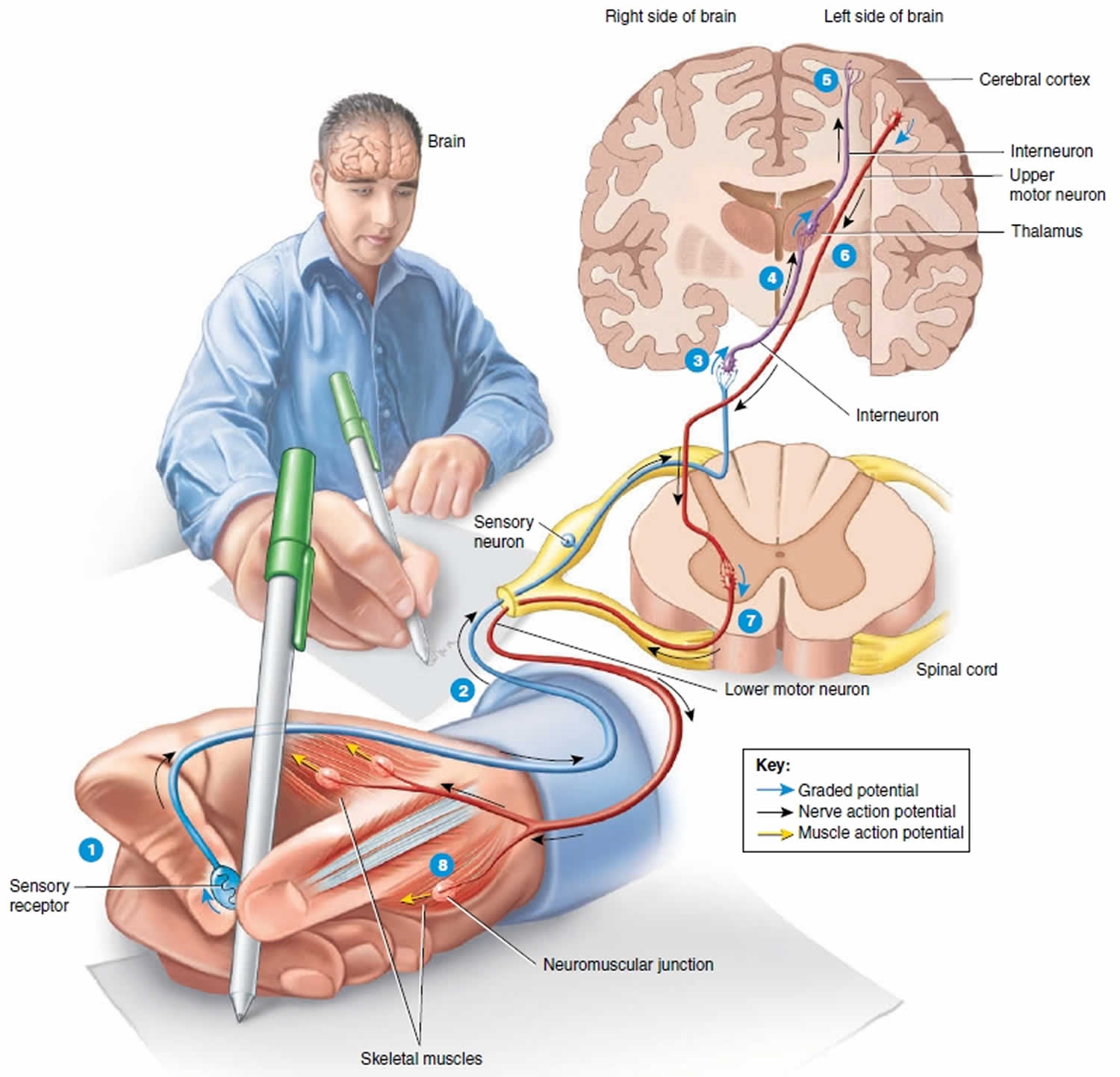
Footnotes: 1) As you touch the pen, a sensory receptor in the skin of the fingers is stimulated. 2) The sensory receptor in the skin of the fingers triggers the axon of the sensory neuron to send electrical signal which travels along the axon into the spinal cord and the brain and ultimately causes the release of neurotransmitter at a synapse with an interneuron. 3) The neurotransmitter stimulates the interneuron to form a graded potential in its dendrites and cell body. 4) In response to the graded potential, the axon of the interneuron forms a nerve action potential. The nerve action potential travels along the axon, which results in neurotransmitter release at the next synapse with another interneuron. 5) This process of neurotransmitter release at a synapse followed by the formation of a graded potential and then a nerve action potential occurs over and over as interneurons in higher parts of the brain (such as the thalamus and cerebral cortex) are activated. Once interneurons in the cerebral cortex, the outer part of the brain, are activated, perception occurs and you are able to feel the smooth surface of the pen touch your fingers. The conscious awareness of a sensation, is primarily a function of the cerebral cortex.
Suppose that you want to use the pen to write a letter.
The nervous system would respond in the following way:
6) A stimulus in the brain causes a graded potential to form in the dendrites and cell body of an upper motor neuron, a type of motor neuron that synapses with a lower motor neuron farther down in the CNS (central nervous system) in order to contract a skeletal muscle. The graded potential subsequently causes a nerve action potential to occur in the axon of the upper motor neuron, followed by neurotransmitter release. 7) The neurotransmitter generates a graded potential in a lower motor neuron, a type of motor neuron that directly supplies skeletal muscle fibers. The graded potential triggers the formation of a nerve action potential and then release of the neurotransmitter at neuromuscular junctions formed with skeletal muscle fibers that control movements of the fingers. 8) The neurotransmitter stimulates the muscle fibers that control finger movements to form muscle action potentials. The muscle action potentials cause these muscle fibers to contract, which allows you to write with the pen.
The axons of upper motor neurons extend from the brain to lower motor neurons via two types of pathways—direct and indirect pathways. Direct motor pathways (pyramidal tracts) provide input to lower motor neurons via axons that extend directly from the cerebral cortex. Indirect motor pathways (extrapyramidal tracts) provide input to lower motor neurons from motor centers in the brainstem. Direct and indirect pathways both govern generation of action potentials in the lower motor neurons, the neurons that stimulate contraction of skeletal muscles.
What is lower motor neuron?
Lower motor neurons have their cell bodies in the lower parts of the central nervous system (brainstem and spinal cord). From the brainstem, axons of lower motor neurons extend through cranial nerves to innervate skeletal muscles of the face and head. From the spinal cord, axons of lower motor neurons extend through spinal nerves to innervate skeletal muscles of the limbs and trunk. Only lower motor neurons provide output from the central nervous system to skeletal muscle fibers. For this reason, they are also called the final common pathway.
Input arrives at lower motor neurons from upper motor neurons and nearby interneurons called local circuit neurons. Most upper motor neurons synapse with local circuit neurons, which in turn synapse with lower motor neurons. The local circuit neurons are located close to the lower motor neuron cell bodies in the brainstem and spinal cord. Local circuit neurons receive input from somatic sensory receptors, such as nociceptors and muscle spindles, as well as from higher centers in the brain. They help coordinate rhythmic activity in specific muscle groups, such as alternating flexion and extension of the lower limbs during walking. A few upper motor neurons synapse directly with lower motor neurons.
Lower motor neurons damage or disease produces flaccid paralysis of muscles on the same side of the body. There is neither voluntary nor reflex action of the innervated muscle fibers, muscle tone is decreased or lost, and the muscle remains limp or flaccid. Injury or disease of upper motor neurons in the cerebral cortex removes inhibitory influences that some of these neurons have on lower motor neurons, which causes spastic paralysis of muscles on the opposite side of the body. In this condition muscle tone is increased, reflexes are exaggerated, and pathological reflexes such as the Babinski sign appear.
The most common causes of lower motor neuron lesions are trauma to peripheral nerves that serve the axons, and viruses that selectively attack ventral horn cells. Disuse atrophy of the muscle occurs i.e., shrinkage of muscle fiber finally replaced by fibrous tissue (fibrous muscle). Other common causes of lower motor neuron degeneration is amyotrophic lateral sclerosis (ALS), Guillain–Barré syndrome, Bell’s palsy, poliomyelitis, spinal muscular atrophy, Clostridium botulinum, and cauda equina syndrome.
Amyotrophic lateral sclerosis types
There are two main types of amyotrophic lateral sclerosis:
- Sporadic ALS (SALS): This type can affect anyone. Sporadic ALS accounts for 90-95% of U.S. cases.
- Familial ALS (FALS): This type is inherited through genetic changes passed down in families. An estimated 5 to 10 percent of ALS is familial and caused by mutations in one of several genes. The pattern of inheritance varies depending on the gene involved. Most familial ALS (FALS) cases are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition. If a parent has familial ALS, each child has a 50% chance of developing it, too. Some people who inherit a familial genetic mutation known to cause ALS never develop features of the condition. This situation is known as reduced penetrance. It is unclear why some people with a mutated gene develop the disease and other people with a mutated gene do not. If you have this familial ALS, your healthcare team may suggest genetic counseling.
- Less frequently, familial ALS is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Because an affected person’s parents are not affected, autosomal recessive ALS is often mistaken for sporadic ALS even though it is caused by a familial genetic mutation.
- Very rarely, familial ALS is inherited in an X-linked dominant pattern. X-linked conditions occur when the gene associated with the condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. In most cases, males tend to develop the disease earlier and have a decreased life expectancy compared with females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
The most common symptoms that appear in both types of ALS are muscle weakness, twitching, and cramping, which eventually can lead to the impairment of muscles 23. In the most advanced stages, ALS patients will develop symptoms of shortness of breath (dyspnea) and difficulty swallowing (dysphagia) 24, 25
Amyotrophic lateral sclerosis (ALS) is also described by how it starts:
- Rapid-onset ALS has symptoms that appear quickly.
- Limb-onset ALS starts with symptoms in arms or legs. Limb onset ALS is the predominant type, presenting in 70% of patients 26. Limb onset ALS can be further classified as flail arm syndrome or brachial amyotrophic diplegia, which is characterized by lower motor neuron weakness and wasting. It usually starts proximally and often symmetrically, then progresses distally to a point where upper extremity function is severely impaired 27. Limb onset ALS can also be classified as flail leg syndrome or pseudopolyneuritic variant, which is also characterized by lower motor neuron weakness and wasting, but of the lower extremities and with distal onset. Patients have a slower rate of progression to the involvement of other body segments and respiratory muscle weakness 28.
- Bulbar-onset ALS starts with trouble swallowing or speaking. Bulbar onset ALS accounts for 25% of patients and is characterized by upper motor neuron and lower motor neuron involvement of the cranial nerves, usually manifesting as speech difficulties and dysphagia followed by limb involvement in later stages 26.
- Although debated, some experts believe progressive muscular atrophy (PMA) represents a form of ALS. The disease is progressive, initially exclusively involving the lower motor neuron. Many patients go on to develop clinical signs and symptoms of upper motor neuron disease, at which point it is called lower motor neuron-onset ALS. Interestingly, even in those patients who never show clinical evidence of upper motor neuron involvement, corticospinal tract involvement is detected at autopsy in up to 50% to 66% of patients with an antemortem diagnosis of progressive muscular atrophy (PMA) 29.
- Primary lateral sclerosis (PLS) is a disorder of initially exclusive upper motor neuron disease. These patients have slower progression, lacking weight loss, and lower motor neuron symptoms/signs in the first four years of the disease. Most will eventually develop them, however, and at this point is known as upper motor neuron-dominant ALS 30. These patients have a better prognosis than typical patients with ALS but worse than patients with primary lateral sclerosis (PLS) 31.
- ALS-plus syndrome. If there are any additional symptoms/signs besides lower motor neuron and upper motor neuron disease, such as dementia (mostly frontotemporal), extrapyramidal, autonomic dysfunction, ocular motility disturbance, and/or sensory loss, these patients are considered to have ALS-plus syndrome 32.
- Approximately 20 percent of individuals with ALS also develop frontotemporal lobe dementia (FTD). Changes in personality and behavior may make it difficult for affected individuals to interact with others in a socially appropriate manner. Communication skills worsen as the disease progresses. It is unclear how the development of ALS and frontotemporal lobe dementia (FTD) are related. Individuals who develop both conditions are diagnosed as having ALS-frontotemporal lobe dementia (FTD).
- A rare form of ALS that often runs in families is known as ALS-parkinsonism-dementia complex (ALS-PDC). This disorder is characterized by the signs and symptoms of ALS, in addition to a pattern of movement abnormalities known as parkinsonism, and a progressive loss of intellectual function (dementia). Signs of parkinsonism include unusually slow movements (bradykinesia), stiffness, and tremors. Affected members of the same family can have different combinations of signs and symptoms. Among the Chamorro people of Guam and people from the Kii Peninsula of Japan, ALS-parkinsonism-dementia complex (ALS-PDC) can be 100 times more frequent than ALS is in other populations 33. ALS-parkinsonism-dementia complex (ALS-PDC) has not been reported outside of these populations 34.
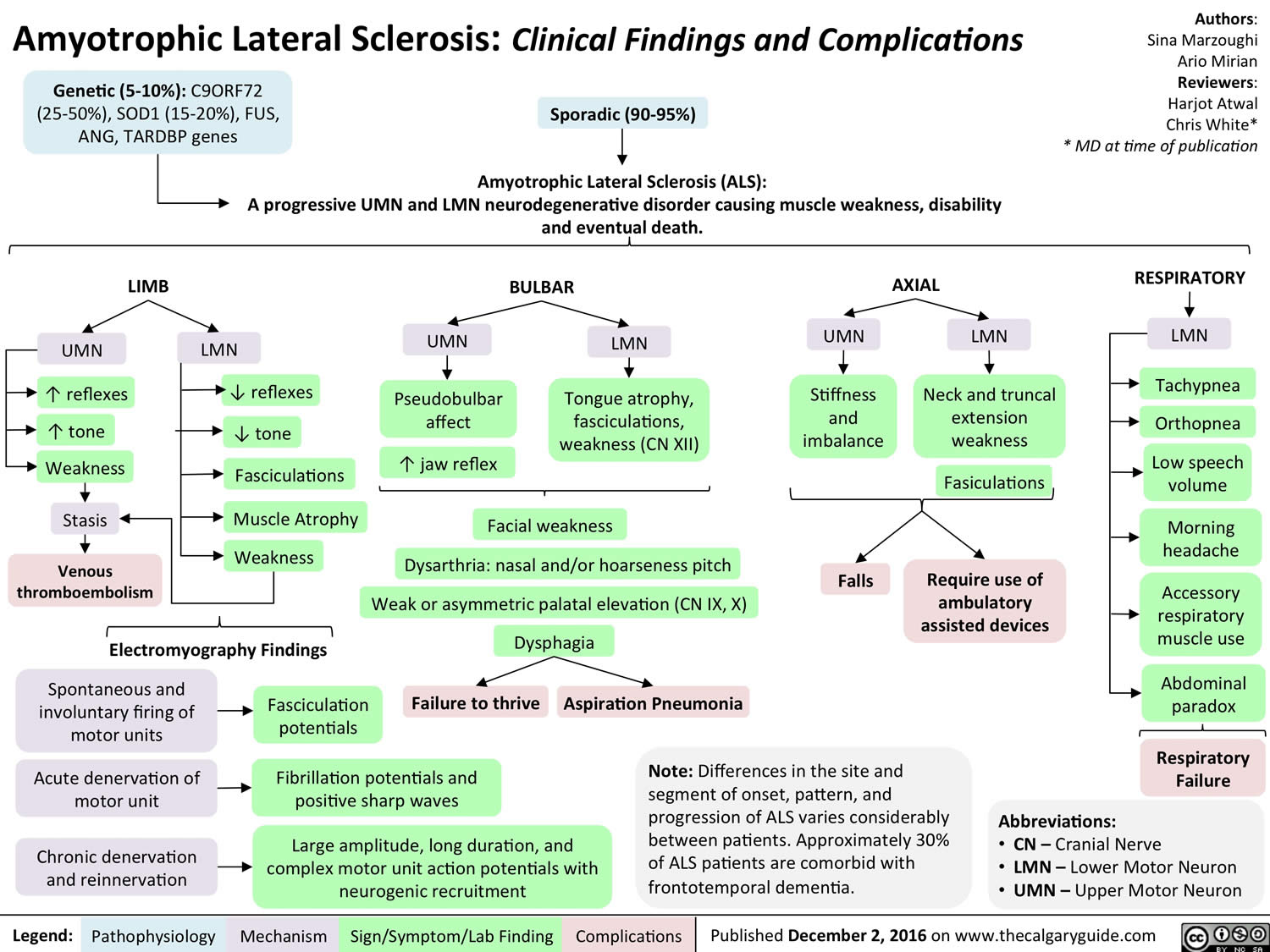
Sporadic ALS and Familial ALS
Nearly all cases of ALS are considered sporadic ALS (sporadic ALS). This means the disease seems to occur at random with no clearly associated risk factors and no family history of the disease. Although family members of people with sporadic ALS are at an increased risk for the disease, the overall risk is very low and most will not develop ALS.
About 5 to 10 percent of all ALS cases are familial (also called inherited or genetic). Mutations in more than a dozen genes have been found to cause familial ALS 35:
- About 25 to 40 percent of all familial cases (and a small percentage of sporadic cases) are caused by a defect in the C9ORF72 gene, which makes a protein that is found in motor neurons and nerve cells in the brain 36. Some people with this gene also develop a type of frontotemporal degeneration (FTD, a form of dementia) caused by atrophy of the brain’s temporal and frontal lobes.
- Another 12 to 20 percent of familial cases result from mutations in the superoxide dismutase 1 (SOD1) gene that is involved in production of the enzyme copper-zinc superoxide dismutase 1 37.
- In 2021, a team of scientists announced it had discovered a unique form of genetic ALS that affects children as early as age 4 years. This childhood form of ALS is linked to the gene SPTLC1 that is part of the body’s fat production system and may be caused by changes in the way the body metabolizes fatty materials called lipids.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Amyotrophic lateral sclerosis causes
Scientists don’t know exactly what causes ALS. Amyotrophic lateral sclerosis (ALS) is a progressive, degenerative disease that affects the nerve cells that control voluntary muscle movements such as walking and talking (motor neurons). These “motor neurons” run from your brain through the brainstem to the spinal cord to muscles that control movement in your arms, legs, chest, throat and mouth. Amyotrophic lateral sclerosis causes the motor neurons to gradually deteriorate, and then die. When motor neurons are damaged, they stop sending messages to the muscles, so the muscles can’t function and the muscle tissues waste away. At autopsy, ALS cases reveal degeneration of motor neurons in the motor cortex, brainstem motor nuclei, and anterior horns in the spinal cord 2.
ALS does not affect a person’s sensory functions or mental faculties. Other, nonmotor neurons, such as sensory neurons that bring information from sense organs to the brain, remain healthy.
Generally, ALS is categorized in one of two ways: Upper motor neuron disease that affects the nerves in your brain, while lower motor neuron disease that affects nerves coming from your spinal cord or brainstem. In both cases, motor neurons are damaged and eventually die.
Recent research suggests that multiple complex factors contribute to the death of motor neurons, one of these include altered mRNA processing leading to prion-like self-aggregation, superoxide dismutase type 1 SOD1 mutations leading to free radical toxicity, cascading inflammatory responses, and excessive concentrations of glutamate, among others 38, 39, 40. Mutations in several genes can cause familial ALS and contribute to the development of sporadic ALS. The rarer entity of familial ALS has numerous genetic mechanisms, most frequently repeat expansion of the C9ORF72 gene and various mutations of the SOD1 gene 41. Mutations in the C9orf72 gene account for 30 to 40 percent of familial ALS in the United States and Europe. Worldwide, SOD1 gene mutations cause 15 to 20 percent of familial ALS, and TARDBP and FUS gene mutations each account for about 5 percent of cases. The other genes that have been associated with familial ALS each account for a small proportion of cases. The C9orf72, SOD1, TARDBP, and FUS genes are key to the normal functioning of motor neurons and other cells. It is unclear how mutations in these genes contribute to the death of motor neurons, but it is thought that motor neurons are more sensitive to disruptions in function because of their large size. Some gene mutations lead to a disruption in the development of axons, the specialized extensions of nerve cells (such as motor neurons) that transmit nerve impulses. The altered axons may impair transmission of impulses from nerves to muscles, leading to muscle weakness and atrophy. Other mutations lead to a slowing in the transport of materials needed for the proper function of axons in motor neurons, eventually causing the motor neurons to die. Additional gene mutations prevent the breakdown of toxic substances, leading to their buildup in nerve cells. The accumulation of toxic substances (aggregates) can damage motor neurons and eventually cause cell death. However, it is unknown whether these toxic substances (aggregates) are involved in causing ALS or are a byproduct of the dying cell. In some cases of ALS, it is unknown how the gene mutation causes the condition.
Mutated SOD1 protein misfolds and forms aggregates, leading to cellular injury and eventually apoptosis. Both genetic aberrations are inherited in a mainly autosomal dominant pattern 42.
Although most cases of ALS are sporadic, about 5% of the cases have a family history. Familial amyotrophic lateral sclerosis (FALS) is inherited through genetic changes that are passed down in families. Familial ALS (FALS), are predominantly autosomal dominant and rarely X-linked or recessive 3. If a parent has familial ALS, each child has a 50% chance of developing it, too. The age of onset for familial amyotrophic lateral sclerosis (FALS) is about a decade earlier than for sporadic cases 43, 44. If you have familial amyotrophic lateral sclerosis (FALS), your healthcare team may suggest genetic counseling.
More than 20 mutated genes have been found to cause familial ALS including SOD1 45, TAR-DNA binding protein (TDP43) 46, FUS 47, OPTN 48, VCP 49, UBQLN2 50, C9orf72 51 and very recently TBK1 52. It is estimated that 60 percent of individuals with familial ALS have an identified genetic mutation. The cause of familial ALS in the remaining individuals is unknown.
Sporadic ALS and familial ALS (FALS) are clinically indistinguishable, and since mutations in familial ALS (FALS) genes are also present in sporadic or isolated cases of ALS, the disease can be interpreted as complex and multi-factorial 53. Nevertheless, clinical variability such as rate of progression, site of onset (limb or bulbar) and survival within patients and even relatives who carry the same gene mutation highlight the importance of external factors which may play a role in the susceptibility and age of onset of the disease 53.
Table 1. Gene mutations that cause ALS
| Gene | Fraction fALS (%) | Locus | Encoded protein | Functionality | Clinical phenotype | Neuropathology |
|---|---|---|---|---|---|---|
| C9ORF72 | 40–50 | 9p21.3 | C9ORF72 | Transcription and pre-mRNA splicing regulation; membrane traffic via Rab GTPase family | ALS; ALS+ FTLD; FTLD | NCI; DN; GCI; intranuclear RNA foci (sense, antisense); cytoplasmic RNA peptide aggregates |
| SOD1 | 20–25 | 21q22 | SOD1 | Major cytosolic antioxidant | ALS; PMA | NCI; NII; DN; GCI; aggregates—p62, C9ORF72, ubiquilin 2, others; impaired axonal transport, mitochondrial function; disturbed dendritic arborization of neurons; oxidative stress-related neuronal toxicity |
| TARDBP | 4–5 | 1p36.2 | TDP-43 | Transcription and pre-mRNA splicing regulation; micRNA biogenesis; RNA transport and stabilization; translational regulation of ApoE-II and CFTR | ALS; ALS+ FTLD; FTLD | NCI; NII; DN; GCI |
| FUS | 4–5 | 16p11.2 | FUS (or TLS) | Transcription and pre-mRNA splicing regulation; micRNA processing; mRNA transport and stabilization; maintenance of genomic integrity; regulating protein synthesis at synapse | ALS; ALS+ FTLD; FTLD | NCI; DN; GCI |
| OPTN | 2–3 | 10p13 | Optineurin | Golgi maintenance; exocytosis; vesicular trafficking; regulator of NF-kB signaling pathway; autophagy process | ALS; ALS+ FTLD | NCI; ↑ TDP-43, FUS, and SOD1 aggregates |
| PFN1 | 1–2 | 17p13 | Profilin 1 | Regulates ATP-mediated actin polymerization | ALS | NCI; ↓ axonal distension and growth cone elongation; co-aggregation with TDP-43 |
| VCP | 1–2 | 9p13 | VCP or p97 | Protein degradation via UPS, autophagy, and the ER; membrane fusion | ALS; ALS+ FTLD; FTLD | NCI; NII; DN; ↑ TDP-43 aggregates; ↓ stress-granule clearance |
| ANG | 1–2 | 14q11.2 | Angiogenin | RNA processing and tRNA modification; vascularization; RNAase activity and assembly of stress granules; neurite outgrowth and pathfinding | ALS; ALS+ FTLD | ↓ Stress-granule formation in motor neurons |
| TUBA4A | 1 | 2q35 | Tubulin α4A | Major component of microtubules; neuronal cell skeleton | ALS; ALS+ FTLD | NCI; destabilized microtubule network; ↓ microtubules repolymerization capability |
| UBQLN2 | <1 | Xp11 | Ubiquilin 2 | Protein degradation via UPS | ALS; ALS+ FTLD; FTLD (Rare) | NCI; ↑ TDP-43, p62, FUS, and OPTN inclusions |
| TAF15 | <1 | 17q11 | TAF15 | Transcription initiation; RNA polymerase II gene component | ALS | NCI |
| EWSR1 | <1 | 22q12.2 | EWSR1 | Transcriptional repressor | ALS | NCI; DN |
| hnRNPA1 | <1 | 12q13 | hnRNPA1 | Packing and transport of mRNA; micRNA biogenesis | Rare ALS; ALS+ FTLDa; FTLD (rare) | NCI |
| hnRNPA2B1 | <1 | 7p15 | hnRNPA2/B1 | Packing and transport of mRNA; micRNA biogenesis | ALS+ FTLD (rare)a; FTLD (rare) | NCI |
| SETX | <1 | 9q34.13 | Senataxin | DNA/RNA helicase activity; DNA/RNA metabolism | ALSb | ↓ neuronal differentiation; ↓ neurite growth |
| CREST | <1 | 20q13.3 | SS18L1 | Ca2+-dependent transcriptional activator | ALS | DN; ↓ dendrite outgrowth; ↑ interaction with FUS |
| MATR3 | <1 | 5q31.2 | Matrin 3 | RNA processing; stabilizing mRNAs; gene silencing; chromatin organization | ALS; ALS+ FTLD (Rare) | NCI; NII; ↑ interaction with TDP-43 |
| ATXN2 | 01/02/23 | 12q24 | Ataxin 2 | RNA processing; regulation of receptor tyrosine kinase endocytosis | ALS; ALS+ FTLD; PMA | NCI; ↑ interaction with TDP-43 |
| ELP3 | <1 | 8p21.1 | ELP3 | RNA processing; transcript elongation; histone acetylation; modification of tRNA wobble nucleosides | ALS | NCI; abnormal branching in motor axons; co-localization with TDP-43 and FUS aggregates |
| SQSTM1 | <1 | 5q35 | p62 or SQSTM1 | Autophagy and UPS degradation; regulator of NF-kB signaling pathway; immune response | ALS; ALS+ FTLD; FTLD | NCI; NII; GCI; ↓ mutSOD1 autophagic degradation |
| CHMP2B | <1 | 3p11 | CHMP2B | MVBs formation; protein trafficking between plasma membrane, trans-Golgi network, and lysosome | ALSc; PMAd; FTLD | NCI; DN; GCI; disrupted endosomal structure; aggregates of autophagosomes and multilamellar structures; ↑ TDP-43, p62, and ubiquitin inclusions |
| ALS2 | <1 | 2q33.1 | Alsin | Activation of the small GTPase Rac1 macropinocytosis-associated endosome fusion and trafficking; neurite outgrowth | ALSe; PLS | ↓ axonal growth; ↓ lysosome-dependent clearance of p62 and LC3-II |
| VAPB | <1 | 20q13 | VAPB | Regulation of ER–Golgi transport and secretion | ALS; PLS; PMA | NCI; ↑ TDP-43 toxicity and inclusions; aberrant synaptic microtubule cytoskeleton; nuclei mispositioning and aberrant architecture |
| SIGMAR1 | <1 | 9p13.3 | SIGMAR1 | Lipid transport through ER; BDNF and EGF signaling | ALS; ALS+ FTLD; FTLD | NCI; ↑ apoptosis induced by ER stress; ↑ interaction with VAPB |
| DCTN1 | <1 | 2p13 | Dynactin | ER–Golgi transport; centripetal movement of lysosomes and endosomes; spindle formation, chromosome movement; nuclear positioning; axonogenesis | ALS | NCI; p150glued aggregation; ↑ SOD1 aggregates |
| FIG4 | <1 | 6q21 | PI3,5P2 | Phosphoinositide phosphatase activity; endosomal vesicle trafficking to the trans-Golgi network; regulation of autophagy | ALS; PLS | NCI; ↑ swollen intracellular vacuoles; ↑ LC3-II, p62, and LAMP-2 aggregates in neurons and astrocytes |
| SPG11 | 1 | 15q21.1 | Spatascin | Neuronal cell skeleton; axonal transport; involved in synaptic vesicles | ALS; HSP | NCI; DN; ↓ acetylated stabilized tubulin; ↓ synaptic vesicles in neurites; disrupted anterograde axonal transport |
| NEFH | <1 | 22q12.2 | NEFH | Maintaining axon diameter | ALS | NCI; ↑ neurofilament aggregates |
| PRPH | <1 | 12q13 | Peripherin | Regulating neurite elongation during development and axonal regeneration after injury | ALS | NCI; DN; ↓ ability of the neurofilament network to assemble; ↑ ubiquitinated inclusions; coaggregation with mutSOD1 |
| NTE | <1 | 19p13 | Neuropathy target esterase | Regulating the neuronal membrane composition | ALS; HSP | Disruption of ER; ↑ reticular aggregates; ↑ vacuolization of nerve cell bodies |
| PON1-3 | <1 | 7q21 | Paraoxonase 1-3 | Enzymatic breakdown of nerve toxins | ALS | Oxidative stress-related neuronal toxicity |
| DAO | <1 | 12q22 | DAO | Regulating levels of D-serine, NMDAR function | ALS | NCI; NII; ↑ D-serine levels in motor neurons and glia; ↑ ubiquitinated inclusions |
| CHRNA3, CHRNA4, CHRNB4 | <1 | 15q24, 20q13, 15q24 | nAChR | Cholinergic neurotransmission | ALS§ | Cationic overloading, Ca+2 toxicity in MNs |
| ERBB4 | <1 | 2q34 | Receptor tyrosine-protein kinase ErbB-4 | Neuronal cell mitogenesis and differentiation | ALS | |
| CHCHD10 | <1 | 22q11 | Mitochondrial protein | Mitochondrial genome stability; cristae integrity and mitochondrial fusion | ALS+ FTLD | Mitochondrial fragmentation and DNA instability; mitochondrial crystalloid inclusions |
| C19orf12 | <1 | 9q12 | Mitochondrial protein | Unknown | ALS | |
| ALS3 | <1 | 18q21 | Disulfide redox protein | Unknown | ALS | |
| ALS7 | <1 | 20p13 | Unknown | Unknown | ALS | |
| ALS6-21 | <1 | 6p25, 21q22 | Unknown | Unknown | ALS§ | |
| ALS-FTD | <1 | 16p12 | Unknown | Unknown | ALS+ FTLD | NCI; DN; Type B TDP pathology; phosphorylated tau pathology |
| Gene variants that influence ALS phenotype | ||||||
| UNC13A | 19p13 | Unc-13 homolog A | Regulating neurite outgrowth and synaptic neurotransmission | ALS; ALS+ FTLD | ↓ synaptogenesis at neuromuscular junction; possible glutamate excitotoxicity | |
| EPHA4 | 2q36.1 | Ephrin receptor A4 | Receptor tyrosine kinase activity Modulation of cell morphology and integrin-dependent cell adhesion; regulation of synaptic plasticity and CNS development | ALS | NII; neurite outgrowth deficits in mutant TDP-43 expressed neurons | |
| CHGB | 20p12.3 | CHGB | Involved in the ER–Golgi system | ALS | NCI; ↓ density of synaptophysin-like immunoreactivity; ↑ interaction with mutSOD1 | |
| KIFAP3 | 1q24.2 | Kinesin-associated protein 3 | Tethering chromosomes to spindle pole; chromosome movement; axonal transport of choline acetyl-transferase | ALS§ | NCI; KIFAP3-SOD1 coaggregation in Lewy-body-like hyaline inclusions | |
| SMN | 5q13 | Germin 1 | Regulating biogenesis of snRNPs | ALS; LMN | NCI; NII; DN; coaggregation with mutFUS, mutSOD1; axonal defects | |
Footnotes:
a As part of multisystem proteinopathy.
b Phenotype more similar to Silver syndrome than to ALS.
c Predominant lower motor neuron (LMN) phenotype.
d As part of ALS.
e Predominant upper motor neuron (UMN) phenotype.
Abbreviations: ALS =amyotrophic lateral sclerosis; BDNF = brain derived neurotrophic factor; C9ORF72 = chromosome 9 open reading frame 72; CHGB = chromogranin B (secretogranin 1); CHMP2B = charged multivesicular body protein 2B; DAO = D-amino acid oxidase; DN = dystrophic neurites; EGF = epidermal growth factor; ELP3 = elongator acetyltransferase complex subunit 3; ER = endoplasmic reticulum; EWSR1 = Ewing sarcoma breakpoint region 1; FTLD = frontotemporal lobe dementia; FUS = fused in sarcoma; GCIs = glial cell inclusions; hnRNPA1 = heterogeneous nuclear ribonucleoprotein A1; hnRNPA2B1 = heterogeneous nuclear ribonucleoproteins A2/B1; HSP = hereditary spastic paraplegia; LAMP-2 = lysosomal-associated membrane protein 2; LC3-II = microtubule-associated protein 1A/1B-light chain 3-II; LMN = lower motor neuron disease; micRNA = micro RNA; mRNA = messenger RNA; mutSOD1 = mutant superoxide dismutase 1; MVBs = multivesicular bodies; nAChR = nicotinic acetylcholine receptor; NCI = neuronal cytoplasmic inclusions; NEFH = neurofilament heavy chain; NII = neuronal intranuclear inclusions; NMDAR = N-methyl-D-aspartate receptor; PI3,5P2 = phosphatidylinositol 3,5-bisphosphate 5-phosphatase; PLS = primary lateral sclerosis; PMA = progressive muscular atrophy; SIGMAR1 = sigma non-opioid intracellular receptor 1; SQSTM1 = sequestosome 1; SS18L1 = synovial sarcoma translocation gene on chromosome 18-Like 1; TAF15 = TATA box binding protein-associated factor 15; TDP-43 = TAR DNA-binding protein; TLS = translocated in liposarcoma; UMN = upper motor neuron; UPS = ubiquitin-proteasome system; VAPB = vesicle-associated membrane protein B; VCP = valosin-containing protein.
[Source 54 ]Risk factors for getting amyotrophic lateral sclerosis
Established risk factors for ALS include:
- Family history. Five to 10 percent of the people with ALS inherited it (familial ALS). In most people with familial amyotrophic lateral sclerosis, their children have a 50-50 chance of developing the disease.
- Age. Although amyotrophic lateral sclerosis can strike at any age, symptoms most commonly develop between the ages of 40 and the mid-60s. The mean age of onset of ALS varies from 50 to 65 years with the median age of onset of 64 years old. Only 5% of the cases have an onset <30 years of age 55, 56. ALS incidence is most pronounced in people 80 years or older (10.2/100,00 in men; 6.1/100,000 in women).
- Sex. Before the age of 65, slightly more men than women develop ALS. This sex difference disappears after age 70.
- Genetics. Some studies examining the entire human genome found many similarities in the genetic variations of people with familial ALS and some people with noninherited ALS (sporadic ALS). These genetic variations might make people more susceptible to ALS.
- Race and ethnicity. Caucasians and non-Hispanics are most likely to develop amyotrophic lateral sclerosis, but ALS affects people of all races and ethnic backgrounds.
Environmental factors, such as the following, might trigger amyotrophic lateral sclerosis:
- Smoking. Smoking is the only likely environmental risk factor for ALS. The risk seems to be greatest for women, particularly after menopause.
- Environmental toxin exposure. Some evidence suggests that exposure to lead, pesticides, fertilizers, herbicides, insecticides, formaldehyde or other substances in the workplace or at home might be linked to ALS 57, 58, 59, 60. Much study has been done, but no single agent or chemical has been consistently associated with ALS.
- Military service. Studies indicate that people who have served in the military are about one and half to two times more likely to develop ALS. It’s unclear what about military service might trigger the development of ALS. It might include exposure to lead, pesticides, beta-N-methylamino-l-alanine (BMAA) and other environmental toxins, traumatic injuries, viral infections, and intense exertion.
Amyotrophic lateral sclerosis pathophysiology
The pathology of amyotrophic lateral sclerosis is characterized by progressive degeneration and gliosis of axons of motor neurons within the anterior and lateral columns of the spinal cord 2. Motor neurons within the spinal cord anterior horns and Betz cells within the motor cortex are also lost 61. First described by French neurologist Jean-Martin Charcot in 1869, the name amyotrophic lateral sclerosis reflects both the degeneration of corticospinal motor neurons, whose descending axons in the lateral spinal cord appear scarred (“lateral sclerosis”), and the demise of spinal motor neurons, with secondary denervation and wasting of muscle (“amyotrophy”) 62. Unique to amyotrophic lateral sclerosis, Bunina bodies are eosinophilic inclusions visible in affected motor cells in many cases 63. Intracellular TDP-43 inclusions are present in most cases, providing a pathologic link between ALS and frontotemporal dementia (FTD), in which they are also found 64.
Several pathophysiological mechanisms have been proposed including: cytoplasmic protein mis-localization and aggregation 65, aberrant protein homeostasis 66, RNA toxicity 67, dysregulation of RNA processing 68, excitotoxicity mediated by excessive glutamate receptor activation 69, mitochondrial dysfunction 70, endoplasmic reticulum stress response and microglial activation 71, abnormal rearrangement of the cytoskeleton with impaired axonal transport 72 and oxidative stress 73. Moreover, the contribution of microglial cells, oligodendrocytes and astrocytes seems to be critical for the development of amyotrophic lateral sclerosis influencing significantly the speed of disease progression after onset 74. Figure 3 summarizes the major pathological mechanisms contributing to motor neuron injury in ALS.
Figure 3. Amyotrophic lateral sclerosis pathophysiology
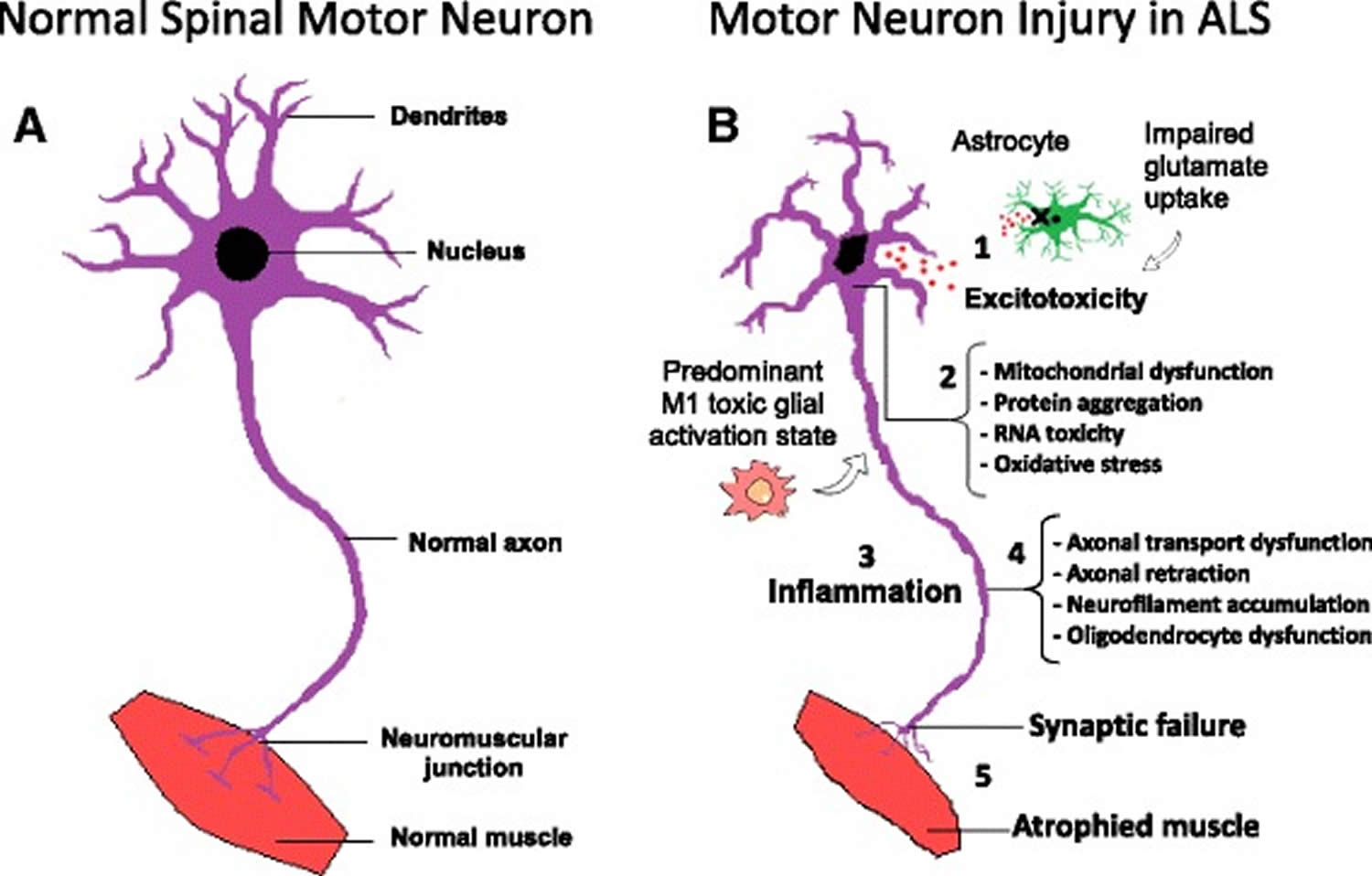
Footnotes: Molecular mechanisms in the pathology of amyotrophic lateral sclerosis. (a) Schematic representation of healthy spinal cord motor neuron. (b) Schematic representation of ALS affected spinal cord motor neuron: (1) Astrocytes are not able to support neuronal functions and impaired glutamate clearance leads to neuronal excitotoxicity; (2) Defects in protein degradation pathways and disturbances in RNA processing result in protein aggregate formation, RNA toxicity and mitochondrial dysfunction; (3) The secretion of pro-inflammatory cytokines by predominant M1 activated microglia contributes to the development of an inflammatory milieu; (4) Failure of axonal architecture and transport functions, together with the alteration of the physiological role of oligodendrocytes results in (5) synaptic failure, denervation and finally, muscle atrophy
[Source 4 ]Amyotrophic lateral sclerosis signs and symptoms
In amyotrophic lateral sclerosis (ALS), as motor neurons degenerate and die, they stop sending messages to the muscles, which causes the muscles to weaken, start to twitch (fasciculations), and waste away (atrophy). Eventually, the brain loses its ability to initiate and control voluntary movements.
Signs and symptoms of amyotrophic lateral sclerosis (ALS) vary greatly from person to person, depending on which neurons are affected. It generally begins with muscle weakness that spreads and gets worse over time. ALS often starts in the hands, feet or limbs, and then spreads to other parts of your body. As the disease advances and nerve cells are destroyed, your muscles get weaker. This eventually affects chewing, swallowing, speaking and breathing.
There’s generally no pain in the early stages of amyotrophic lateral sclerosis (ALS), and pain is uncommon in the later stages. ALS doesn’t usually affect your bladder control or your senses.
Early symptoms of amyotrophic lateral sclerosis (ALS) include:
- Muscle twitches in the arm, leg, shoulder, or tongue
- Muscle cramps
- Tight and stiff muscles (spasticity)
- Muscle weakness affecting an arm, a leg, the neck, or diaphragm
- Slurred and nasal speech
- Difficulty chewing or swallowing
As the disease progresses, muscle weakness and atrophy (muscle wasting) spread to other parts of your body. You may develop problems with:
- People with ALS eventually will not be able to stand or walk, get in or out of bed on their own, or use their hands and arms
- Chewing food and swallowing (dysphagia)
- Speaking or forming words (dysarthria)
- Shortness of breath (dyspnea); individuals with amyotrophic lateral sclerosis eventually lose the ability to breathe on their own and must depend on a ventilator
- Maintaining weight and malnourishment
- Muscle cramps and neuropathy (nerve damage or disease)
- Pseudobulbar affect (episodes of uncontrollable laughter or crying) can affect up to 50% of patients with ALS, especially those with the bulbar form.
- Anxiety and depression, because people with amyotrophic lateral sclerosis usually remain able to reason, remember, understand, and are aware of their progressive loss of function
Although not as common, people with ALS may also:
- Experience problems with language or decision-making
- Develop a form of dementia over time
ALS doesn’t affect your ability to taste, touch, or smell, or hear.
Amyotrophic lateral sclerosis complications
As the disease progresses, ALS causes complications, such as:
Breathing problems
Over time, amyotrophic lateral sclerosis paralyzes the muscles you use to breathe. You might need a device to help you breathe at night, similar to what someone with sleep apnea might wear. For example, you may be given a bilevel positive airway pressure (BiPAP) device to help with your breathing at night. This type of device supports your breathing through a mask worn over your nose, your mouth or both.
Some people with advanced ALS choose to have a tracheostomy — a surgically created hole at the front of the neck leading to the windpipe (trachea) — for full-time use of a respirator that inflates and deflates their lungs.
The most common cause of death for people with amyotrophic lateral sclerosis is respiratory failure. On average, death occurs within 3 to 5 years after symptoms begin. However, some people with amyotrophic lateral sclerosis live 10 or more years.
Speaking problems
Most people with amyotrophic lateral sclerosis develop trouble speaking. This usually starts as occasional, mild slurring of words, but becomes more severe. Speech eventually becomes difficult for others to understand, and people with ALS often rely on other communication technologies to communicate.
Eating problems
People with ALS can develop malnutrition and dehydration from damage to the muscles that control swallowing. They are also at higher risk of getting food, liquids or saliva into the lungs, which can cause pneumonia. A feeding tube can reduce these risks and ensure proper hydration and nutrition.
Dementia
Some people with amyotrophic lateral sclerosis have problems with memory and decision-making, and some are eventually diagnosed with a form of dementia called frontotemporal dementia.
Amyotrophic lateral sclerosis and frontotemporal degeneration
Frontotemporal degeneration has a significant coincidence with ALS, with approximately 20% of patients with ALS demonstrating criteria for frontotemporal dementia (FTD). Individuals who develop both conditions are diagnosed as having ALS-frontotemporal dementia (FTD). It theorized that chromosome 9 open-reading-frame 72 (C9orf72) gene mutation is a continuum between frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) and is responsible for the occurrence of both entities 75, 76, 77, 78. Many patients (about 30% to 50%) diagnosed with ALS will go on to develop varying degrees of cognitive impairment. While not overt dementia, patients can experience changes related to executive function and fluency, as well as behavioral changes such as apathy and disinhibition.
Frontotemporal dementia (FTD) is an umbrella term for a group of brain disorders that primarily affect the frontal and temporal lobes of the brain 79, 80. These areas of the brain are generally associated with personality, behavior and language. Frontotemporal lobe dementia (FTD) is characterized by loss of intellectual functions, such as memory problems, impaired abstract thinking, reasoning, and executive function, that are severe enough to hamper activities of daily living.
Signs and symptoms of frontotemporal dementia (FTD) vary, depending on which part of the brain is affected. Some people with frontotemporal dementia (FTD) have dramatic changes in their personalities and become socially inappropriate, impulsive or emotionally indifferent, while others lose the ability to use language properly.
Behavioral changes
The most common signs of frontotemporal dementia involve extreme changes in behavior and personality. These include:
- Increasingly inappropriate social behavior
- Loss of empathy and other interpersonal skills, such as having sensitivity to another’s feelings
- Lack of judgment
- Loss of inhibition
- Lack of interest (apathy), which can be mistaken for depression
- Repetitive compulsive behavior, such as tapping, clapping or smacking lips
- A decline in personal hygiene
- Changes in eating habits, usually overeating or developing a preference for sweets and carbohydrates
- Eating inedible objects
- Compulsively wanting to put things in the mouth
Speech and language problems
Some subtypes of frontotemporal dementia lead to language problems or impairment or loss of speech. Primary progressive aphasia, semantic dementia and progressive agrammatic (nonfluent) aphasia are all considered to be frontotemporal dementia.
Problems caused by these conditions include:
- Increasing difficulty in using and understanding written and spoken language, such as having trouble finding the right word to use in speech or naming objects
- Trouble naming things, possibly replacing a specific word with a more general word such as “it” for pen
- No longer knowing word meanings
- Having hesitant speech that may sound telegraphic
- Making mistakes in sentence construction
Motor disorders
Rarer subtypes of frontotemporal dementia are characterized by problems with movement similar to those associated with Parkinson’s disease or amyotrophic lateral sclerosis (ALS).
Motor-related problems may include:
- Tremor
- Rigidity
- Muscle spasms or twitches
- Poor coordination
- Difficulty swallowing
- Muscle weakness
- Inappropriate laughing or crying
- Falls or walking problems
Amyotrophic lateral sclerosis diagnosis
Amyotrophic lateral sclerosis (ALS) is difficult to diagnose early because it can mimic other neurological diseases. Furthermore, there is no single test that can definitely diagnose ALS.
Your doctor will conduct a physical exam and review your full medical history. A neurologic examination will test your reflexes, muscle strength, and other responses and will be held at regular intervals to assess whether symptoms such as muscle weakness, muscle wasting, and spasticity are progressively getting worse.
The most distinctive feature of amyotrophic lateral sclerosis is the coexistence of upper motor neuron and lower motor neuron signs and symptoms. Upper motor neuron (UMN) findings include hyperreflexia and spasticity, lower motor neuron (LMN) findings include muscle wasting and fasciculations.
Tests to rule out other conditions might include:
- Electromyogram (EMG). Your doctor inserts a needle electrode through your skin into various muscles. The test evaluates the electrical activity of your muscles when they contract and when they’re at rest. Abnormalities in muscles seen in an EMG can help doctors diagnose or rule out amyotrophic lateral sclerosis. An EMG can also help guide your exercise therapy.
- Nerve conduction study (NCS). This study measures your nerves’ ability to send impulses to muscles in different areas of your body. This test can determine if you have nerve damage or certain muscle or nerve diseases.
- Magnetic resonance imaging (MRI). Using radio waves and a powerful magnetic field, an MRI produces detailed images of your brain and spinal cord. An MRI can reveal spinal cord tumors, herniated disks in your neck or other conditions that might be causing your symptoms.
- Blood and urine tests. Analyzing samples of your blood and urine in the laboratory might help your doctor eliminate other possible causes of your signs and symptoms.
- Spinal tap (lumbar puncture). This involves removing a sample of your spinal fluid for laboratory testing using a small needle inserted between two vertebrae in your lower back.
- Muscle biopsy. If your doctor believes you may have a muscle disease rather than amyotrophic lateral sclerosis, you might undergo a muscle biopsy. While you’re under local anesthesia, a small portion of your muscle is removed and sent to a lab for analysis.
The World Federation of Neurology has also set forth categories that aid in the description of amyotrophic lateral sclerosis. These categories reflect the degree of clinical involvement evident at the time of an examination. They are as follows 6:
- Clinically definite ALS: upper motor neuron and lower motor neuron signs in at least 3 body segments.
- Clinically probable ALS: upper motor neuron and lower motor neuron signs in at least 2 body segments with some upper motor neuron signs in a segment above the lower motor neuron signs.
- Clinically probable, laboratory-supported ALS: upper motor neuron and lower motor neuron signs in 1 segment or upper motor neuron signs in 1 region coupled with lower motor neuron signs by EMG in at least two limbs.
- Clinically possible ALS: upper motor neuron and lower motor neuron signs in 1 body segment, upper motor neuron signs alone in at least 2 segments, or lower motor neuron signs in segments above upper motor neuron signs.
- Clinically suspected ALS: Pure lower motor neuron syndrome with other causes of lower motor neuron disease adequately excluded.
While the diagnosis of ALS has historically been primarily clinical, electrodiagnostic studies can further support the diagnosis if the clinical picture is unclear. Electromyography (EMG) is useful in detecting the findings of acute denervation (fibrillation and positive sharp waves), chronic denervation (long-duration, complex motor unit action potentials [MUAP]), and chronic reinnervation (large amplitude MUAP) 81. Nerve conduction studies will show normal sensory action potentials.
The Awaji criteria also include fasciculation potentials as evidence of acute denervation, along with the previously established electrodiagnostic signs, fibrillation, and sharp waves. In addition, electrodiagnostic evidence is considered of equal weight to clinical exam findings of lower motor neuron abnormality. These changes increase sensitivity while preserving specificity from the revised El Escorial criteria 82.
Motor nerve action potential amplitudes may be low before weakness is clinically evident 83. To be indicative of ALS, needle EMG must show signs of acute or chronic denervation in at least three spinal levels (bulbar, cervical, thoracic, and/or lumbosacral). If three spinal levels are not abnormal, acute or, chronic denervation needs to be evident in three extremities with the involvement of at least two muscles supplied by two different roots and two different nerves in each extremity.
Neuroimaging of ALS relies solely on magnetic resonance imaging (MRI) 6. Studies have shown patients with ALS to demonstrate iron accumulation within the precentral gyrus 84. As a result, on susceptibility-weighted imaging, the decreased signal is visible across the precentral gyrus, which is known as the “motor band sign” 85. On conventional MRI, decreased signal intensity within the motor cortex on T2 weighted images has been associated with ALS and may be used to support the diagnosis. In addition, well-defined lesions of increased signal intensity can be visible within the corticospinal tracts on T2 weighted images.
Findings indicative of upper motor neuronal disease have also been elucidated utilizing advanced MRI techniques such as spectroscopy and diffusion tensor imaging (DTI) 86. MR spectroscopy can detect and quantify chemical concentrations, specifically of N-acetyl aspartate (NAA), choline, and creatine within imaged tissues. Multiple studies have demonstrated decreased absolute and relative quantities of NAA in patients with ALS 87.
Once the diagnosis has been established, genetic testing is encouraged, particularly for the SOD1 and C9ORF72 genotypes, due to emerging genotype-specific therapies in clinical trials. For patients who have a family history of the autosomal dominant disease, genetic testing can be used both as a screening tool and to provide prognostic information. For patients who have a family history of unclear inheritance, genetic testing can be useful to find reduced penetrance or when information about family members is limited.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Amyotrophic lateral sclerosis differential diagnosis
Multiple diseases can mimic amyotrophic lateral sclerosis including, but not limited to 88, 89, 90, 91:
- Cervical myelopathy,
- Multifocal motor neuropathy,
- Myasthenia gravis,
- Lambert-Eaton syndrome,
- Inclusion body myositis,
- Benign fasciculation,
- Monomelic amyotrophy,
- Spinal muscular atrophy,
- Spinal bulbar muscular atrophy,
- Poliomyelitis
- Post-poliomyelitis syndrome,
- Late-onset Tay-Sachs disease,
- Motor neuron syndromes secondary to lymphoproliferative disorders and other cancers,
- Intraspinal lesions,
- Radiation spinal myelopathy,
- Hexosaminidase A deficiency,
- Cramp-fasciculation syndrome,
- Neuromyotonia,
- Radiculoplexopathy,
- Thyrotoxicosis,
- Myopathies.
Amyotrophic lateral sclerosis treatment
Amyotrophic lateral sclerosis (ALS) has no cure and there is no effective treatment to reverse the damage of amyotrophic lateral sclerosis, but treatments can slow the progression of symptoms, prevent complications, and make you more comfortable and independent. You might need an integrated team of doctors trained in many areas and other health care professionals to provide your care. This might prolong your survival and improve your quality of life.
Your team will help you select the right treatments for you. You have the right to choose or refuse any of the treatments suggested.
Medications
The United States Food and Drug Administration (FDA) has approved three medicines for treating ALS:
- Riluzole (Rilutek, Exservan, Tiglutik kit). Riluzole (Rilutek) is an oral medication believed to reduce damage to motor neurons by decreasing levels of glutamate, which transports messages between nerve cells and motor neurons. Taken orally, riluzole can increase life expectancy by 3 to 6 months. The thickened liquid form (Tiglutik) or the tablet (Exservan) that dissolves on the tongue may be preferred if you have swallowing difficulties. Riluzole (Rilutek) can cause side effects such as dizziness, gastrointestinal conditions and liver function changes. Your health care provider will monitor your blood counts and liver function while you’re taking the medicine. It is important to note that the elimination of riluzole will be affected by CYP1A2 inhibitors like caffeine and theophylline.
- Edaravone (Radicava). Edaravone (Radicava) is given intravenously through a vein in your arm or orally as a pill, can reduce the decline in daily functioning in people with ALS. Edaravone is a free radical scavenger that is thought to reduce oxidative stress and has proven to be beneficial on a subset of patients with early-stage (less than two years of symptoms, independently living, forced vital capacity (FVC) >80% and scores of 2 or more in all items of Revised Amyotrophic Lateral Sclerosis Functional Rating Scale [ALSFRS-R]) probable or definite ALS. One study showed approximately 33% slower functional decline in patients at 24 weeks follow up 92. Its effect on life span isn’t yet known. Side effects can include bruising, headache and shortness of breath. This medicine is given daily for two weeks a month. Edaravone has the limitation of being very expensive at around $146,000 per year and is administered in 60 mg daily infusions for 14 days followed by 14 days off for the first cycle, and then 60 mg/day for 10 days and 14 days off for the subsequent cycles 6. Edaravone should be used with caution in patients with asthma as it can cause serious asthmatic reactions in up to 5%.
- Sodium phenylbutyrate and taurursodiol (Relyvrio). This medicine, recently approved by the FDA, can slow the rate of decline in people with ALS. In particular, it may help people with performing daily tasks. It also may help people with ALS live longer, but more study is needed. Potential side effects of the medicine include diarrhea, belly pain, nausea and upper respiratory infection. People with disorders that affect bile acid circulation may experience diarrhea that gets worse when taking this medicine.
Your doctor might also prescribe medications to provide relief from other symptoms, including:
- Muscle cramps and muscle spasms. Frequent and painful muscle spasms can be treated with mexiletine, which was well-tolerated and demonstrated good symptomatic response at a dose of 150 mg oral twice daily, in a small sample study 93, 94. Other options are levetiracetam and with less efficacy gabapentin, baclofen, and tizanidine 95. The latter two of which have shown efficacy in the management of spasticity. When oral therapy is not effective or well-tolerated, botulinum toxin injections into the spastic muscles can be useful. As weakness and functional decline inevitably progress, patients should be provided assistive devices (canes, orthoses, crutches, and eventually wheelchairs), removable headrests in those with neck weakness, specialized utensils, and holders, and eventually a pressure-relieving mattress with frequent repositioning to prevent pressure ulcers 96.
- Constipation
- Fatigue
- Excessive saliva (sialorrhea) and phlegm. Excessive saliva is very common and can be treated with atropine (0.4 mg every 4-6 hour), hyoscyamine, amitriptyline (10 to 150 mg every night at bedtime), glycopyrrolate (1 mg three times a day), botulinum toxin injections into salivary glands, and even low-dose radiation therapy in those with refractory symptoms 97, 98, 97.
- Pain is a commonly reported symptom of patients with ALS, arising from numerous causes including muscle cramping, spasticity, and as a result of decreased mobility. Assistive devices such as special mattresses, pillows, and wheelchairs may help to prevent pain. Ultimately many patients require nonopioid analgesics and anti-inflammatory drugs, and when these fail, opioids become the mainstay of pain treatment 99.
- Depression has a significant effect on the quality of life in patients with ALS, and studies have shown that treatment can improve quality of life. While no controlled trials have evaluated the treatment of depression in patients with ALS, amitriptyline is commonly used as it can also treat other symptoms such as insomnia, excessive saliva (sialorrhea), and pseudobulbar affect.
- Sleep problems
- Uncontrolled outbursts of laughing or crying (pseudobulbar affect). In these patients, AVP-923 (dextromethorphan-quinidine 20mg/10mg) has proven to be effective in a multicenter, randomized, double-blind, controlled clinical trial 100. Less rigorous evidence has supported the use of amitriptyline and fluvoxamine in smaller trials.
Therapies
- Breathing support. As your muscles responsible for breathing start to weaken, you may have shortness of breath during physical activity and difficulty breathing at night or when lying down. Your doctors might test your breathing regularly and provide you with devices to assist your breathing at night.
- Noninvasive ventilation (NIV) refers to breathing support that is usually delivered through a mask over the nose and/or mouth. Initially, noninvasive ventilation (NIV) may only be necessary at night but may eventually be used full time. Because the muscles that control breathing become weak, you also may have trouble generating a strong cough. There are several techniques to increase forceful coughing, including mechanical cough assistive devices.
- As the disease progresses, you might need mechanical ventilation (respirators) to inflate and deflate your lungs. Doctors may place a breathing tube through your mouth or may surgically create a hole at the front of your neck and insert a tube leading to the windpipe (tracheostomy) that connects to a respirator.
- Physical therapy. A physical therapist can address pain, walking, mobility, bracing and equipment needs that help you stay independent. Practicing low-impact exercises can help maintain your cardiovascular fitness, muscle strength and range of motion for as long as possible. Regular exercise can also help improve your sense of well-being. Appropriate stretching can help prevent pain and help your muscles function at their best. A physical therapist can also help you adjust to a brace, walker or wheelchair and might suggest devices such as ramps that make it easier for you to get around.
- Occupational therapy. An occupational therapist can help you find ways to remain independent despite hand and arm weakness. Adaptive equipment can help you perform activities such as dressing, grooming, eating and bathing. An occupational therapist can also help you modify your home to allow accessibility if you have trouble walking safely.
- Speech therapy. A speech therapist can teach you adaptive techniques to make your speech more understandable. Speech therapists can also help you explore other methods of communication, such as an alphabet board or pen and paper. Ask your therapist about the possibility of borrowing or renting devices such as tablet computers with text-to-speech applications or computer-based equipment with synthesized speech that can help you communicate.
- Devices such as computer-based speech synthesizers use eye-tracking technology and can help you develop ways to respond to yes-or-no questions with your eyes or by other nonverbal means. Some people with ALS may choose to use voice banking while they are still able to speak as a process of storing their own voice for future use in computer-based speech synthesizers.
- A brain-computer interface (BCI) is a system that allows individuals with ALS to communicate or control equipment such as a wheelchair using only brain activity. Researchers are developing more efficient, mobile, and even some auditory-based brain-computer interfaces (BCIs) for those with severe paralysis and/or visual impairments.
- Nutrition support. Nutritionists can teach you to plan and prepare small meals throughout the day that provide enough calories, fiber, and fluid and how to avoid foods that are difficult to swallow. Suction devices can remove excess fluids or saliva and prevent choking. When you can no longer eat with help, doctors may advise inserting a feeding tube, which reduces your risk of choking and pneumonia that can result from inhaling liquids into your lungs.
- Psychological and social support. Your team might include a social worker to help with financial issues, insurance, and getting equipment and paying for devices you need. Psychologists, social workers and others may provide emotional support for you and your family.
- Rehabilitation. A treatment plan for ALS usually includes rehabilitation. Rehabilitation is different for everyone and a very important part of management.
Coping and support
Learning you have ALS can be devastating. The following tips may help you and your family cope:
- Take time to grieve. The news that you have a fatal condition that will reduce your mobility and independence is difficult. You and your family will likely go through a period of mourning and grief after diagnosis.
- Be hopeful. Your team will help you focus on your abilities and healthy living. Some people with ALS live much longer than the 3 to 5 years usually associated with this condition. Some live 10 years or more. Maintaining an optimistic outlook can help improve quality of life for people with ALS.
- Think beyond the physical changes. Many people with amyotrophic lateral sclerosis lead rewarding lives despite physical limitations. Try to think of ALS as only one part of your life, not your entire identity.
- Join a support group. You might find comfort in a support group with others who have ALS. Loved ones helping with your care also might benefit from a support group of other ALS caregivers. Find support groups in your area by talking to your doctor or by contacting the ALS Association.
Make decisions now about your future medical care. Planning for the future allows you to be in control of decisions about your life and your care. With the help of your doctor, hospice nurse or social worker, you can decide whether you want certain life-extending procedures.
You can also decide where you want to spend your final days. You might consider hospice care options. Planning for the future can help you and your loved ones calm anxieties.
Clinical trials
Consider participating in a clinical trial so clinicians and scientists can learn more about ALS. Clinical research uses human volunteers to help researchers learn more about a disorder and perhaps find better ways to safely detect, treat, or prevent disease.
All types of volunteers are needed—those who are healthy or may have an illness or disease—of all different ages, sexes, races, and ethnicities to ensure that study results apply to as many people as possible, and that treatments will be safe and effective for everyone who will use them.
For information about participating in clinical research visit NIH Clinical Research Trials and You (https://www.nih.gov/health-information/nih-clinical-research-trials-you). Learn about clinical trials currently looking for people with ALS at Clinicaltrial.gov (https://clinicaltrials.gov).
Potential future treatments
Clinical studies on promising medications and treatments are occurring in ALS all the time.
Whether you are eligible for a clinical study depends on many factors related to your ALS and also whether there are ongoing studies enrolling patients. While many of these studies are promising, they are still only studies, and it isn’t yet clear if these treatments will help people with ALS. Talk to your doctor about what might be available for you.
Cellular defects
Scientists are seeking to understand the mechanisms that selectively trigger motor neurons to degenerate in ALS, and to find effective approaches to halt the processes leading to cell death. Using both animal models and cell culture systems, scientists are trying to determine how and why ALS-causing gene mutations lead to the destruction of neurons. These animal models include fruit flies, zebrafish, and rodents.
Initially, genetically modified animal models focused on mutations in the SOD1 gene but more recently, models have been developed for defects in the C9ORF72, TARDP, FUS, PFN1, TUBA4A, and UBQLN2 genes. Research in these models suggests that, depending on the gene mutation, motor neuron death is caused by a variety of cellular defects, including in the processing of RNA molecules and recycling of proteins, and structural impairments of motor neurons. Increasing evidence also suggests that various types of glial support cells and inflammation cells of the nervous system may play an important role in the disease.
Stem Cells
In addition to animal models, scientists are also using innovative stem cells models to study ALS. Scientists have developed ways to take skin or blood cells from individuals with ALS and turn them into stem cells, which are capable of becoming any cell type in the body, including motor neurons and other cell types that may be involved in the disease. NINDS is supporting research on the development of stem cell lines for a number of neurodegenerative diseases, including ALS.
Familial versus sporadic ALS
Overall, the work in familial ALS is already leading to a greater understanding of the more common sporadic form of the disease. Because familial ALS and sporadic ALS show many of the same signs and symptoms, some researchers believe that some familial ALS genes may also be involved in sporadic ALS.
Clinical research studies are looking into how ALS symptoms change over time in people with C9ORF72 mutations. Other research studies are working to identify additional genes that may cause or put a person at risk for either familial or sporadic ALS.
Additionally, researchers are looking at the potential role of epigenetics in the development of ALS. Epigenetic changes can switch genes on and off, and thus can profoundly affect the human condition in both health and disease. These changes can occur in response to multiple factors, including external or environmental conditions and events. Although this research is still at a very exploratory stage, scientists hope that understanding epigenetics can offer new information about how ALS develops.
Biomarkers
Biomarkers are biological measures that help to identify the presence or rate of progression of a disease or the effectiveness of a therapeutic intervention. Since ALS is difficult to diagnose, biomarkers could potentially help clinicians diagnose ALS earlier and faster.
Additionally, biomarkers are needed to help predict and accurately measure disease progression and enhance clinical studies aimed at developing more effective treatments. Biomarkers can be molecules derived from a bodily fluid (such as those in the blood and cerebrospinal fluid), an image of the brain or spinal cord, or a measure of the ability of a nerve or muscle to process electrical signals.
New treatment options
Potential therapies for ALS are being investigated in a range of disease models. This work involves tests of drug-like compounds, gene therapy approaches, antibodies, and cell-based therapies. Scientists are currently investigating whether lowering levels of the SOD1 enzyme in the brain and spinal cord of individuals with SOD1 gene mutations would slow the rate of disease progression. Other scientists are studying the use of glial-restricted progenitor cells (which have the ability to develop into other support cells) to slow disease progression and improve respiratory function. Additionally, a number of exploratory treatments are being tested in people with ALS. Investigators are optimistic that these and other basic, translational, and clinical research studies will eventually lead to new and more effective treatments for ALS.
Amyotrophic lateral sclerosis life expectancy
The median survival of people with amyotrophic lateral sclerosis is between 3 to 5 years; however, around 30% of patients are alive after five years of diagnosis and 10% to 20% after ten years 6. Factors associated with better survival include increased weight at diagnosis (mild obesity by body mass index [BMI)], younger age at onset, higher ALS functional rating scale score, forced vital capacity (FVC) >50% at presentation, and limb rather than bulbar symptoms 101, 102.
References- Walling AD. Amyotrophic lateral sclerosis: Lou Gehrig’s disease. Am Fam Physician. 1999 Mar 15;59(6):1489-96. https://www.aafp.org/pubs/afp/issues/1999/0315/p1489.html
- Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016 Nov 10;539(7628):197-206. doi: 10.1038/nature20413
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7(11):603–615. doi: 10.1038/nrneurol.2011.150
- Ciervo Y, Ning K, Jun X, Shaw PJ, Mead RJ. Advances, challenges and future directions for stem cell therapy in amyotrophic lateral sclerosis. Mol Neurodegener. 2017 Nov 13;12(1):85. doi: 10.1186/s13024-017-0227-3
- Bello-Haas VD. Physical therapy for individuals with amyotrophic lateral sclerosis: current insights. Degener Neurol Neuromuscul Dis. 2018 Jul 16;8:45-54. doi: 10.2147/DNND.S146949
- Brotman RG, Moreno-Escobar MC, Joseph J, et al. Amyotrophic Lateral Sclerosis. [Updated 2022 Aug 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK556151
- Amyotrophic Lateral Sclerosis. https://www.mayoclinic.org/departments-centers/amyotrophic-lateral-sclerosis-subspecialty-group/overview/ovc-20443595
- Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012 Mar 14;2012(3):CD001447. doi: 10.1002/14651858.CD001447.pub3
- Mehta P, Raymond J, Punjani R, Larson T, Bove F, Kaye W, Nelson LM, Topol B, Han M, Muravov O, Genson C, Davis B, Hicks T, Horton K. Prevalence of amyotrophic lateral sclerosis (ALS), United States, 2016. Amyotroph Lateral Scler Frontotemporal Degener. 2022 May;23(3-4):220-225. https://doi.org/10.1080/21678421.2021.1949021
- National Amyotrophic Lateral Sclerosis (ALS) Registry. https://www.cdc.gov/als/National_disease_estimates.html
- National Amyotrophic Lateral Sclerosis (ALS) Registry. https://www.cdc.gov/als/dashboard/index.html
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006 Dec 22;351(3):602-11. doi: 10.1016/j.bbrc.2006.10.093
- Bigio EH, Wu JY, Deng HX, Bit-Ivan EN, Mao Q, Ganti R, Peterson M, Siddique N, Geula C, Siddique T, Mesulam M. Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid. Acta Neuropathol. 2013 Mar;125(3):463-5. doi: 10.1007/s00401-013-1089-6
- Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR Jr, Parisi JE, Lucas JA, Petersen RC, Rademakers R. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012 Mar;135(Pt 3):765-83. doi: 10.1093/brain/aws004
- Zhang JV, Irwin DJ, Blennow K, Zetterberg H, Lee EB, Shaw LM, Rascovsky K, Massimo L, McMillan CT, Chen-Plotkin A, Elman L, Lee VM, McCluskey L, Toledo JB, Weintraub D, Wolk D, Trojanowski JQ, Grossman M. Neurofilament Light Chain Related to Longitudinal Decline in Frontotemporal Lobar Degeneration. Neurol Clin Pract. 2021 Apr;11(2):105-116. doi: 10.1212/CPJ.0000000000000959. Erratum in: Neurol Clin Pract. 2021 Dec;11(6):e980-e981.
- Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J, Muravov O, Horton K. Prevalence of Amyotrophic Lateral Sclerosis – United States, 2015. MMWR Morb Mortal Wkly Rep. 2018 Nov 23;67(46):1285-1289. doi: 10.15585/mmwr.mm6746a1
- Jordan H, Rechtman L, Wagner L, Kaye WE. Amyotrophic lateral sclerosis surveillance in Baltimore and Philadelphia. Muscle Nerve. 2015 Jun;51(6):815-21. doi: 10.1002/mus.24488
- Jordan H, Fagliano J, Rechtman L, Lefkowitz D, Kaye W. Population-based surveillance of amyotrophic lateral sclerosis in New Jersey, 2009-2011. Neuroepidemiology. 2014;43(1):49-56. doi: 10.1159/000365850
- Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci. 2001 Oct 15;191(1-2):3-9. doi: 10.1016/s0022-510x(01)00630-x
- Cronin S, Hardiman O, Traynor BJ. Ethnic variation in the incidence of ALS: a systematic review. Neurology. 2007 Mar 27;68(13):1002-7. doi: 10.1212/01.wnl.0000258551.96893.6f
- Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118-30. doi: 10.1159/000351153
- Armon C. Smoking may be considered an established risk factor for sporadic ALS. Neurology. 2009 Nov 17;73(20):1693-8. doi: 10.1212/WNL.0b013e3181c1df48
- Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009 Feb 3;4:3. doi: 10.1186/1750-1172-4-3
- Leigh PN, Abrahams S, Al-Chalabi A, Ampong MA, Goldstein LH, Johnson J, Lyall R, Moxham J, Mustfa N, Rio A, Shaw C, Willey E; King’s MND Care and Research Team. The management of motor neurone disease. J Neurol Neurosurg Psychiatry. 2003 Dec;74 Suppl 4(Suppl 4):iv32-iv47. doi: 10.1136/jnnp.74.suppl_4.iv32
- Polkey MI, Lyall RA, Moxham J, Leigh PN. Respiratory aspects of neurological disease. J Neurol Neurosurg Psychiatry. 1999 Jan;66(1):5-15. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1736177/pdf/v066p00005.pdf
- Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015 Nov 16;6:171. doi: 10.4103/2152-7806.169561
- Couratier, P., Truong, C., Khalil, M., Devière, F. and Vallat, J.-M. (2000), Clinical features of flail arm syndrome. Muscle Nerve, 23: 646-647. https://doi.org/10.1002/(SICI)1097-4598(200004)23:4<646::AID-MUS26>3.0.CO;2-E
- Wijesekera LC, Mathers S, Talman P, Galtrey C, Parkinson MH, Ganesalingam J, Willey E, Ampong MA, Ellis CM, Shaw CE, Al-Chalabi A, Leigh PN. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology. 2009 Mar 24;72(12):1087-94. doi: 10.1212/01.wnl.0000345041.83406.a2
- Rowland, L.P. (2010), Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve, 41: 161-165. https://doi.org/10.1002/mus.21565
- Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, Rowland LP. The natural history of primary lateral sclerosis. Neurology. 2006 Mar 14;66(5):647-53. doi: 10.1212/01.wnl.0000200962.94777.71
- Singer, M.A., Statland, J.M., Wolfe, G.I. and Barohn, R.J. (2007), Primary lateral sclerosis. Muscle Nerve, 35: 291-302. https://doi.org/10.1002/mus.20728
- McCluskey LF, Elman LB, Martinez-Lage M, Van Deerlin V, Yuan W, Clay D, Siderowf A, Trojanowski JQ. Amyotrophic lateral sclerosis-plus syndrome with TAR DNA-binding protein-43 pathology. Arch Neurol. 2009 Jan;66(1):121-4. doi: 10.1001/archneur.66.1.121
- McGeer PL, Steele JC. The ALS/PDC syndrome of Guam: potential biomarkers for an enigmatic disorder. Prog Neurobiol. 2011 Dec;95(4):663-9. doi: 10.1016/j.pneurobio.2011.04.001
- Amyotrophic lateral sclerosis. https://medlineplus.gov/genetics/condition/amyotrophic-lateral-sclerosis/#frequency
- Amyotrophic Lateral Sclerosis (ALS). https://www.ninds.nih.gov/health-information/disorders/amyotrophic-lateral-sclerosis-als
- Gossye H, Engelborghs S, Van Broeckhoven C, et al. C9orf72 Frontotemporal Dementia and/or Amyotrophic Lateral Sclerosis. 2015 Jan 8 [Updated 2020 Dec 17]. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK268647
- Dangoumau A, Verschueren A, Hammouche E, Papon MA, Blasco H, Cherpi-Antar C, Pouget J, Corcia P, Andres CR, Vourc’h P. Novel SOD1 mutation p.V31A identified with a slowly progressive form of amyotrophic lateral sclerosis. Neurobiol Aging. 2014 Jan;35(1):266.e1-4. doi: 10.1016/j.neurobiolaging.2013.07.012
- Honig LS, Chambliss DD, Bigio EH, Carroll SL, Elliott JL. Glutamate transporter EAAT2 splice variants occur not only in ALS, but also in AD and controls. Neurology. 2000 Oct 24;55(8):1082-8. doi: 10.1212/wnl.55.8.1082
- Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011 Oct 28;147(3):498-508. doi: 10.1016/j.cell.2011.10.011
- Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011 Mar;10(3):253-63. doi: 10.1016/S1474-4422(11)70015-1
- Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, Heverin M, Jordan N, Kenna K, Lynch C, McLaughlin RL, Iyer PM, O’Brien C, Phukan J, Wynne B, Bokde AL, Bradley DG, Pender N, Al-Chalabi A, Hardiman O. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012 Mar;11(3):232-40. doi: 10.1016/S1474-4422(12)70014-5. Epub 2012 Feb 3. Erratum in: Lancet Neurol. 2012 May;11(5):388.
- Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017 Jul;88(7):540-549. doi: 10.1136/jnnp-2016-315018
- Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology. 2008 Jan 8;70(2):144-52. doi: 10.1212/01.wnl.0000296811.19811.db
- Ticozzi, N., Vance, C., LeClerc, A.L., Keagle, P., Glass, J.D., McKenna-Yasek, D., Sapp, P.C., Silani, V., Bosco, D.A., Shaw, C.E., Brown, R.H., Jr and Landers, J.E. (2011), Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet., 156: 285-290. https://doi.org/10.1002/ajmg.b.31158
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, YF X, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4(9):e1000193. doi: 10.1371/journal.pgen.1000193
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–226. doi: 10.1038/nature08971
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864. doi: 10.1016/j.neuron.2010.11.036
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. doi: 10.1038/nature10353
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.01
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436–1441. doi: 10.1126/science.aaa3650
- Poppe L, Rué L, Robberecht W, Van Den Bosch L. Translating biological findings into new treatment strategies for amyotrophic lateral sclerosis (ALS). Exp Neurol. 2014 Dec;262 Pt B:138-51. doi: 10.1016/j.expneurol.2014.07.001
- Ghasemi M, Brown RH Jr. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb Perspect Med. 2018 May 1;8(5):a024125. doi: 10.1101/cshperspect.a024125
- Logroscino G, Traynor BJ, Hardiman O, Chio’ A, Couratier P, Mitchell JD, Swingler RJ, Beghi E; EURALS. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry. 2008 Jan;79(1):6-11. doi: 10.1136/jnnp.2006.104828
- Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E; EURALS. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010 Apr;81(4):385-90. doi: 10.1136/jnnp.2009.183525
- Kamel F, Umbach DM, Munsat TL, Shefner JM, Hu H, Sandler DP. Lead exposure and amyotrophic lateral sclerosis. Epidemiology. 2002 May;13(3):311-9. doi: 10.1097/00001648-200205000-00012
- Valerie McGuire, W. T. Longstreth, Jr., Lorene M. Nelson, Thomas D. Koepsell, Harvey Checkoway, Michael S. Morgan, Gerald van Belle, Occupational Exposures and Amyotrophic Lateral Sclerosis. A Population-based Case-Control Study, American Journal of Epidemiology, Volume 145, Issue 12, 15 June 1997, Pages 1076–1088, https://doi.org/10.1093/oxfordjournals.aje.a009070
- Weisskopf MG. Enironmental and Molecular Mutagensis. Vol. 55. NJ USA: Wiley-Blackwell; 2014. Formaldehyde exposure and amyotrophic lateral sclerosis; p. S25.
- Yu Y, Hayashi S, Cai X, Fang C, Shi W, Tsutsui H, Sheng J. Pu-erh tea extract induces the degradation of FET family proteins involved in the pathogenesis of amyotrophic lateral sclerosis. Biomed Res Int. 2014;2014:254680. doi: 10.1155/2014/254680
- Saberi S, Stauffer JE, Schulte DJ, Ravits J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol Clin. 2015 Nov;33(4):855-76. doi: 10.1016/j.ncl.2015.07.012
- Goetz, C.G. (2000), Amyotrophic lateral sclerosis: Early contributions of Jean-Martin Charcot. Muscle Nerve, 23: 336-343. https://doi.org/10.1002/(SICI)1097-4598(200003)23:3<336::AID-MUS4>3.0.CO;2-L
- Piao YS, Wakabayashi K, Kakita A, Yamada M, Hayashi S, Morita T, Ikuta F, Oyanagi K, Takahashi H. Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 2003 Jan;13(1):10-22. doi: 10.1111/j.1750-3639.2003.tb00002.x
- Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ, Xie SX, Kwong LK, Elman L, McCluskey L, Clark CM, Malunda J, Miller BL, Zimmerman EA, Qian J, Van Deerlin V, Grossman M, Lee VM, Trojanowski JQ. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. 2009 Feb;66(2):180-9. doi: 10.1001/archneurol.2008.558
- Lagier-Tourenne C, Polymenidou M, Hutt KR, AQ V, Baughn M, Huelga SC, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15(11):1488–1497. doi: 10.1038/nn.3230
- Kitamura A, Inada N, Kubota H, Matsumoto G, Kinjo M, Morimoto RI, et al. Dysregulation of the proteasome increases the toxicity of ALS-linked mutant SOD1. Genes Cells. 2014;19(3):209–224. doi: 10.1111/gtc.12125
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011
- Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR, et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol. 2014;40(6):670–685. doi: 10.1111/nan.12148
- Alexander GM, Deitch JS, Seeburger JL, Del Valle L, Heiman-Patterson TD. Elevated cortical extracellular fluid glutamate in transgenic mice expressing human mutant (G93A) cu/Zn superoxide dismutase. J Neurochem. 2000;74(4):1666–1673. doi: 10.1046/j.1471-4159.2000.0741666.x
- Shi P, Gal J, Kwinter DM, Liu X, Zhu H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2010;1802(1):45–51. doi: 10.1016/j.bbadis.2009.08.012
- Saxena S, Cabuy E, Caroni PA. Role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12(5):627–636. doi: 10.1038/nn.2297
- Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol. 1997;139(5):1307–1315. doi: 10.1083/jcb.139.5.1307
- Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med. 2010;48(5):629–641. doi: 10.1016/j.freeradbiomed.2009.11.018
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013 May;16(5):571-9. doi: 10.1038/nn.3357
- Borroni B, Benussi A, Premi E, Alberici A, Marcello E, Gardoni F, Di Luca M, Padovani A. Biological, Neuroimaging, and Neurophysiological Markers in Frontotemporal Dementia: Three Faces of the Same Coin. J Alzheimers Dis. 2018;62(3):1113-1123. doi: 10.3233/JAD-170584
- Grossman M, Elman L, McCluskey L, McMillan CT, Boller A, Powers J, Rascovsky K, Hu W, Shaw L, Irwin DJ, Lee VM, Trojanowski JQ. Phosphorylated tau as a candidate biomarker for amyotrophic lateral sclerosis. JAMA Neurol. 2014 Apr;71(4):442-8. doi: 10.1001/jamaneurol.2013.6064
- Junttila A, Kuvaja M, Hartikainen P, Siloaho M, Helisalmi S, Moilanen V, Kiviharju A, Jansson L, Tienari PJ, Remes AM, Herukka SK. Cerebrospinal Fluid TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis Patients with and without the C9ORF72 Hexanucleotide Expansion. Dement Geriatr Cogn Dis Extra. 2016 Apr 16;6(1):142-9. doi: 10.1159/000444788
- Bretag-Norris R, Gallur L, Flynn P. Heterogeneity in the psychiatric presentation of behavioural variant frontotemporal dementia (bvFTD). Australas Psychiatry. 2019 Oct;27(5):491-495. doi: 10.1177/1039856219860031
- Mulkey M. Understanding Frontotemporal Disease Progression and Management Strategies. Nurs Clin North Am. 2019 Sep;54(3):437-448. doi: 10.1016/j.cnur.2019.04.011
- Kurz A, Kurz C, Ellis K, Lautenschlager NT. What is frontotemporal dementia? Maturitas. 2014 Oct;79(2):216-9. doi: 10.1016/j.maturitas.2014.07.001
- Krivickas LS. Amyotrophic lateral sclerosis and other motor neuron diseases. Phys Med Rehabil Clin N Am. 2003 May;14(2):327-45. doi: 10.1016/s1047-9651(02)00119-5
- Costa J, Swash M, de Carvalho M. Awaji Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis: A Systematic Review. Arch Neurol. 2012;69(11):1410–1416. doi:10.1001/archneurol.2012.254
- Gooch CL, Shefner JM. ALS surrogate markers. MUNE. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004 Sep;5 Suppl 1:104-7. doi: 10.1080/17434470410019889
- Kwan JY, Jeong SY, Van Gelderen P, Deng HX, Quezado MM, Danielian LE, Butman JA, Chen L, Bayat E, Russell J, Siddique T, Duyn JH, Rouault TA, Floeter MK. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS One. 2012;7(4):e35241. doi: 10.1371/journal.pone.0035241
- Chakraborty S, Gupta A, Nguyen T, Bourque P. The “Motor Band Sign:” Susceptibility-Weighted Imaging in Amyotrophic Lateral Sclerosis. Can J Neurol Sci. 2015 Jul;42(4):260-3. doi: 10.1017/cjn.2015.40
- Wang S, Melhem ER, Poptani H, Woo JH. Neuroimaging in amyotrophic lateral sclerosis. Neurotherapeutics. 2011 Jan;8(1):63-71. doi: 10.1007/s13311-010-0011-3
- Verma G, Woo JH, Chawla S, Wang S, Sheriff S, Elman LB, McCluskey LF, Grossman M, Melhem ER, Maudsley AA, Poptani H. Whole-brain analysis of amyotrophic lateral sclerosis by using echo-planar spectroscopic imaging. Radiology. 2013 Jun;267(3):851-7. doi: 10.1148/radiol.13121148
- Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017 Oct 5;3:17071. doi: 10.1038/nrdp.2017.71. Erratum in: Nat Rev Dis Primers. 2017 Oct 20;3:17085.
- Pradhan S. Bilaterally symmetric form of Hirayama disease. Neurology. 2009 Jun 16;72(24):2083-9. doi: 10.1212/WNL.0b013e3181aa5364
- Tiryaki E, Horak HA. ALS and other motor neuron diseases. Continuum (Minneap Minn). 2014 Oct;20(5 Peripheral Nervous System Disorders):1185-207. doi: 10.1212/01.CON.0000455886.14298.a4
- Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011 Mar 12;377(9769):942-55. doi: 10.1016/S0140-6736(10)61156-7
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017 Jul;16(7):505-512. doi: 10.1016/S1474-4422(17)30115-1
- Weiss MD, Macklin EA, Simmons Z, Knox AS, Greenblatt DJ, Atassi N, Graves M, Parziale N, Salameh JS, Quinn C, Brown RH Jr, Distad JB, Trivedi J, Shefner JM, Barohn RJ, Pestronk A, Swenson A, Cudkowicz ME; Mexiletine ALS Study Group. A randomized trial of mexiletine in ALS: Safety and effects on muscle cramps and progression. Neurology. 2016 Apr 19;86(16):1474-81. doi: 10.1212/WNL.0000000000002507
- Oskarsson B, Moore D, Mozaffar T, Ravits J, Wiedau-Pazos M, Parziale N, Joyce NC, Mandeville R, Goyal N, Cudkowicz ME, Weiss M, Miller RG, McDonald CM. Mexiletine for muscle cramps in amyotrophic lateral sclerosis: A randomized, double-blind crossover trial. Muscle Nerve. 2018 Mar 6:10.1002/mus.26117. doi: 10.1002/mus.26117
- Bedlack RS, Pastula DM, Hawes J, Heydt D. Open-label pilot trial of levetiracetam for cramps and spasticity in patients with motor neuron disease. Amyotroph Lateral Scler. 2009 Aug;10(4):210-5. doi: 10.1080/17482960802430773
- Borasio GD, Voltz R, Miller RG. Palliative care in amyotrophic lateral sclerosis. Neurol Clin. 2001 Nov;19(4):829-47. doi: 10.1016/s0733-8619(05)70049-9
- Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, Katz JS, Mitsumoto H, Rosenfeld J, Shoesmith C, Strong MJ, Woolley SC; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009 Oct 13;73(15):1227-33. doi: 10.1212/WNL.0b013e3181bc01a4
- Stone CA, O’Leary N. Systematic review of the effectiveness of botulinum toxin or radiotherapy for sialorrhea in patients with amyotrophic lateral sclerosis. J Pain Symptom Manage. 2009 Feb;37(2):246-58. doi: 10.1016/j.jpainsymman.2008.02.006
- Chiò A, Mora G, Lauria G. Pain in amyotrophic lateral sclerosis. Lancet Neurol. 2017 Feb;16(2):144-157. doi: 10.1016/S1474-4422(16)30358-1
- Brooks BR, Thisted RA, Appel SH, Bradley WG, Olney RK, Berg JE, Pope LE, Smith RA; AVP-923 ALS Study Group. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004 Oct 26;63(8):1364-70. doi: 10.1212/01.wnl.0000142042.50528.2f
- Westeneng HJ, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, Calvo A, Martin S, McDermott CJ, Thompson AG, Pinto S, Kobeleva X, Rosenbohm A, Stubendorff B, Sommer H, Middelkoop BM, Dekker AM, van Vugt JJFA, van Rheenen W, Vajda A, Heverin M, Kazoka M, Hollinger H, Gromicho M, Körner S, Ringer TM, Rödiger A, Gunkel A, Shaw CE, Bredenoord AL, van Es MA, Corcia P, Couratier P, Weber M, Grosskreutz J, Ludolph AC, Petri S, de Carvalho M, Van Damme P, Talbot K, Turner MR, Shaw PJ, Al-Chalabi A, Chiò A, Hardiman O, Moons KGM, Veldink JH, van den Berg LH. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018 May;17(5):423-433. doi: 10.1016/S1474-4422(18)30089-9
- Limousin N, Blasco H, Corcia P, Gordon PH, De Toffol B, Andres C, Praline J. Malnutrition at the time of diagnosis is associated with a shorter disease duration in ALS. J Neurol Sci. 2010 Oct 15;297(1-2):36-9. doi: 10.1016/j.jns.2010.06.028





{kind=link}