Angiofibroma
Solitary cutaneous angiofibroma may also be called fibrous papule or perifollicular fibroma, is a term used to define a group of lesions with different clinical presentations but with the same histologic findings 1. Cutaneous angiofibromas are benign fibrous neoplasms comprised of a proliferation of stellate and spindled cells, thin-walled blood vessels with dilated lumina in the dermis, and concentric collagen bundles. Cutaneous angiofibromas can be located on different areas of the body including the face where they are commonly called fibrous papules or adenoma sebaceum; on the penis where they are called pearly penile papules, underneath the nail where they are called periungual angiofibromas or Koenen tumors, and in the mouth where they are called oral fibromas. Angiofibromas are very common the nose and look like intradermal nevi. As a solitary lesion, there are no associated systemic implications.
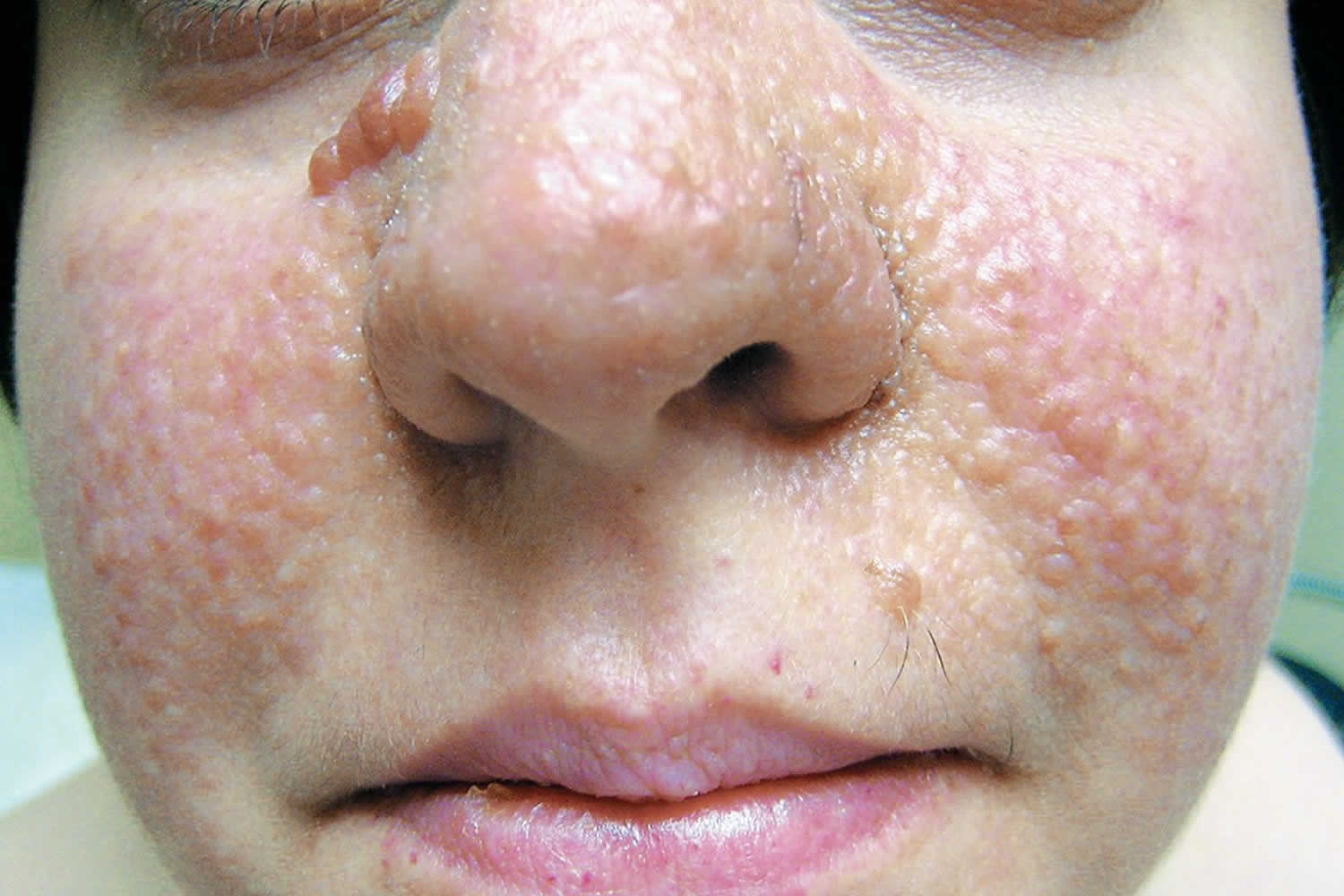
When multiple angiofibromas are seen in an individual, particularly clustered over the central face, around the nose and cheeks, the differential diagnosis should include the tuberous sclerosis complex, and appropriate evaluation should ensue to assess the patient for tuberous sclerosis. Adenoma sebaceum is the term for the multiple angiofibromas distributed on the central face and nasolabial grooves in patients with tuberous sclerosis (Figure 1). Adenoma sebaceum is a misnomer, as the lesions are not adenomas or related to sebaceous glands. Multiple facial angiofibromas may also be seen as the presenting sign or in association with multiple endocrine neoplasia type 1 (MEN-1) and Birt-Hogg-Dube syndrome.
Multiple angiofibromas around the nose and cheeks are associated with tuberous sclerosis, an autosomal dominant hamartomatous disorder in which mutations in tumor suppressor gene TSC1 or TSC2 result in the formation of benign hamartomas throughout the body. Hamartoma is a benign (not cancer) growth made up of an abnormal mixture of cells and tissues normally found in the area of the body. Multiple hamartomas affects the skin, kidneys, heart, brain, and lungs. With tuberous sclerosis, multiple angiofibromas typically arise on the face during childhood and early adulthood and become increasingly prominent. These patients may also have periungual fibromas. Both facial angiofibromas (greater than or equal to 3 needed) and periungual angiofibroma (greater than or equal to 2 needed) are 2 of the major criteria for tuberous sclerosis. Tuberous sclerosis complex affects about 1 in 6,000 people 2.
Tuberous sclerosis complex often affects the brain, causing seizures, behavioral problems such as hyperactivity and aggression, and intellectual disability or learning problems. Some affected children have the characteristic features of autism, a developmental disorder that affects communication and social interaction. Benign brain tumors can also develop in people with tuberous sclerosis complex; these tumors can cause serious or life-threatening complications.
Kidney tumors are common in people with tuberous sclerosis complex; these growths can cause severe problems with kidney function and may be life-threatening in some cases. Additionally, tumors can develop in the heart, lungs, and the light-sensitive tissue at the back of the eye (the retina).
Tuberous sclerosis is caused by mutations in tuberous sclerosis complex 1 (TSC-1) which encodes the protein hamartin and tuberous sclerosis complex 2 (TSC-2) which encodes the protein tuberin. These proteins normally suppress the activation of mammalian target of rapamycin (mTOR), however, when mutated, they cause unregulated proliferation of cell growth forming multi-organ hamartomas. mTOR is activated in the proliferating fibroblast-like cells within facial angiofibromas. The cells produce an epidermal growth factor called epiregulin, which stimulates epidermal cell proliferation so that they are produced at a faster rate. Angiofibromas of tuberous sclerosis also have vascular proliferation from increased expression of angiogenic factors such as vascular endothelial growth factor (VEGF). VEGF stimulates mTOR 3.
There are numerous distinct clinical forms of angiofibromas which share an identical histopathology:
- Fibrous papule
- Pearly penile papules. Pearly penile papules are chronic, asymptomatic, papules found on the coronal margin and sulcus of the penis. They are more common in uncircumcised men 4.
- Adenoma sebaceum (facial angiofibomas associated with tuberous sclerosis)
- Syndromal angiofibroma
Current treatments for angiofibromas include shave excision, cryotherapy, electrodessication, radiofrequency ablation, dermabrasion, lasers such as ablative fractional laser resurfacing and pulsed dye laser and topical podophyllotoxin 1. Although these treatments have proven to be successful, they can result in scarring, post-inflammatory hyperpigmentation, and pain. The recurrence rate can be up to 80%, necessitating follow-up treatments. Topical rapamycin, a mTOR inhibitor, seems to be a safe and effective treatment for angiofibromas however long-term studies still need to be conducted. Combination treatments, such as fractional laser resurfacing and pulsed dye laser, can be employed with topical medications, such as timolol or rapamycin, to effectively treat these lesions. Because the lesions are benign, they should only be removed for cosmesis or compression on adjacent structures resulting in pain.
Figure 1. Multiple angiofibromas (adenoma sebaceum) in an individual with tuberous sclerosis
Angiofibroma causes
Mutations in the tuberous sclerosis complex 1 (TSC1) or tuberous sclerosis complex 2 (TSC2) gene can cause tuberous sclerosis complex. The TSC1 and TSC2 genes provide instructions for making the proteins hamartin and tuberin, respectively. Within cells, these two proteins likely work together to help regulate cell growth and size. The proteins act as tumor suppressors, which normally prevent cells from growing and dividing too fast or in an uncontrolled way.
People with tuberous sclerosis complex are born with one mutated copy of the TSC1 or TSC2 gene in each cell. This mutation prevents the cell from making functional hamartin or tuberin from the altered copy of the gene. However, enough protein is usually produced from the other, normal copy of the gene to regulate cell growth effectively. For some types of tumors to develop, a second mutation involving the other copy of the TSC1 or TSC2 gene must occur in certain cells during a person’s lifetime.
When both copies of the TSC1 gene are mutated in a particular cell, that cell cannot produce any functional hamartin; cells with two altered copies of the TSC2 gene are unable to produce any functional tuberin. The loss of these proteins allows the cell to grow and divide in an uncontrolled way to form a tumor. In people with tuberous sclerosis complex, a second TSC1 or TSC2 mutation typically occurs in multiple cells over an affected person’s lifetime. The loss of hamartin or tuberin in different types of cells leads to the growth of tumors in many different organs and tissues.
MEN-1 is due to a mutation in the MEN1 gene which encodes menin.
Birt-Hogg-Dube syndrome is caused by a mutation in the FLCN gene which encodes folliculin.
Angiofibroma symptoms
Fibrous papules are solitary, dome-shaped, skin-colored to red papules located on the central face, usually around the nose and on the malar eminences. They can have tiny telangiectatic vessels located on the surface of the papule. In tuberous sclerosis, angiofibromas typically arise symmetrically on the cheeks, nasolabial folds, nose, and chin. They can start off as erythematous macules that form into the red to red-brown papules that can coalesce into plaques.
Pearly penile papules are pearly, white, dome-shaped, closely aggregated small papules located on the glans penis in a multilayered and circumferential manner on the corona.
Periungual angiofibromas begin to occur in tuberous sclerosis in late childhood to early adulthood. They arise from the lateral or proximal nail fold and more commonly occur on the toes. They can be painful and can distort the shape of the nail.
Oral fibromas occur most commonly on the gingiva, but lesions can also occur on buccal or labial mucosa and occasionally the tongue.
Clinical findings in Birt-Hogg-Dube include fibrofolliculomas, perifollicular fibromas (some authorities consider perifollicular fibroma to be related to angiofibroma), and trichodiscomas. These are all present as skin colored to hypopigmented papules on the head and neck or upper trunk.
Angiofibroma diagnosis
The diagnosis of angiofibroma is made by history, physical exam, and skin biopsy. If tuberous sclerosis, MEN-1, or Birt-Hogg-Dube syndrome are suspected, genetic testing should be completed as well as an extensive workup searching for tumors for their respected conditions.
All cutaneous angiofibromas are composed of a dermal proliferation of fibroblasts in a collagenous stroma with an increase in the number of thin-walled, dilated blood vessels. Collagen fibers are concentrically arranged around hair follicles and blood vessels. Elastic fibers can be decreased, and the epidermis can be atrophic. Fibroblasts can be stellate in shape and can be multinucleated. Immunohistochemistry for these cells will show positivity for factor XIIa and negative for S100 protein.
Angiofibroma treatment
Current treatments for angiofibromas include shave excision, cryotherapy, electrodessication, radiofrequency ablation, dermabrasion, lasers such as ablative fractional laser resurfacing (AFR) and pulsed dye laser (PDL), and topical podophyllotoxin. Although these treatments have proven to be successful, they can result in scarring, post-inflammatory hyperpigmentation, and pain. The recurrence rate can be up to 80%, necessitating follow-up treatments. Topical rapamycin, a mTOR inhibitor, seems to be a safe and effective treatment for angiofibromas however long-term studies still need to be conducted. Combination treatments, such as fractional laser resurfacing and pulsed dye laser, can be employed with topical medications, such as timolol or rapamycin, to effectively treat these lesions 5.
Rapamycin has recently gained popularity in the treatment of angiofibromas. After binding to mTOR, it inhibits its activity which downregulates cell proliferation. It also decreases VEGF production by downregulating VEGF- stimulated endothelial cell proliferation (Habib). Several case series, case reports, and one randomized controlled trial have been published verifying the effectiveness of topical rapamycin used as .1% once or twice daily, as well as .2%, used 5 times a week and .4% used 3 times a week. The angiofibromas cleared as long as the medication was being used. The longest reported follow up has been 3 years. Many have used crushed rapamycin tablets and mixed them in Vaseline to obtain the desired concentration which was not a standardized dose. DeKlotz et al. proposed a standardized formulation on how to make .1% topical rapamycin in 2011. Few if any side effects occur from the topical medication including mild irritation and erythema. Park et al. showed that topical rapamycin was enough to treat the lesions when they were small in size, that is less than 4 mm. However, if they were larger than 4 mm, ablative resurfacing was needed in conjunction with rapamycin. It can also be costly to use topical rapamycin in the treatment of angiofibromas due to the length of treatment that is necessary to obtain sufficient results costing several hundred to several thousand dollars out of pocket.
Beta-blockers have been used for many years now in the treatment of vascular lesions. Oral propranolol has been successful in the treatment of hemangiomas in the pediatric population. However, side effects such as hypoglycemia limit its use in certain patients. Topical timolol .5% solution or gel used 2 or 3 times a day has proven to be very successful in the treatment of superficial hemangiomas. The mechanism of action of beta-blockers is thought to be due to its role in blocking the formation of renin to angiotensin II. In doing so, angiotensin II does not form VEGF, which converts endothelial stem cells to endothelial cells, leading to decreased capillary development. Matrix metalloproteinase-9 is involved in angiogenesis, and its activity is thought to be inhibited by beta blockers. One other way beta- blockers work to decrease angiogenesis is by producing osteoprotegerin. This causes tumor necrosis factor apoptosis of mesenchymal and endothelial cells 6.
Angiofibroma prognosis
Angiofibromas are benign prolifferations however they do not spontaneous improve and, when multiple, can cause significant disfigurement, bleeding, itch, and redness emanating the need for an effective treatment.
Juvenile nasopharyngeal angiofibroma
Juvenile nasopharyngeal angiofibroma also called nasopharyngeal angiofibroma or fibromatous or angiofibromatous hamartoma of the nasal cavity 7. Nasopharyngeal may not be entirely accurate, as some sources state that it arises from the sphenopalatine foramen and the posterior nasal cavity 8, while others proffer that it has more of a choanal and nasopharyngeal origin 9. What research does agree upon is that juvenile nasopharyngeal angiofibroma is a benign, highly vascular lesion that comprises approximately 0.05 to 0.5% of all head and neck masses 10. Though histologically benign, it often demonstrates aggressive features with local invasion into the nasal turbinates, nasal septum, and medial pterygoid lamina. It commonly extends into the nasal cavity, nasopharynx, and pterygopalatine fossa, with larger lesions extending into the sphenoid, maxillary, and ethmoid sinuses. They can also demonstrate extension through the inferior orbital fissure, and into the masticator space through the infratemporal fossa. Severe disease is likened to have orbital and intracranial involvement, seen in approximately 10 to 37% of cases 11.
Juvenile nasopharyngeal angiofibroma is a highly vascular lesion, with one or more arterial vascular pedicles. The most common primary arterial supply is the internal maxillary artery, a branch of the external carotid artery 12. Larger lesions may invoke multiple feeding arteries, with even bilateral involvement. The ascending pharyngeal artery is the second most common sizeable supplying branch of the external carotid artery, with additional accessory arteries including the middle meningeal, accessory meningeal, and facial artery branches. Research has also described the recruitment of internal carotid artery branches, most commonly the vidian artery, and to a slightly lesser extent the ophthalmic artery 13.
Nasopharyngeal angiofibroma is seen almost exclusively in adolescent males, accounting for approximately 0.05 to 0.5% of head and neck tumors, and a reported incidence ranging from 1 in 150,000 to 1 in 1,500,000 9. There are also reports that individuals from India and the Middle East appear to have an increased incidence when compared to those of European descent 7. The typical age range of juvenile nasopharyngeal angiofibroma is 9 to 25 years, and though there have been case reports of nasopharyngeal angiofibroma diagnoses in older males, this is still considered a rare occurrence 14. Ther are also rare reports in females. In these cases, further immunohistochemical and genetic testing should be pursued given its rarity, as genetic mosaicism could serve as a potential predisposing factor in these women 15.
Juvenile nasopharyngeal angiofibroma causes
The cause of juvenile nasopharyngeal angiofibroma is not well understood, and still often debated. There are a few different hypotheses currently in the literature, primarily revolving around a vascular source, such as an arteriovenous malformation, or remnant of the first branchial arch 16. This first branchial arch remnant could also help explain the typical location of the nasopharyngeal angiofibroma, as incomplete regression can leave remnants in or near the sphenopalatine foramen. The expression of vascular growth receptors, primarily vascular endothelial growth factor (VEGFR-2) also helps explain the highly vascular nature of the mass.
Others have connected juvenile nasopharyngeal angiofibroma to a hormonal influence affecting the proliferation of vascular erectile tissues following repeated microhemorrhages and repair 17. Previous studies have reported the presence of androgen, estrogen, and progesterone receptors. Some hypothesize this is the reason for the predominant adolescent male incidence, as the increase in androgen production in puberty stimulates the growth and vascular expansion of the tumor. Others have shown that tumor growth can occur at any time, even after treatment, from testosterone administration. This concept has support from case reports of nasopharyngeal angiofibroma in older women that have since downregulated their estrogen and progesterone production, suggesting that estrogen has a protective effect; but further confounded by case reports of nasopharyngeal angiofibroma being discovered in pregnant females, suggesting that androgen influence is not important. Thus, the importance of hormonal influence remains unclear 15.
Nasopharyngeal angiofibroma has also correlated with genetic anomalies and other disorders. There have been reports of chromosome 17 deletions 18. The most significant interest in these deletions is their association with the TP53 suppressor gene, as well as the human epidermal growth factor receptor 2 (HER2), involving the HER2/NEU oncogene, both well known in the realm of tumor growth and malignancy. Other nasopharyngeal angiofibroma associations reported include familial adenomatous polyposis (FAP) and Gardner syndrome, with an altered APC gene expression in this subset of nasopharyngeal angiofibroma 19.
Another association with recent documentation is with human papillomavirus (HPV) infection. HPV is known to be associated with head and neck squamous cell carcinoma. It is also known for its tumorigenic effect, similar to Epstein-Barr virus (EBV), raising the question if there could be an association with nasopharyngeal angiofibroma. A small study demonstrated a strong association between juvenile nasopharyngeal angiofibroma and HPV, with the presence of HPV specific proteins and DNA within the nasopharyngeal angiofibroma tissue, without concomitant infection in the control group’s adenoidal tissue 20. Given the rising incidence of HPV infection worldwide, this raises concern for an increase in juvenile nasopharyngeal angiofibroma, with implications in developing possible preventative practices. However, given the limited amount of current evidence, further study is recommended.
Juvenile nasopharyngeal angiofibroma symptoms
The most common presentation is an adolescent male with chronic unilateral nasal obstruction. Also, painless, unprovoked epistaxis is common. Other presenting complaints consist of headache and rhinorrhea, and when the mass is large and invasive, proptosis, visual disturbance, cranial nerve palsy, Eustachian tube dysfunction, and facial deformity may occur. On physical exam, a mass is generally visible within the nasal cavity.
Nasopharyngeal angiofibroma staging
Various systems have been proposed for staging and classifying nasopharyngeal angiofibroma to aid in decision making for surgery and adjunctive treatment. The most commonly used today are the Radkowski and Andrews-Fisch staging systems, primarily for their impact on surgical approach and recurrence/outcome 16. Most recently, the University of Pittsburgh Medical College (UPMC) system was proposed focusing on endoscopic staging, considering current surgical approaches and the nature of the tumor. Anatomic extent is the main determining factor utilized by all staging studies.
The Radkowski system is a three-level staging system, each stage being multipartite. Stage Ia disease is confined to the nasal cavity and nasopharyngeal vault, with extension into the sinuses classifying stage Ib disease. Extension into the pterygopalatine fossa is the discriminating factor for stage II, with minimal extension determining stage IIa, and stage IIb when it involves the entirety of the fossa. Stage IIc is when it extends into the infratemporal fossa or posterior to the pterygoid plates. Advanced stage III disease involves the skull base and intracranial extension. Stage IIIa is minimal involvement of the middle cranial fossa or pterygoid plates, with stage IIIb when it demonstrates intracranial extension with or without cavernous sinus invasion.
The Andrews-Fisch system is similar, with an extra stage delineating intracranial extent. Stage I is limited to the nasal cavity/nasopharyngeal vault. Stage II includes pterygopalatine fossa or any sinus invasion. Stage IIIa involves extension in the infratemporal fossa or orbital invasion, whereas stage IIIb describes intracranial, extradural extension into the parasellar region. Intradural extension denotes more advanced stage IV disease, with cavernous sinus, pituitary fossa or optic chiasm invasion separating stage IVa from stage IVb.
There are various other proposed classification systems less commonly used, or too new to fully understand their effectiveness relative to the others. Some examples of these include the Sessions, Chandler, and INCan staging systems. The most recent proposed system was introduced by a group from the University of Pittsburgh Medical College (UPMC) in 2010, based on endoscopic staging. It stages juvenile nasopharyngeal angiofibroma in five stages, with more focus on residual vascularity, primarily from the ICA after preoperative embolization. It also de-emphasizes size and paranasal sinus invasion, considered limited factors in outcome predictions following resection. As with the other systems, stage I is limited to the nasal cavity and nasopharynx. Stage II includes paranasal sinus and lateral pterygopalatine fossa invasion, but without residual vascularity. Stage III and IV disease consist of skull base erosion and involvement of the orbit and infratemporal fossa, only separated by residual vascular supply from the ICA in stage IV. Stage V disease is subdivided based on laterality, with medial (stage V(M)) vs. lateral (stage V(L)) intracranial extension and residual vascularity.
Juvenile nasopharyngeal angiofibroma diagnosis
Nasopharyngeal angiofibroma is both a clinical and imaging diagnosis. There is no beneficial laboratory evaluation for diagnosing juvenile nasopharyngeal angiofibroma.
Clinical exam with nasal endoscopy will demonstrate a firm, friable, reddish, or reddish-purple mass within the nasal cavity.
Imaging evaluation is primarily performed with either computed tomography (CT) or magnetic resonance imaging (MRI), though radiographs can demonstrate a bowing or anterior displacement of the posterior wall of the maxillary sinus from mass effect.
Contrast-enhanced CT will demonstrate an avidly enhancing soft tissue mass within the posterior nasal cavity near the sphenopalatine foramen with extension to and/or beyond the nasopharynx, pterygopalatine fossa, and adjacent sinuses. Depending on tumor size, the opacification of the sphenoid sinus, or even the maxillary sinus(es) and ethmoid air cells can be seen either from obstruction or infiltration. Bone kernel postprocessing will aid in the evaluation of osseous remodeling or destruction. Expansion of the nasal cavity, pterygopalatine fossa, and anterior bowing of the posterior wall maxillary sinus can present secondary to mass effect. Osseous destruction also occurs in more aggressive tumors, especially those involving the skull base and intracranial extension. Further evaluation with CT angiography can help determine the extent of vascularity and vascular supply for preoperative planning. Examination for unilateral enlargement of the external carotid and/or internal maxillary artery can help delineate the situs of the vascular source.
Traditional cerebral angiography can also help with determining the vascular source, especially if there are multiple feeding arteries. The ability to select the internal and external carotid arteries, as well as subselect different branches can help direct preoperative management and planning.
Though CT is better for evaluation of osseous involvement and demonstrates better spatial resolution, MRI is a useful adjunct in the fact that it offers superior contrast resolution. On non-contrast sequences, NA will be heterogeneous, with intermediate T1-weighted, and intermediate to high T2-weighted signal intensity. Flow voids will be present on both T1 and T2 sequences. Areas of prior hemorrhage can be seen, accentuated on susceptibility-weighted imaging. Diffusion-weighted imaging (DWI) with associated apparent diffusion coefficient (ADC) values tend to show facilitated diffusion, with elevated ADC values.[20] And as in CT, MR demonstrates intense enhancement following gadolinium contrast administration. MR is also good for distinguishing extension into the cavernous sinus, sphenoid sinus, or perineural extension through the skull base or into the orbit. MR angiography, as with CT angiography, can show an enlarged ipsilateral external carotid or internal maxillary artery but is less effective regarding evaluating vascular source.
Given the extensive vascularity of the tumor, a biopsy should not be a consideration in the diagnostic evaluation of nasal angiofibroma, especially in a clinic setting. An unwarranted biopsy can result in significant morbidity and even mortality from blood loss following the procedure.
Juvenile nasopharyngeal angiofibroma treatment
Treatment of choice and standard of care is surgical resection of the tumor. Due to the extensive vascularity of the tumor, many patients are treated with preoperative embolization to minimize the risk of intraoperative hemorrhage and related complications; this also allows for improved identification of vascular supply, especially delineation of external and internal carotid artery feeding vessels and is also beneficial if a staged or segmented procedure is under consideration. Commonly after femoral artery access, digital subtraction angiography is performed with a 5F catheter of the common, internal, and external carotid arteries. The artery or arteries that are best seen supplying the tumor are then sub-selected with a 3F microcatheter. Embolization materials include gelatin sponge, particles, and micro coils. Particle size varies, but the preferred size is 300 to 500 micrometers to reduce the risk of nontarget embolization with smaller particles in the event of an external-internal carotid artery communication within the tumor. Micro coils are useful if a more precise placement is required, such as in the setting of an external to internal carotid artery communication, or a branch artery from the internal carotid artery where nontarget embolization may have more severe complications. Some branches are avoided altogether based on their proximity to cranial nerves to minimize the risk of iatrogenic cranial nerve injury. Preoperative embolization appears to be most effective for higher stage tumors with a more robust vascular supply, with a less additional benefit in tumors at a lower stage.
Early on, the most common open surgical approach performed was a lateral rhinotomy 21. Now, many avenues of approach exist for removal of nasopharyngeal angiofibroma, and in some instances, multiple may need to be used, such as transpalatal, transfacial, transnasal, sublabial/Le Fort I, transmaxillary, and infratemporal surgical corridors 22. When possible, many cases now are treated endoscopically with an endonasal approach and may be augmented with an anterior maxillotomy or other craniotomy approaches, as needed 23. Controlled hypotension while under general anesthesia is typically employed, with epinephrine to contract the nasal mucosa and improve the operative view. During the endonasal approach, parts of the inferior and middle turbinates may need to be resected to improve visualization. Also, depending on tumor size, the ethmoid air cells and maxillary ostium may need to be opened for further access to tumor boundaries, especially when removal of the posterior wall of the maxillary sinus is needed to access the pterygopalatine or infratemporal fossae 24. The strategy regarding these masses is to determine the periphery and extent of the tumor by dissecting around the margins, with as little manipulation as possible of the tumor itself. This approach allows identification of key anatomic landmarks while keeping the tumor en bloc. Upon stripping the tumor away from the periosteum, the embolized and thrombosed feeding vessel can be dissected away and separated, allowing the delivery of the resected mass. There are a few instruments that aid in this process. Monopolar and bipolar electrocautery can help decrease blood loss from minute feeding vessels as it cauterizes while it cuts, but it also leads to damage to surrounding tissue. To offset this, ultrasonic cautery is an option as it causes less thermal damage to surrounding tissue, which requires an additional sublabial or transmaxillary approach due to the larger size of the instrument. Laser-assisted endonasal cautery and dissection have also been demonstrated, using an Nd: YAG laser as an additional option for more focused cautery 25. Another option for a dissection instrument that does not generate as much heat is to use radiofrequency energy to disrupt the immediately surrounding tissues to develop the dissection plane 23.
In the setting of large tumors, it may be beneficial to remove it in a staged, segmented approach based on vascular territory. Some of the larger tumors can have bilateral external carotid artery branch vessel involvement, or even internal carotid artery supply, especially if there is an intracranial extension of the tumor. Performing a staged procedure to segment each of these vascular territories can help minimize excessive blood loss, by a “one bleed at a time” approach 23. The external carotid artery branches are usually resected first, with the intracranial extension, and internal carotid artery supplied segments performed last. These are more involved procedures with a potentially higher risk for bleeding, especially if the branch of the internal carotid artery does not lend itself to preoperative embolization. Occasionally resection or drilling of portions of the skull base may be required depending on the invasive extent and course of the tumor, expanding the skull base foramina. An example of this is expanding the vidian, or pterygoid, canal to dissect out the vidian artery. If there is a dural tear or communication, then special care must be taken not to allow blood and cerebrospinal fluid to mix, minimizing the risk of intracranial vasospasm or possibly even infective meningitis given the potential communication with the nasal cavity. Even with these extensive endoscopic combination approaches, the occurrence of facial deformity is decreased compared to the solely open techniques. As discussed, surgical resection is the primary treatment of choice, but radiation therapy can be an adjuvant therapy for residual or recurrent disease; this primarily takes place in the setting of advanced tumors that involve critical areas, or those with intracranial extension that may not be completely resectable. To some this remains controversial owing to concerns over long term morbidity or the possibility of secondary neoplasm from the high doses of radiation. In some groups there has been documented local control rates of 85 to 91%, showing success with adjuvant radiotherapy. These groups also demonstrated a low risk for severe late complications following this treatment plan 7. Additional case series have demonstrated effective rates around 80% from primary radiotherapy, showing a viable alternative in cases where complete surgical excision is not possible or prudent. Some advocate for the use of definitive or primary radiotherapy in the setting of these inoperable cases. This approach can result in long term disease control of 80 to 88% with regression in tumor size, but persistent tumor burden is a common, and an expected finding, while long term radiation-related complications remain a concern 11. Various external radiotherapy treatments are available, including stereotactic radiosurgery (gamma knife surgery), intensity-modulated radiotherapy (IMRT) using a conformal technique, and conventional external beam radiotherapy. Reported success rates for all appear similar, with most occurring in the setting of advanced-stage disease (Radkowski stage III), with various long-term complications. The conformal technique seems to offer a better option when comparing disease control balanced with limited long-term morbidity 11. Besides long-term complications from radiation, re-operative, or post-operative morbidity is also a concern following radiation therapy. Changes in the surgical bed with increased tissue friability following radiation can lead to increased morbidity and surgical complications. One example of this is delayed CSF leak with rhinorrhea following surgical resection and stereotactic radiosurgery requiring multiple additional endoscopic procedures for surgical repair of the CSF leak 26. Though radiation therapy can result in tumor regression, initial pre-operative radiotherapy can lead to increased surgical morbidity.
Hormonal therapy in the way of androgen receptor blockers such as flutamide is an additional possible adjuvant therapy. These have been shown to help reduce tumor size prior to surgical resection or in the setting of recurrence, but do not have curative results on their own 27.
Limited evidence is available regarding the use of chemotherapy/cytotoxic drugs in the treatment of juvenile nasopharyngeal angiofibroma. Of the little data that is available, patient responses range from no recurrence to partial response, advocating for additional research into the use of cytotoxic drugs as a therapy regimen 27.
Complications
The most significant complication of juvenile nasopharyngeal angiofibroma is blood loss, primarily in the operative/procedural setting, and can be fatal absent proper precautions. Exophthalmos, facial/orbital deformity, vision loss, and loss of extraocular movements can occur from orbital invasion by the tumor. Vision loss can also be a potential complication of nontarget embolization if there is internal carotid artery branch involvement. Other severe complications of preoperative embolization include arterial vasospasm, facial palsy, infarction, or cranial nerve injury. More self-limiting, less severe complications include facial swelling, pain or abnormal sensation, headache, or nausea/vomiting. Surgical complications can include much of the same, with the addition of scarring and facial deformity. Hormonal therapy, if employed, can lead to feminization as a complication or at least undesirable side effect in adolescent boys.
Postoperative and rehabilitation care
Imaging and clinical surveillance is warranted to evaluate for residual versus recurrent disease. Evaluating for residual disease can be difficult, as the imaging characteristic of importance is tumor/tissue enhancement, which is visible in granulation tissue, especially in early postoperative imaging. Clinical exam and symptoms will play a primary role during this stage of recovery, if not as an adjunct to imaging. Most recurrences are reported to occur within the initial 6 to 36 months 22. This situation leads to performing annual, or as often as biannually, follow-up for at least four years following surgery.
Radiotherapy
As described above, radiotherapy is primarily an adjuvant treatment in the setting of residual or recurrent disease. It can, however, be the primary treatment modality if a tumor is deemed unresectable based on its extent of invasion and the critical structures that it involves. Reports of local control rates with radiation alone are around 73 to 87.5% 11.
External beam radiation is used in the form of intensity-modulated radiation therapy (IMRT) in a conformal technique to limit radiation exposure and doses to nearby optic nerves/optic chiasm, lens, retina, brain/brainstem, spinal cord, and salivary glands as compared to conventional radiotherapy. Various case series involving radiotherapy treatments prescribe a typical dose range of 30 to 50 Gy, at 1.8 to 2 Gy per daily fraction.
Stereotactic radiotherapy/radiosurgery has also been used but is believed to be better suited for well-defined residual or recurrent disease. It has a proposed higher risk of late complications given its single high dose of radiation over a tighter dose distribution area 7. There are case reports of successful use of gamma knife surgery, one such describing 17 Gy supplied to the 50% isodense line of tumor margin in a focus of recurrence that had a volume of 6.8 cm 22. Still, limited data exist regarding treatment efficacy and long-term complications of stereotactic radiosurgery.
Juvenile nasopharyngeal angiofibroma prognosis
Nasopharyngeal angiofibroma is a benign entity, and in that regard portends a good prognosis. The main concern with juvenile nasopharyngeal angiofibroma is an advanced disease that does not allow total resection or disease recurrence. Literature has reported as many as 33% of advanced (Radkowski stage III) disease are unresectable, and in those that undergo resection, recurrence can occur in 30 to 38%, which can lead to additional morbidity from residual/recurrent tumor growth and invasion. Some that have residual/recurrent disease also undergo adjuvant radiotherapy, with subsequent, though uncommon, accounts of likely radiation-induced secondary malignancies such as basal cell and squamous cell carcinoma within the radiation port 11. Rare malignant transformation of juvenile nasopharyngeal angiofibroma has also been reported, primarily into well-differentiated tumors following radiotherapy, but reports of undifferentiated sarcomatous transformation also exist, leading to a poorer prognosis 10.
References- Macri A, Tanner LS. Cutaneous Angiofibroma. [Updated 2019 Dec 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482470
- Tuberous sclerosis complex. https://ghr.nlm.nih.gov/condition/tuberous-sclerosis-complex
- Garcia NG, de Carli ML, Oliveira DT, Soares CT, Ribeiro Júnior NV, Sperandio FF, Hanemann JA. Tuberous Sclerosis with Severe Cutaneous Manifestation and Multiples Facial Angiofibromas. Head Neck Pathol. 2016 Dec;10(4):542-546.
- Bakić M, Ratković M, Gledović B, Vujović B, Radunović D, Babić V, Prelević V. Cutaneous Manifestations of Tuberous Sclerosis. Acta Dermatovenerol Croat. 2018 Apr;26(1):73-74.
- Salomon FF, Barreto MM, Zanetti G, Rodrigues RS, Gasparetto EL, Marchiori E. CNS and cutaneous involvement in tuberous sclerosis complex. Arq Neuropsiquiatr. 2015 Sep;73(9):813.
- Park J, Yun SK, Cho YS, Song KH, Kim HU. Treatment of angiofibromas in tuberous sclerosis complex: the effect of topical rapamycin and concomitant laser therapy. Dermatology (Basel). 2014;228(1):37-41.
- López F, Triantafyllou A, Snyderman CH, Hunt JL, Suárez C, Lund VJ, Strojan P, Saba NF, Nixon IJ, Devaney KO, Alobid I, Bernal-Sprekelsen M, Hanna EY, Rinaldo A, Ferlito A. Nasal juvenile angiofibroma: Current perspectives with emphasis on management. Head Neck. 2017 May;39(5):1033-1045.
- Makhasana JA, Kulkarni MA, Vaze S, Shroff AS. Juvenile nasopharyngeal angiofibroma. J Oral Maxillofac Pathol. 2016 May-Aug;20(2):330.
- McKnight CD, Parmar HA, Watcharotone K, Mukherji SK. Reassessing the Anatomic Origin of the Juvenile Nasopharyngeal Angiofibroma. J Comput Assist Tomogr. 2017 Jul/Aug;41(4):559-564.
- Allensworth JJ, Troob SH, Lanciault C, Andersen PE. High-grade malignant transformation of a radiation-naïve nasopharyngeal angiofibroma. Head Neck. 2016 Apr;38 Suppl 1:E2425-7.
- Mallick S, Benson R, Bhasker S, Mohanti BK. Long-term treatment outcomes of juvenile nasopharyngeal angiofibroma treated with radiotherapy. Acta Otorhinolaryngol Ital. 2015 Apr;35(2):75-9.
- Overdevest JB, Amans MR, Zaki P, Pletcher SD, El-Sayed IH. Patterns of vascularization and surgical morbidity in juvenile nasopharyngeal angiofibroma: A case series, systematic review, and meta-analysis. Head Neck. 2018 Feb;40(2):428-443.
- Mehan R, Rupa V, Lukka VK, Ahmed M, Moses V, Shyam Kumar NK. Association between vascular supply, stage and tumour size of juvenile nasopharyngeal angiofibroma. Eur Arch Otorhinolaryngol. 2016 Dec;273(12):4295-4303.
- McGarey PO, David AP, Payne SC. Nasopharyngeal angiofibroma in a 32-year-old man. BMJ Case Rep. 2018 Feb 08;2018
- Ralli M, Fusconi M, Visconti IC, Martellucci S, de Vincentiis M, Greco A. Nasopharyngeal angiofibroma in an elderly female patient: A rare case report. Mol Clin Oncol. 2018 Dec;9(6):702-704.
- Alshaikh NA, Eleftheriadou A. Juvenile nasopharyngeal angiofibroma staging: An overview. Ear Nose Throat J. 2015 Jun;94(6):E12-22.
- Marshall AH, Bradley PJ. Management dilemmas in the treatment and follow-up of advanced juvenile nasopharyngeal angiofibroma. ORL J. Otorhinolaryngol. Relat. Spec. 2006;68(5):273-8.
- Schick B, Veldung B, Wemmert S, Jung V, Montenarh M, Meese E, Urbschat S. p53 and Her-2/neu in juvenile angiofibromas. Oncol. Rep. 2005 Mar;13(3):453-7.
- Guertl B, Beham A, Zechner R, Stammberger H, Hoefler G. Nasopharyngeal angiofibroma: an APC-gene-associated tumor? Hum. Pathol. 2000 Nov;31(11):1411-3.
- Mishra A, Sachadeva M, Jain A, Shukla NM, Pandey A. Human Papilloma virus in Juvenile Nasopharyngeal Angiofibroma: possible recent trend. Am J Otolaryngol. 2016 Jul-Aug;37(4):317-22.
- Gołąbek W, Szymańska A, Morshed K. Transnasal microscopic approach for juvenile nasopharyngeal angiofibroma. Otolaryngol Pol. 2018 Aug 06;72(5):31-36.
- Park CK, Kim DG, Paek SH, Chung HT, Jung HW. Recurrent juvenile nasopharyngeal angiofibroma treated with gamma knife surgery. J. Korean Med. Sci. 2006 Aug;21(4):773-7.
- Snyderman CH, Pant H. Endoscopic Management of Vascular Sinonasal Tumors, Including Angiofibroma. Otolaryngol. Clin. North Am. 2016 Jun;49(3):791-807.
- Ye D, Shen Z, Wang G, Deng H, Qiu S, Zhang Y. Analysis of factors in successful nasal endoscopic resection of nasopharyngeal angiofibroma. Acta Otolaryngol. 2016;136(2):205-13.
- Mair EA, Battiata A, Casler JD. Endoscopic laser-assisted excision of juvenile nasopharyngeal angiofibromas. Arch. Otolaryngol. Head Neck Surg. 2003 Apr;129(4):454-9.
- Min HJ, Chung HJ, Kim CH. Delayed cerebrospinal fluid rhinorrhea four years after gamma knife surgery for juvenile angiofibroma. J Craniofac Surg. 2014 Nov;25(6):e565-7.
- Scholfield DW, Brundler MA, McDermott AL, Mussai F, Kearns P. Adjunctive Treatment in Juvenile Nasopharyngeal Angiofibroma: How Should We Approach Recurrence? J. Pediatr. Hematol. Oncol. 2016 Apr;38(3):235-9.





{kind=link}