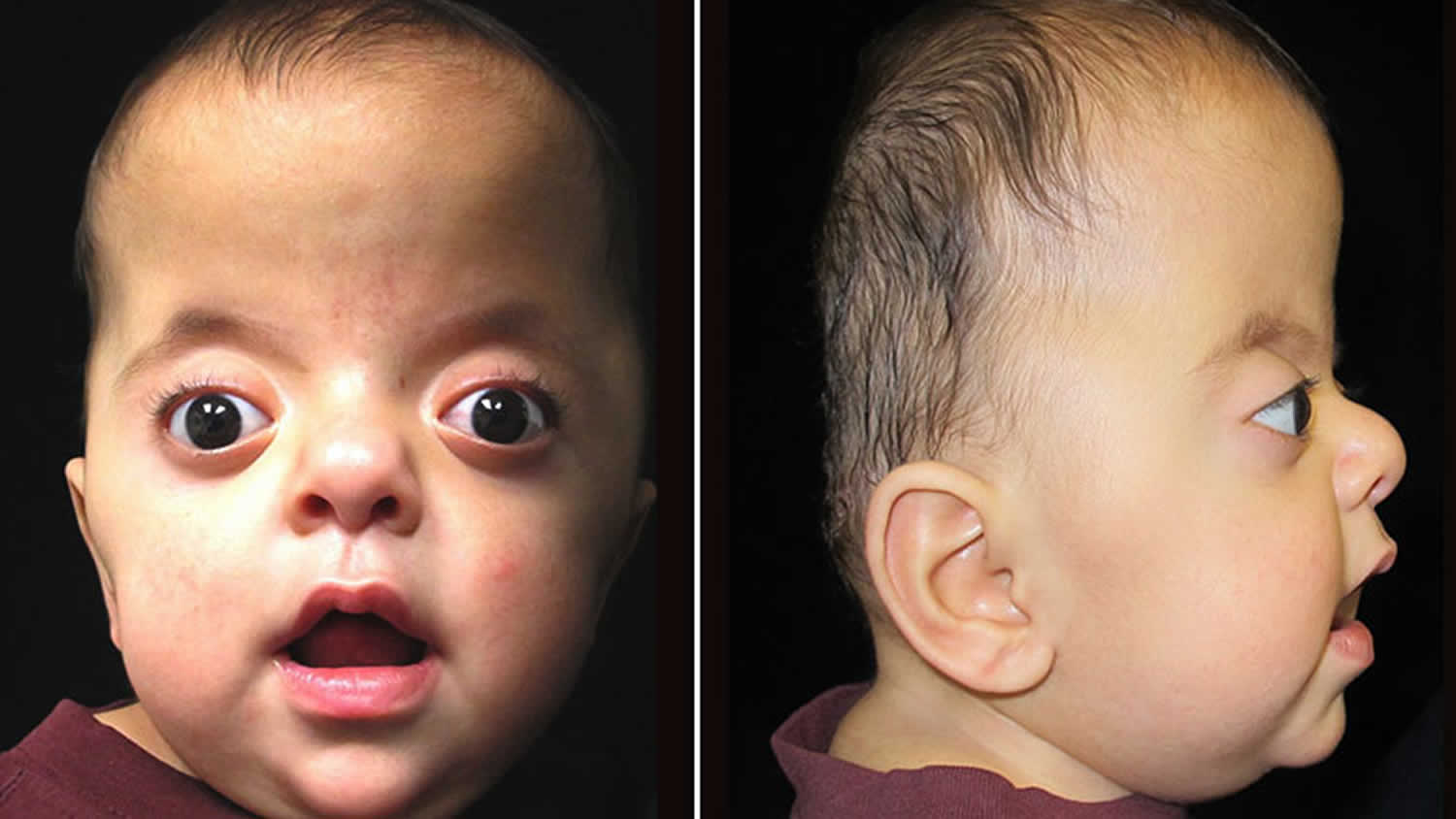
What is Apert syndrome
Apert syndrome also called acrocephalosyndactyly, is a congenital (present at birth) genetic disorder characterized by the premature fusion of certain sutures of the skull bones (craniosynostosis) 1. This early fusion prevents the skull from growing normally and affects the shape of the head and face. Children with Apert syndrome have protruding eyes that are usually wide-set and tilted down at the sides. They usually have problems with teeth alignment due to the underdevelopment of the upper jaw. Some have cleft palate. In addition, Apert syndrome causes abnormal growth of several bones in the body, primarily a varied number of fingers and toes are fused together (syndactyly). Apert syndrome affects an estimated 1 in 160,000 to 200,000 newborns 2.. Apert syndrome is named for the French physician who first described it, E. Apert, in 1906 2.
Many of the characteristic facial features of Apert syndrome result from the premature fusion of the skull bones. The head is unable to grow normally, which leads to a sunken appearance in the middle of the face, bulging and wide-set eyes, a beaked nose, and an underdeveloped upper jaw leading to crowded teeth and other dental problems. Shallow eye sockets can cause vision problems. Early fusion of the skull bones also affects the development of the brain, which can disrupt intellectual development. Cognitive abilities in people with Apert syndrome range from normal to mild or moderate intellectual disability.
Individuals with Apert syndrome have webbed or fused fingers and toes. The severity of the fusion varies; at a minimum, three digits on each hand and foot are fused together. In the most severe cases, all of the fingers and toes are fused. Less commonly, people with this condition may have extra fingers or toes (polydactyly). Additional signs and symptoms of Apert syndrome can include hearing loss, unusually heavy sweating (hyperhidrosis), oily skin with severe acne, patches of missing hair in the eyebrows, fusion of spinal bones in the neck (cervical vertebrae), and recurrent ear infections that may be associated with an opening in the roof of the mouth (a cleft palate).
Apert syndrome can be passed down through families (inherited) as an autosomal dominant trait. This means that only one parent needs to pass on the faulty gene for a child to have the condition. However, almost all cases of Apert syndrome result from new mutations in the gene, and occur in people without a known family history.
Apert syndrome is caused by one of two changes to the FGFR2 gene. This gene defect causes some of the bony sutures of the skull to close too early. This condition is called craniosynostosis.
Treatment consists of surgery to correct abnormal bone growth of the skull, as well as for the fusion of the fingers and toes. Children with Apert syndrome should be examined by a specialized craniofacial surgery team at a children’s medical center. A hearing specialist should be consulted if there are hearing problems.
The long-term outlook for a person with Apert syndrome can be improved with prompt diagnosis and medical attention 3.
Normal newborn skull anatomy and physiology
It is important to have an understanding of skull anatomy and growth in order to understand craniosynostosis. An infant’s skull has 7 bone plates that relate to each other through specialized joints called “cranial sutures”. Sutures are made of tough, elastic fibrous tissue and separate the bones from one another. Sutures meet up (intersect) at two spots on the skull called fontanelles, which are better known as an infant’s “soft spots”. Although there are several major and minor sutures, the sutures that potentially have the most clinical significance are the singular metopic and sagittal sutures, as well as the paired (right and left) coronal and lambdoid sutures (see Figure 1). The seven bones of an infant’s skull normally do not fuse together until around age two or later. The sutures normally remain flexible until this point. In infants with primary craniosynostosis, the sutures abnormally stiffen or harden causing one or more of the bones of the skull to prematurely fuse together. This in turn, may lead to asymmetric skull growth.
Cranial sutures are very unique and specialized joints (syndesmosis joints). Their primary purpose is to grow bone in response to the rapidly developing brain within the protective skull compartment (cranial cavity or calvarium). The skull not only needs to be firm to protect the brain from accidental blows, but should also be expansile to accommodate its rapid growth. The brain doubles in volume in the first year of life and almost triples in volume by the age of three. The sutures of the skull allow for this important but almost contradictory balance of protection and growth.
Each cranial suture is designed to generate growth in the skull in a very specific area and configuration, ultimately reflecting the size and shape of the underlying brain structure. The overall bone development from cranial sutures occurs in a direction perpendicular to the long axis of the suture. Understanding these two facts, it makes sense that a fusion of each suture independently would cause a unique head shape.
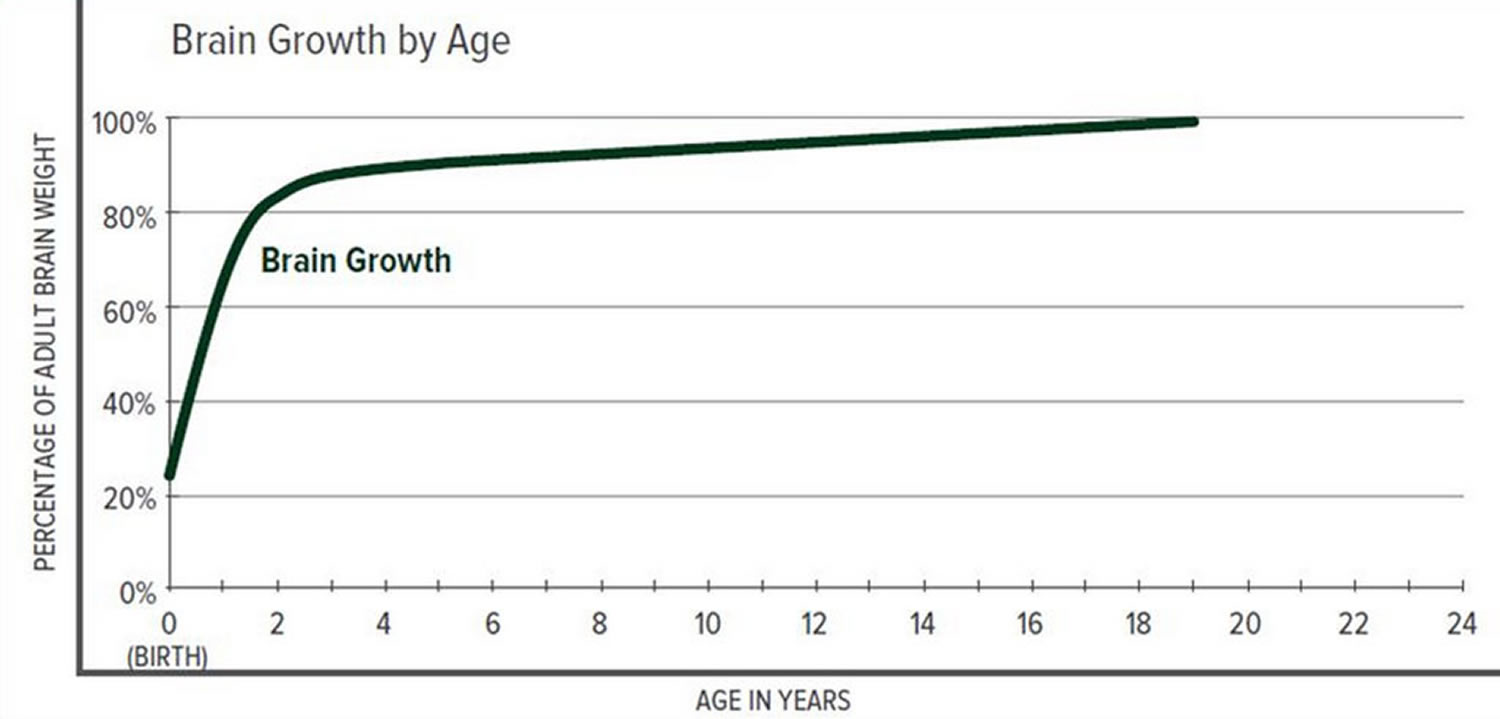
The greatest increase in brain volume (brain growth) occurs from 0 to 14 months of age. The size of a child’s brain typically reaches 80% of adult size by the age of 2. The growth in head circumference after that age is more related to growth in the thickness of the skull and scalp but not actual brain growth. “Hat size” increases but not necessarily “brain size”.
The various cranial sutures close at different ages. The metopic suture closes earliest, around 6 months to 2 years. The rest of the sutures stay open into the 20’s and 30’s. The brain and fluid cavities of the brain do continue to grow in volume as you go into early adulthood, albeit not nearly as rapidly as the first couple years of life. Since the skull is much firmer (calcified or “rock-like”) and thicker, the skull needs the sutures to grow bone for any increase in volume.
Interestingly, there is a lot of variability here. Doctors have operated on adults in their 30’s for reasons unrelated to their skull sutures and have coincidentally found open metopic sutures. They have also seen young adults with closed coronal, lambdoid, and sagittal sutures, but with normal head shapes and often, no indication or symptoms of high pressure.
Scientists have learned that a cranial suture’s purpose is to grow bone to accommodate a growing brain, and that most brain growth occurs in the first two years of life. The brain reaches 85% of adult size by age 3 years (see Figure 2. Brain size vs. age diagram).
So it makes sense that the sutures are vitally important in the first two years of life. The earlier the fusion, the more severe the restriction in growth and, consequently, volume provided to the brain.
Figure 1. Normal skull of a newborn
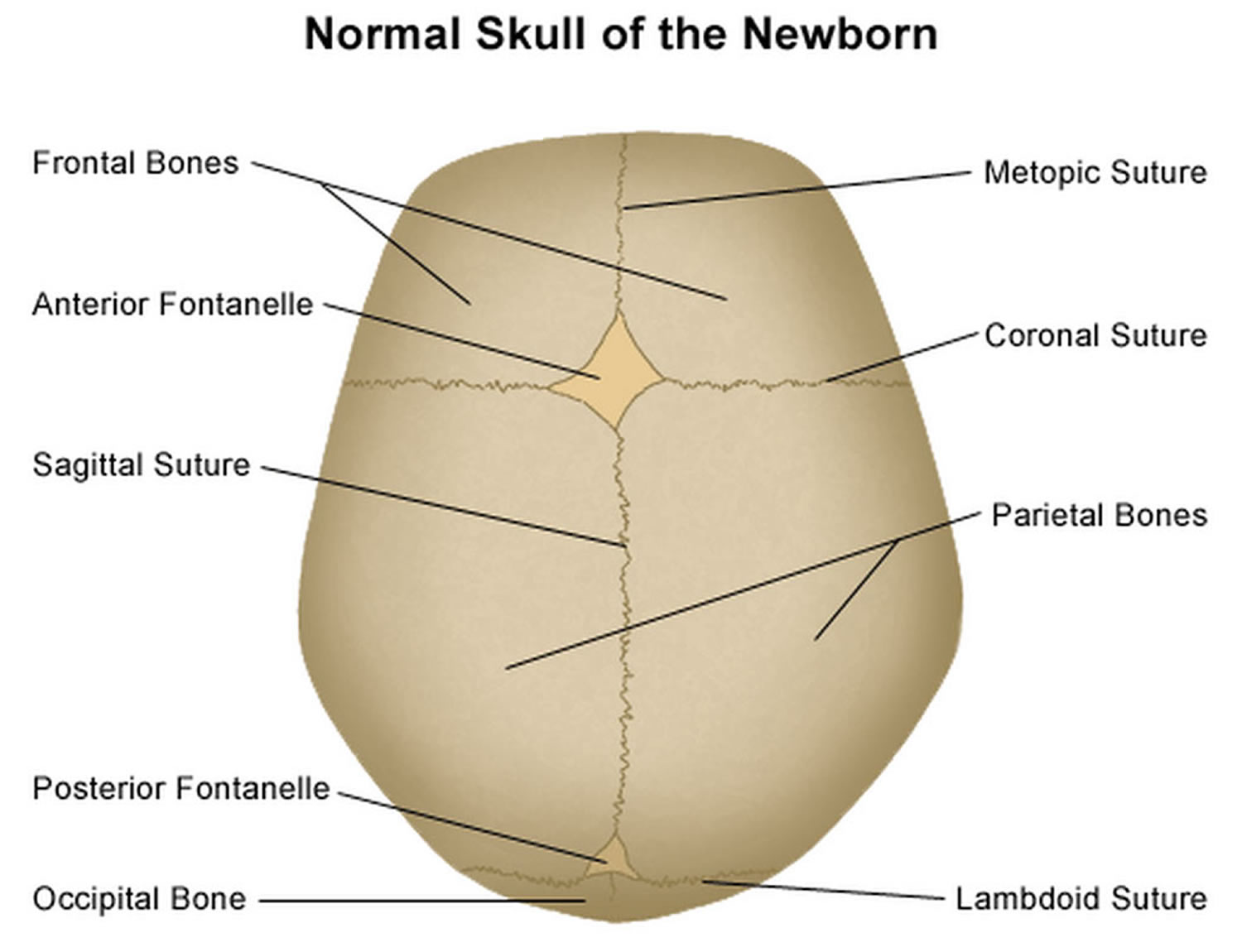
Figure 2. Brain size versus age diagram
 Apert syndrome baby
Apert syndrome baby
The skull is usually severely affected, as well as the entire face, especially the eyes and jaw. The skull bones are prematurely fused and the skull is unable to grow normally; the midface (that area of the face from the middle of the eye socket to the upper jaw) appears retruded or sunken; as a result the eyes bulge out and the eyelids tilt downward. The face can have acne. The hair may be unruly. Cleft palate and deafness are common. Patients also have syndactyly, which means joining of fingers and toes. Many children also have mental delays that can vary from mild to severe. In many cases, speech development and behavioral problems become more obvious as the child gets older.
The following problems have been observed in some children with Apert syndrome. However, whether or not they were caused by Apert syndrome is uncertain 2.
- Various heart defects
- Cleft palate
- Dextrorotation
- Pulmonary Artresia
- Patent Ductus Arteriosus (PDA)
- Tracheoesophageal Fistula
- Pyloric stenosis
- Polycystic kidneys
- Bicornate uterus
- Hydrocephalus
- Ear infections which can cause hearing loss
- Sleep Apnea, small nose and airway passage make breathing difficult
- Severe acne, hyperactive sweat glands
- Increased incidence of eye injuries, imbalance of eye muscles
Figure 3. Apert syndrome baby
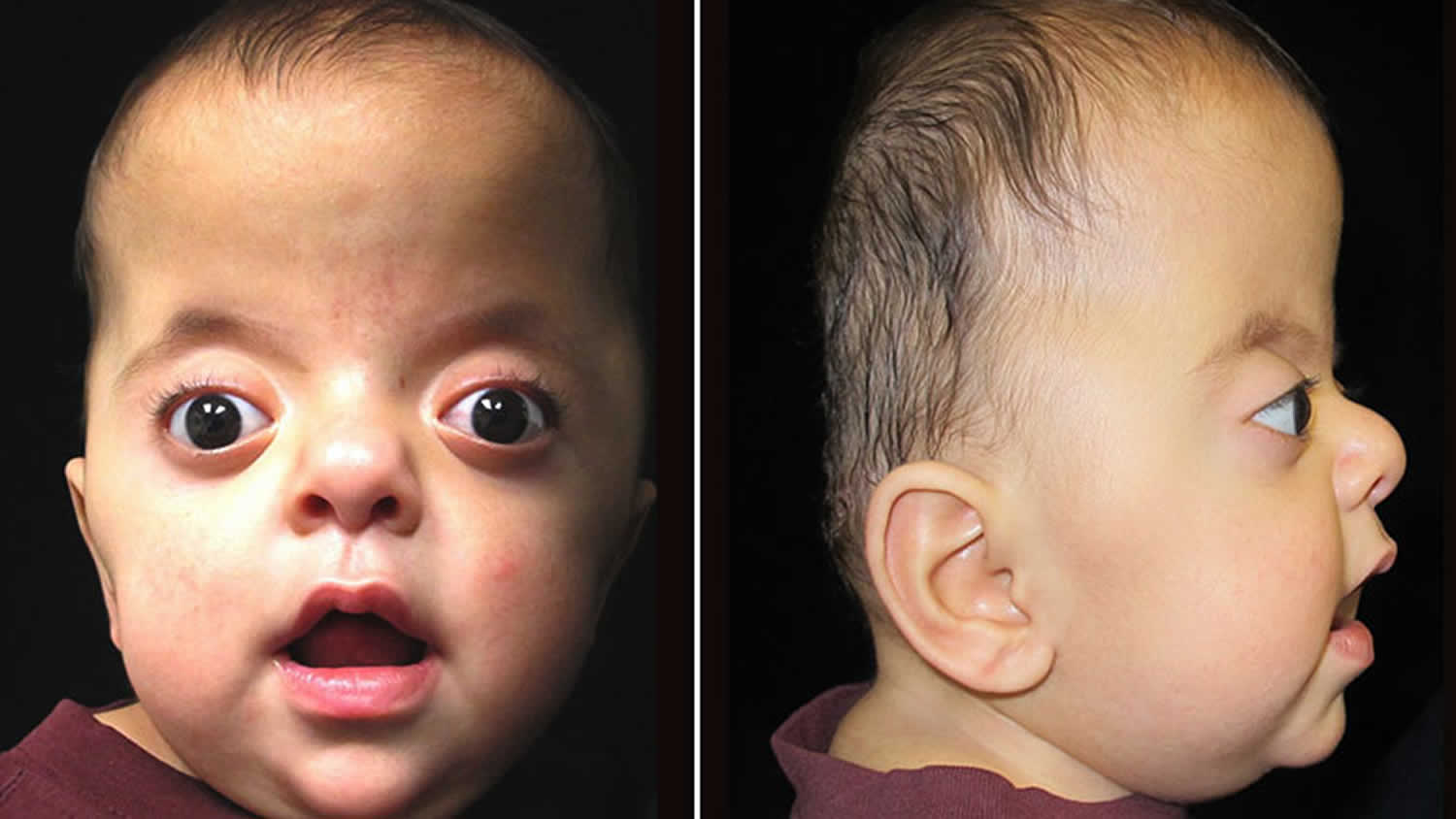
Footnote: 6-month-old male with Apert syndrome. Note tall shape and characteristic flatness of the back of the head. Eyes that appear wide set and have a bulging appearance are also a characteristic of this syndrome.
[Source 4]Figure 4. Apert syndrome baby
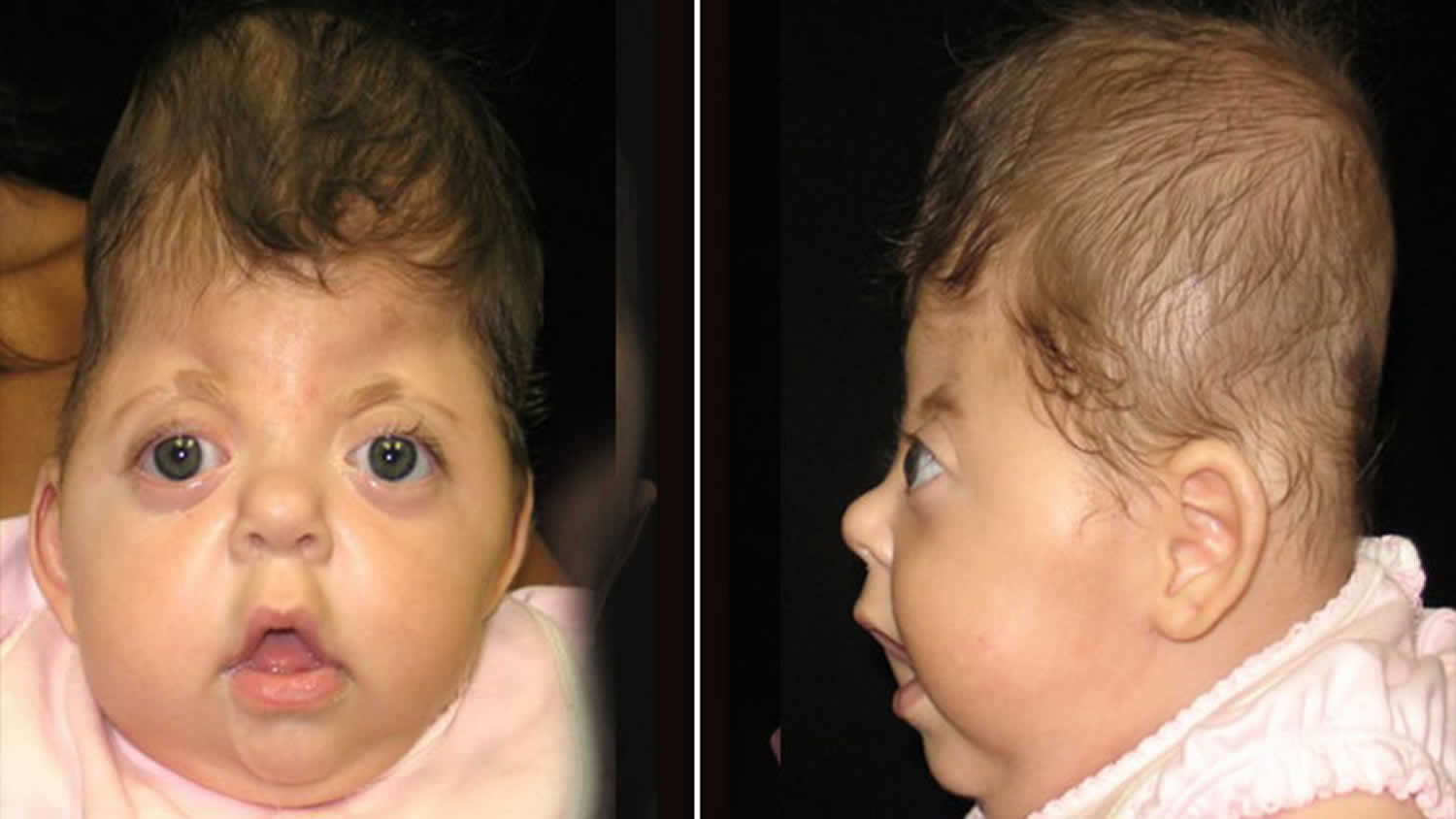
Footnote: 4-month-old female with Apert syndrome. Note the retruded brow and flattened forehead as well as sunken appearance of the face from the middle of the eyes to the upper jaw.
[Source 4]How are the fingers and toes affected?
The fusion of the fingers and toes along with the craniofacial problems mentioned above is what really separates Apert syndrome from other similar syndromes. This condition is called syndactyly. It always involves fusion of the soft tissues of the first, middle, and ring fingers, and often there is fusion of the bones themselves. The joints usually do not move well, if at all. The thumb may be fused into the hand, or may be free. Surgery is used to separate the fingers as needed to obtain better function. The feet and toes are affected similarly, but surgery is usually only done in cases where the ability to walk would be impaired.
Figure 5. Apert syndrome hands
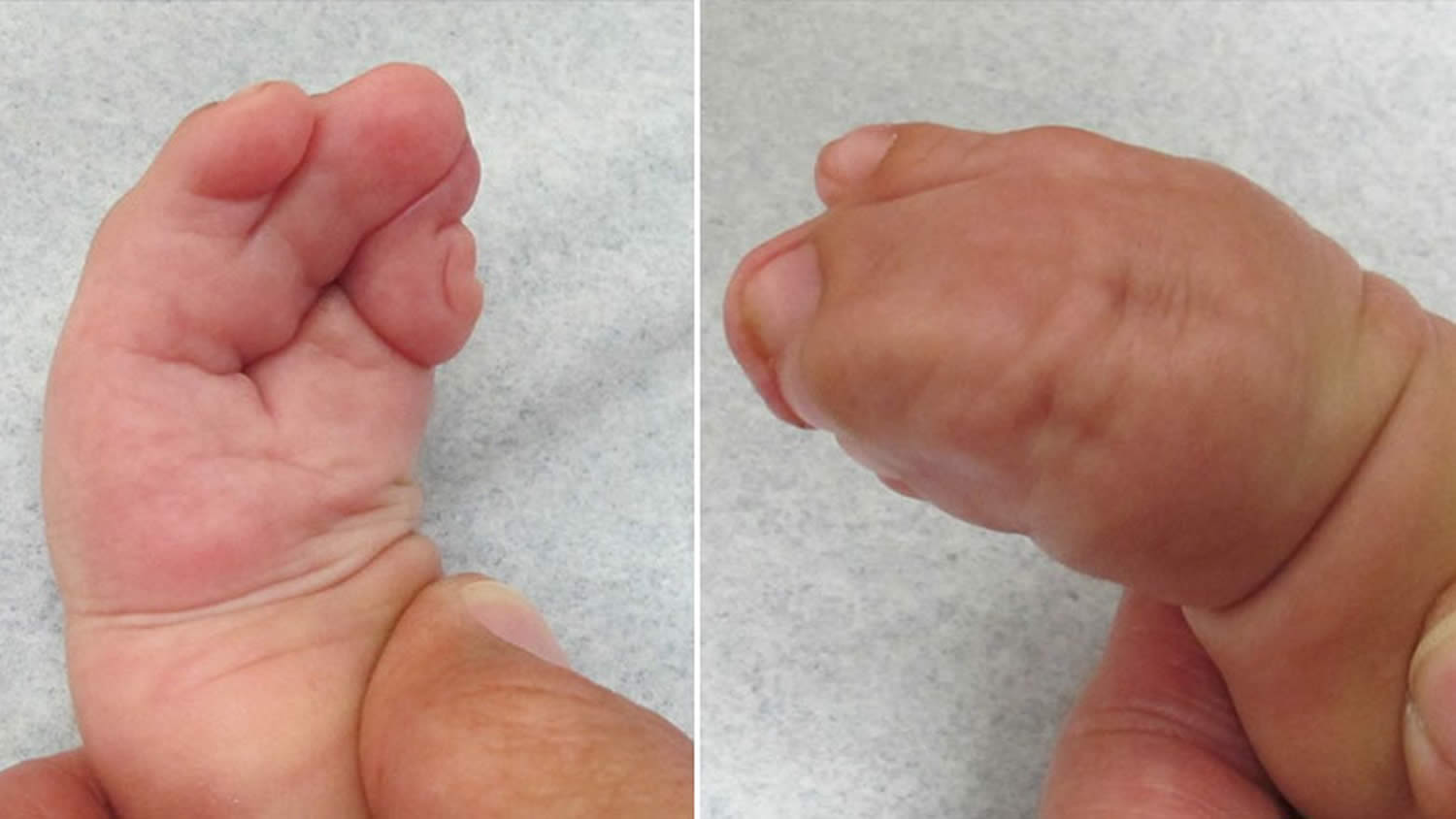
Footnote: Complex syndactyly, when fingers are webbed together, benefit from specialty surgical care to release and result in the greatest degree of function that can be achieved.
[Source 4]Apert syndrome possible complications
Minor complications include infections of the skin, around the stitches, collections of blood under the skin, and hair loss. Orthodontic care is almost always required especially if the jaw is moved. Tooth loss may occur. Scalp and face numbness is common, especially after surgery. This may or may not get better with time. Bruising and swelling always occur to some degree.
Major complications may require surgery or a hospital stay. More severe infections may occur, especially if distractors to move bone are used. Severe bleeding may need blood transfusions or more surgery. Double vision and other vision problems may occur that may need eye surgery. Blindness is extremely rare. Brain damage and death are also extremely rare. Fortunately, with the multi-team approach that most craniofacial centers have, complications are kept to a minimum.
Remember, new advances and procedures concerning Apert syndrome are constantly being developed, so ask questions and be an advocate for your child.
Apert syndrome symptoms
Apert syndrome is primarily characterized by a fusion of the skull bones that occurs too early during development (craniosynostosis) and webbing of the fingers and toes (syndactyly). Individuals with Apert syndrome typically have a flat, elongated forehead causing the head to be cone-shaped (acrocephaly), bulging and wide-set eyes due to shallow orbits (eye sockets), recession or underdevelopment of the midface (maxillary hypoplasia), a “beaked” nose and syndactyly or fusion of the digits.
An underdeveloped upper jaw and shallow eye sockets can cause dental and vision problems. Craniosynostosis can also affect the development of the brain, disrupting intellectual development. Cognitive abilities in people with Apert syndrome range from normal to mild or moderate intellectual or developmental disabilities. Specific cognitive deficits that may impact children with craniosynostosis include impairments in domains such as visuospatial functioning, executive functioning, attention, memory, or general developmental delay.
Additional signs and symptoms of Apert syndrome may include hearing loss, extra fingers or toes (polydactyly), heavy sweating (hyperhidrosis), oily skin with severe acne, patches of missing hair in the eyebrows, and spinal bones in the neck (cervical vertebrae) that are fused. Recurrent ear infections may be associated with an opening in the roof of the mouth (a cleft palate).
In addition, Apert Syndrome can be associated with hydrocephalus, sleep apnea, eye exposure issues when the eyelids can’t close completely and airway compromise that may require tracheostomy 4.
Apert syndrome symptoms include:
- Early closure of sutures between bones of the skull, noted by ridging along sutures (craniosynostosis)
- Frequent ear infections
- Fusion or severe webbing of the 2nd, 3rd, and 4th fingers, often called “mitten hands”
- Hearing loss
- Large or late-closing soft spot on a baby’s skull
- Possible, slow intellectual development (varies from person to person)
- Prominent or bulging eyes
- Severe under-development of the midface
- Skeletal (limb) abnormalities
- Short height
- Webbing or fusion of the toes
Several other syndromes can lead to a similar appearance of the face and head, but do not include the severe hand and foot features of Apert syndrome. These similar syndromes include:
- Carpenter syndrome (kleeblattschadel, cloverleaf skull deformity)
- Crouzon disease (craniofacial dysostosis)
- Pfeiffer syndrome
- Saethre-Chotzen syndrome
The characteristic feature that distinguishes Apert syndrome from other types of syndromic craniosynostosis is the presence of hand anomalies, most commonly fused or webbed fingers (syndactyly). These hand deformities can be quite severe and prevent your child from functioning normally.
In addition to these characteristic signs and symptoms, Apert syndrome affects several other organ systems of the body. The database, Online Mendelian Inheritance in Man, presents the following as a Clinical Synopsis of Apert syndrome 5:
Growth
- Decrease in the rate of growth leading to short stature, in spite of normal birth weight and birth length
Head and Neck
- Head:
- Pointed but broad skull
- Large, late-closing “soft spot” on the skull (fontanelle)
- Face:
- High, broad forehead
- Flat face
- Jutting jaw (mandibular prognathism)
- Right and left asymmetry
- Ears:
- Hearing loss
- Chronic ear infections (otitis media)
- Abnormal semicircular canals
- Eyes:
- Shallow orbits in which the eyes sit
- Wide separation between eyes (hypertelorism)
- Down-slanting eyelids
- Bulging eyes (proptosis)
- Cross-eyes (strabismus)
- Nose:
- Flattened nose with low bridge
- The openings between the nose and throat (choana) may be blocked or narrowed, interfering with breathing and swallowing.
- Mouth:
- Narrow “roof” of the mouth (palate)
- Cleft palate especially of the form known as “bifid uvula”
- Teeth:
- Bad “bite” (malocclusion)
- Delayed dentition teeth are late in coming
Cardiovascular:
- Hole(s) in ventricular wall
- Overriding aorta develops when the aorta is positioned directly over a hole in ventricular wall, instead of over the left ventricle. As a result, the aorta contains some blood from the right ventricle reducing the amount of oxygen transported.
Respiratory
- Malformed cartilage supports of the trachea, interfering with breathing and swallowing
Gastrointestinal
- Narrowing of the opening between the lower part of the stomach and the upper part of the small intestine (duodenum).
- Blockage of the esophagus
- Anus out of position
Genitourinary
- Failure of the testicles to fall in boys (cryptorchidism)
- Blockage of vagina in girls (vaginal atresia)
- Enlarged kidneys due to blockage (hydronephrosis)
Skeletal
- Fusion of cervical vertebrae (C5 and C6)
- Fusion of the two bones of the arms
- Fusion of bones of the wrist
- Fusion (syndactyly) of the bones and/or skin of hands and feet
- Spatulate end joint of the thumb
Skin, Nails and Hair
- Moderate to severe acne
- Single nail across the fused digits from 2-4
Neurologic
- Varying degrees of mental retardation
- Failure to form the fibrous tissue that joins the cerebral hemispheres of the brain (agenesis of the corpus callosum)
- Enlarged brain cavity (ventriculomegaly)
- Failure to form the membranes that separate the various cavities of the brain (absent septum pellucidum)
- Malformations of parts of the brain’s limbic system that deals in part with the autonomous nervous system.
- Chiari I malformation
- Low-lying cerebellar tonsils
- Posterior fossa arachnoid cyst
- Hydrocephalus
Apert syndrome causes
Mutations in the Fibroblast Growth Factor Receptor 2 (FGFR2) gene cause Apert syndrome. This gene produces a protein called fibroblast growth factor receptor 2. Among its multiple functions, this protein signals immature cells to become bone cells during embryonic development. A mutation in a specific part of the FGFR2 gene alters the protein and causes prolonged signaling, which can promote the premature fusion of bones in the skull, hands, and feet.
Apert syndrome inheritance pattern
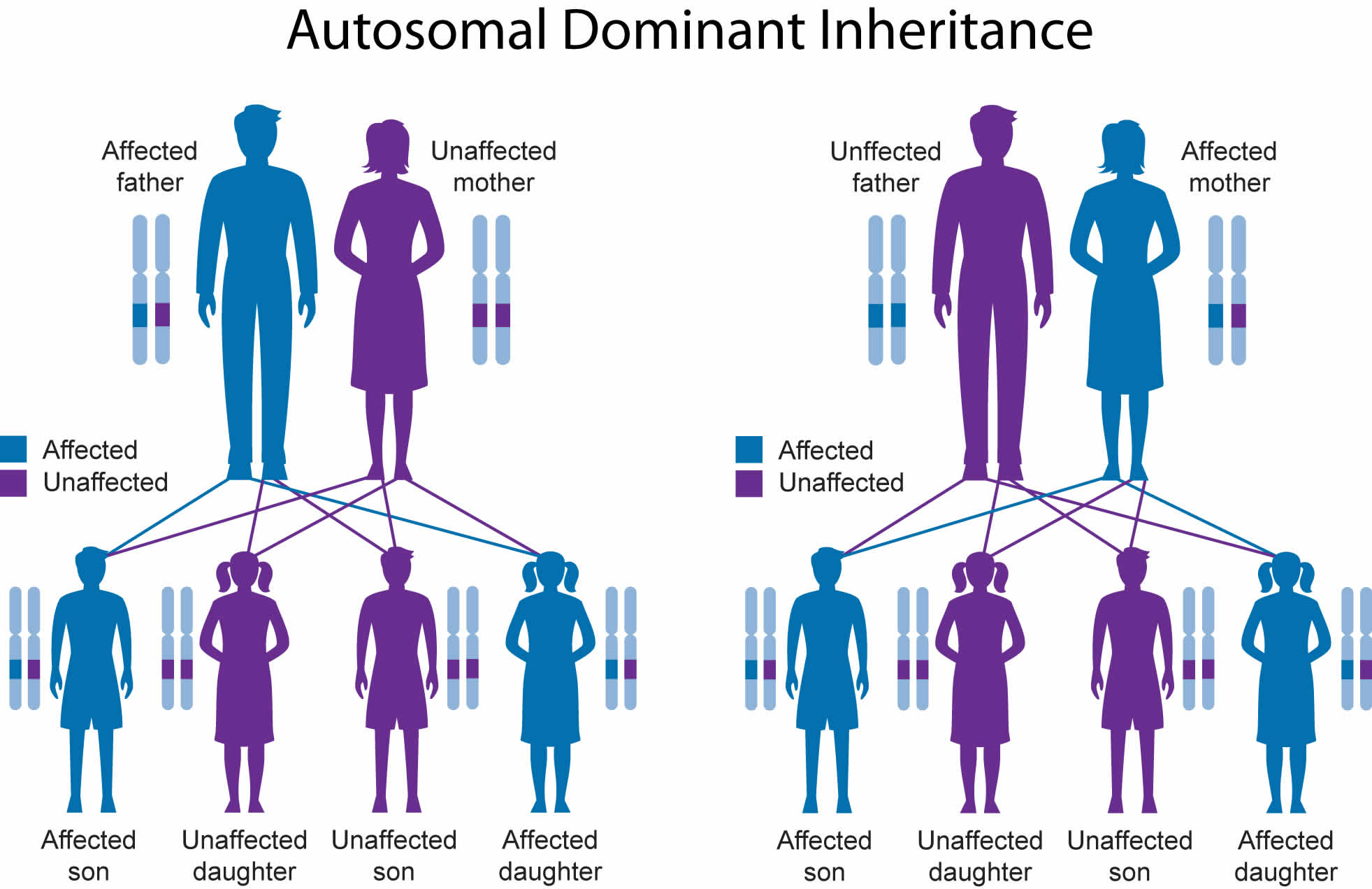
Apert syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Almost all cases of Apert syndrome result from new mutations in the gene, and occur in people with no history of the disorder in their family. Individuals with Apert syndrome, however, can pass along the condition to the next generation. Studies have shown that Apert syndrome occurs more often in children of older fathers.
- When you have Apert syndrome, you have a 1 in 2 (50%) chance of passing this condition to your child. This is because each of us gets 1/2 of our genetic makeup from each parent.
Genetic counseling may be helpful if you have a family history of this disorder and are planning to become pregnant. Your provider can test your baby for this disease during pregnancy.
Genetic counseling may help you understand the risks of passing Apert syndrome on to any children you have.
- To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. Online directories are provided by the American College of Medical Genetics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) and the National Society of Genetic Counselors (https://www.findageneticcounselor.com/).
Figure 6. Apert syndrome autosomal dominant inheritance pattern
Apert syndrome diagnosis
The health care provider will perform a physical exam. Hand, foot, and skull x-rays will be done. Hearing tests should always be performed.
Apert syndrome can be diagnosed based on the presence of the following features 6:
- Turribrachycephalic skull shape (cone-shaped or towering skull) which is visibly apparent and can be confirmed by skull radiograph or head CT examination
- Characteristic facial features including moderate-to-severe underdevelopment of the midface, bulging and wide-set eyes, “beaked” nose, underdeveloped jaw and shallow eye sockets
- Variable hand and foot findings such as syndactyly of the fingers and toes and polydactyly
Genetic testing can confirm the diagnosis of Apert syndrome 7. More than 98% of Apert syndrome cases are caused by specific missense substitution mutations, involving adjacent amino acids (Ser252Trp, Ser252Phe, or Pro253Arg) in exon 7 of FGFR2. The remaining cases are due to Alu-element insertion mutations in or near exon 9.
Genetic counseling may help you understand the risks of passing Apert syndrome on to any children you have.
- To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. Online directories are provided by the American College of Medical Genetics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) and the National Society of Genetic Counselors (https://www.findageneticcounselor.com/).
Apert syndrome treatment
Treatment for Apert syndrome may vary depending on the specific symptoms in each person and is dependent upon both functional and appearance-related needs, and should be addressed immediately after your child is born. Brain surgery may help to relieve the pressure created by the craniosynostosis. Craniosynostosis and, in some cases, associated hydrocephalus may result in abnormally increased pressure within the skull (intracranial pressure) and on the brain. In such cases, early surgery (within 2 to 4 months after birth) may be advised to correct craniosynostosis and, for those with hydrocephalus, to insert a tube (shunt) to drain excess cerebrospinal fluid (CSF) away from the brain and into another part of the body where the CSF (cerebrospinal fluid) can be absorbed. Early corrective and reconstructive surgery may also be performed in some infants with Apert syndrome to help correct certain associated craniofacial abnormalities.
The “retrusion” or lack of development of the midface is what could be described as concave or dished in profile. As the skull grows, the middle third of the face grows slower, resulting in a more pronounced retrusion over time. A surgical procedure known as the LeFort III can be used to correct this condition. The procedure is usually done after substantial growth is complete (preadolescence) and may be repeated as necessary. The LeFort procedure involves detaching the facial bones from mid eye to upper jaw and spacing this area out with bone grafts so that a proper alignment is made. If the forehead has not grown well either, a procedure called “monoblock” may be used.
In the last few years, many surgeons have come to prefer “distraction” of the bones using either the Rigid External Distraction (RED) system or internally placed distractors. With this procedure, the operation remains the same but now the bone is gradually pulled forward instead of moved at once during surgery. This leads to formation of new bone over time.
In addition, your child may need a frontal-orbital advancement within the first twelve months to increase space within the skull and the size of both orbits (the part of the skull which holds the eyeball), a facial bi-partition to widen the upper jaw, derotate the orbits, and to narrow the upper face, and/or during the teen years an osteotomy (cutting through the bone of the upper and lower jaw) to correct further problems.
The severity of your child’s Apert syndrome determines whether he/she needs some or all the procedures described above.
Additional surgical and/or medical treatment may be required to treat and correct other symptoms. A team of specialists may work with a child with Apert syndrome to plan other surgeries to help treat the facial abnormalities and the fusion of the fingers or toes. People with Apert syndrome will need to be closely monitored throughout life to treat any symptoms that arise 8.
Because of the complex issues that can be associated with Apert syndrome, your child should be treated at a medical center where he/she will have access to pediatric specialists across the many clinical areas he/she may need. A medical team may consist of a craniofacial surgeon, neurosurgeon, ENT, audiologist, speech pathologist, oral surgeon, psychologist, ophthalmologist, and an orthodontist. The team approach is used by these physicians to determine the best collaborative corrective plan for the deficiencies of your child.
Children with Apert Syndrome benefit from routine psychosocial support to address any behavioral, developmental, academic, emotional or social difficulties associated with their condition, appearance differences, and surgical treatments throughout childhood.
References- Apert syndrome. https://ghr.nlm.nih.gov/condition/apert-syndrome
- https://www.ccakids.org/assets/syndromebk_apert.pdf
- Apert syndrome. https://rarediseases.info.nih.gov/diseases/5833/apert-syndrome
- Apert syndrome. https://www.chop.edu/conditions-diseases/apert-syndrome
- Apert syndrome. http://omim.org/clinicalSynopsis/101200?highlight=syndromic%20apert%20syndrome
- Robin NH, Falk MJ, Haldeman-Englert CR. FGFR-Related Craniosynostosis Syndromes. 1998 Oct 20 [Updated 2011 Jun 7]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1455
- Apert Syndrome Workup. https://emedicine.medscape.com/article/941723-workup
- Apert Syndrome. https://rarediseases.org/rare-diseases/apert-syndrome/





{kind=link}