
Arginase deficiency
Arginase deficiency also called hyperargininemia, argininemia, arginase deficiency disease or arginase-1 deficiency, is an inherited disorder characterized by complete or partial lack of the enzyme arginase in the liver and red blood cells that causes the amino acid arginine (a building block of proteins) and ammonia to accumulate gradually in the blood 1. Ammonia, which is formed when proteins are broken down in the body, is toxic if levels become too high. The nervous system is especially sensitive to the effects of excess ammonia (hyperammonemia).
Arginase is one of six enzymes that play a role in the breakdown and removal of nitrogen from the body, a process known as the urea cycle. The lack of the arginase enzyme results in excessive accumulation of nitrogen, in the form of ammonia (hyperammonemia), in the blood and arginine (hyperarginemia) in the blood and cerebrospinal fluid 2.
Arginase deficiency usually becomes evident by about the age of 3. It most often appears as stiffness, especially in the legs, caused by abnormal tensing of the muscles (spasticity). Other symptoms may include slower than normal growth, developmental delay and eventual loss of developmental milestones, intellectual disability, seizures, tremor, and difficulty with balance and coordination (ataxia). Occasionally, high protein meals or stress caused by illness or periods without food (fasting) may cause ammonia to accumulate more quickly in the blood. This rapid increase in ammonia may lead to episodes of irritability, refusal to eat, and vomiting.
In some affected individuals, signs and symptoms of arginase deficiency may be less severe, and may not appear until later in life.
Arginase deficiency is a very rare disorder; it has been estimated to occur once in every 300,000 to 1,000,000 individuals 3. Arginase-1 deficiency is among the least common of all the disorders of the urea cycle. The estimated frequency of urea cycle disorders collectively is one in 30,000. However, because urea cycle disorders like arginase-1 deficiency often go unrecognized, these disorders are under-diagnosed, making it difficult to determine the true frequency of urea cycle disorders in the general population. This is likely to change now that arginase-1 deficiency can be diagnosed by newborn screening, affected newborns are found to have elevated levels (up to 4 times) of arginine.
Management should closely mirror that for urea cycle disorders, except that individuals with arginase deficiency are not as likely to have episodes of hyperammonemia; if present, such episodes respond to conservative management (e.g., intravenous fluid administration). Treatment should involve a team coordinated by a metabolic specialist. Routine outpatient management includes restriction of dietary protein and consideration of oral nitrogen-scavenging drugs (in those who have chronic or recurrent hyperammonemia). Treatment of an acutely ill (comatose and encephalopathic) individual requires: rapid reduction of plasma ammonia concentration; use of pharmacologic agents (sodium benzoate and/or sodium phenylbutyrate/phenylacetate) to promote excretion of excess nitrogen through alternative pathways; and introduction of calories supplied by carbohydrates and fat to reduce catabolism and the amount of excess nitrogen in the diet while avoiding overhydration and resulting cerebral edema. Standard treatment for seizures, spasticity, developmental delay / intellectual disability, and joint contractures. In those with persistent hepatic synthetic function abnormalities, fresh-frozen plasma should be considered prior to surgical procedures. In the rare instance of progression to hepatic fibrosis and cirrhosis, liver transplantation can be considered.
Arginase deficiency causes
Mutations in the ARG1 gene cause arginase deficiency. Arginase deficiency belongs to a class of genetic diseases called urea cycle disorders. The urea cycle is a sequence of reactions that occurs in liver cells. This cycle processes excess nitrogen, generated when protein is used by the body, to make a compound called urea that is excreted by the kidneys.
The ARG1 gene provides instructions for making an enzyme called arginase. This enzyme controls the final step of the urea cycle, which produces urea by removing nitrogen from arginine. In people with arginase deficiency, arginase is damaged or missing, and arginine is not broken down properly. As a result, urea cannot be produced normally, and excess nitrogen accumulates in the blood in the form of ammonia. The accumulation of ammonia and arginine are believed to cause the neurological problems and other signs and symptoms of arginase deficiency.
Arginase deficiency inheritance pattern
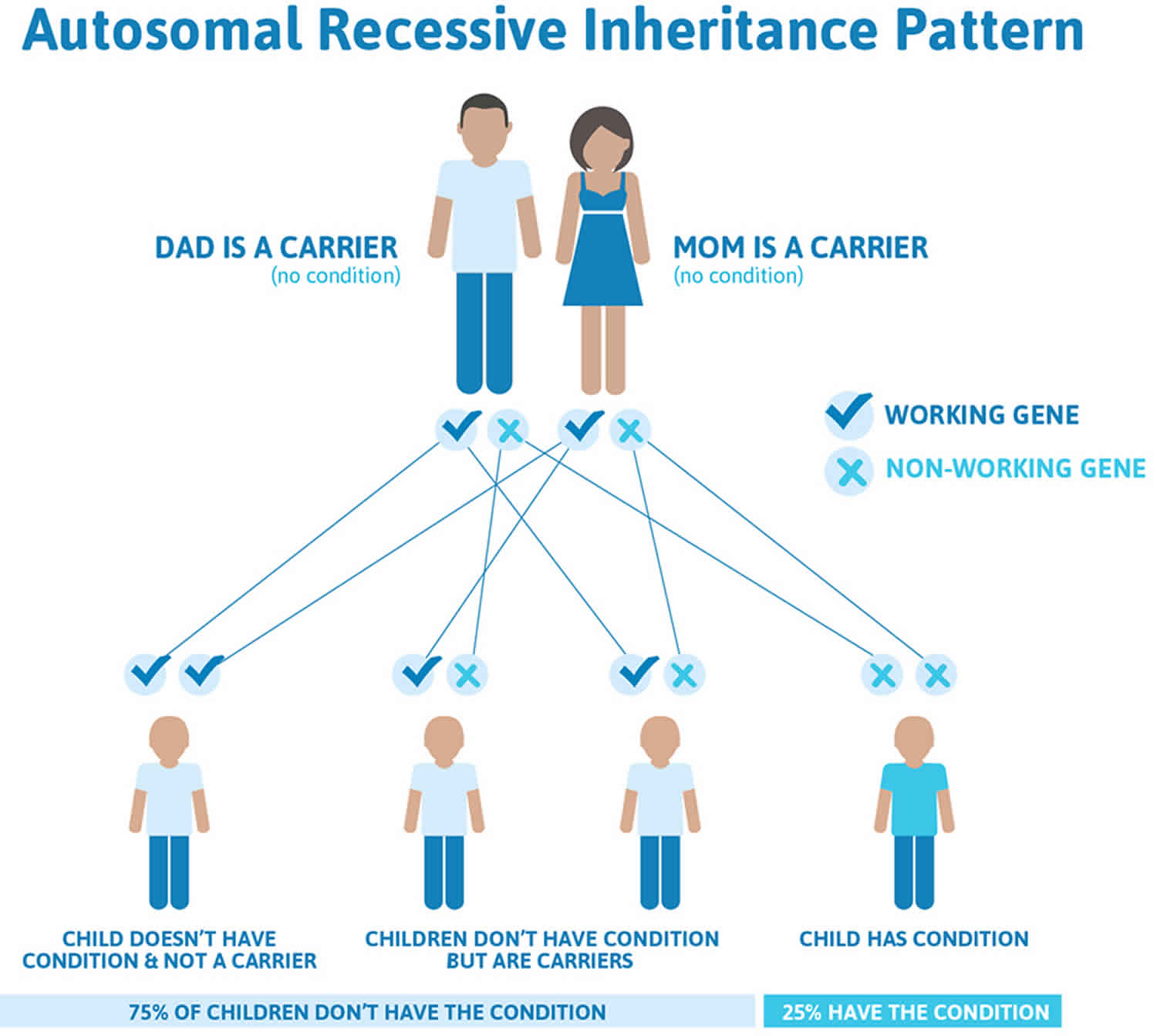
Arginase deficiency is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Arginase deficiency autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Arginase deficiency symptoms
Symptoms associated with arginase-1 deficiency differ from those associated with other disorders of the urea cycle. Arginase deficiency (argininemia) rarely presents in the newborn/infant period. Most infants with arginase-1 deficiency do not exhibit any symptoms during the first few months to a year of life. Infants with arginase-1 deficiency infrequently experience severe hyperammonemia or hyperammonemic coma, which are characteristic of the other urea cycle disorders.
After 1 to 3 years of life, patients develop intermittent episodic hyperammonemia, which can be induced by catabolic states (infections), high dietary protein intake, or medications (valproate). This status can only be recognized while the patient is presenting the acute injury 4.
Affected children may experience a lag in growth between one and three years and may walk on their toes and develop progressive stiffness and lack of control of voluntary movements of the legs (spastic diplegia). Cognitive development slows or stops and if untreated, children develop severe spasticity, an inability to walk, loss of bowel and bladder control and severe intellectual disability.
Almost all affected children have growth deficiency and many also experience seizures.
Arginase deficiency diagnosis
Most affected infants are now identified at birth through newborn screening. Initial steps to be performed after a positive newborn screen are plasma ammonia levels, plasma amino acids, and urine organic acids. Arginase enzyme activity is usually not detectable in red blood cells from affected individuals.
Elevated arginine levels (which can rise 4-fold), and ammonia levels (if present, above 200 micrograms/dL) along with increased orotic acid are suggestive. Subsequent arginase enzyme analysis on red blood cells (less than 1%) or molecular genetic testing of ARG1 to confirm the diagnosis; however, the latter is considered the first confirmatory step due to feasibility to perform enzyme analysis 5. If two mutations are not found, red blood cell enzyme testing is used to confirm the diagnosis.
This initial stage is critical, as there are other types of urea cycle disorders that benefit from arginine administration to reduce ammonia levels by indirectly increasing citrulline levels at the same time it attaches ammonia molecules for excretion.
Arginase deficiency treatment
Treatment should be coordinated by a metabolic specialist and is based on reducing plasma ammonia and arginine concentration, preventing excess ammonia from being formed, and reducing the amount of nitrogen in the diet.
Reduction of plasma ammonia concentration is accomplished by dialysis, either through ECMO or hemodialysis. This should be used only when the high levels are producing severe symptoms. These should be stopped once ammonia levels reach 250 micrograms/dL or lower.
The use of nitrogen scavengers like sodium phenylacetate or sodium benzoate provide an alternative pathway for removing excess nitrogen for severe or moderate cases, along with with restriction of protein intake, and the introduction of non-protein calorie sources like fats and carbohydrates should be considered. Intravenous and oral forms of these medications are available (Ammonul). Phenylbutyrate (Buphenyl) has a less offensive odor than the other medications but is available as oral therapy only. Ravicti is a form of phenylbutyrate that is less irritating to the gastrointestinal track and easier to take. When using carbohydrates with intravenous fluids, use dextrose 10% and appropriate electrolytes (sodium and/or potassium). Avoid overhydration as cerebral edema can occur. Abstinence from protein should not last more than 24-48 hours, as further catabolism can occur.
Dietary restrictions in individuals with arginase-1 deficiency are aimed at limiting the amount of arginine and protein intake. Children with arginase-1 deficiency are placed on a low-protein, arginine-restricted diet supplemented by essential amino acids.
For maintenance, protein restriction should be on the minimal protein intake range to help basic functions and development. With half of the dietary protein free of arginine, a total absence of this amino acid cannot be accepted given its essential role for T cell and endothelial function. Ideal protein intake in infants ranges from 1 to 1.5 gm/kg. As the child grows, the restriction can be tolerated on lower levels. Daily administration of nitrogen scavengers on maintenance dosing is sodium phenylbutyrate 350 to 600 mg/kg per day.
Other authors advocate for ornithine supplementation, which could replenish hepatic ornithine and prevent hyperammonemia 6, while also inhibiting the formation of neurotoxic guanidino compounds 7.
Seizures are treated with phenobarbital or carbamazepine. Valproic acid (valproate) should be avoided, as it can increase blood ammonia levels 4.
Affected individuals should receive periodic blood tests (at least monthly for the first year of life and as determined by a metabolic specialist after the first year of life) to determine the levels of ammonia and arginine in the blood and guanidinoacetate and liver function tests every six to 12 months to be sure that liver function is not impaired with monitoring of growth and developmental progress at each visit. Excessive levels of ammonia or arginine should be promptly treated.
Liver transplantation eliminates hyperargininemia and presumably the risk for hyperammonemia but is rarely necessary in arginase deficiency.
Genetic counseling is recommended for affected individuals and their families.
Arginase deficiency prognosis
In view of the relatively subtle and progressive presentation, patient rarely escape irreversible damage to the central nervous system. Nonetheless, early diagnosis in the clinical course allows for improved outcome.
Even in patients who receive a late diagnosis, treatment from birth in a subsequent infant of an affected family should prevent the developmental delay and the spasticity, based on more recent experience.
References- Morales JA, Sticco KL. Arginase Deficiency (Argininemia) [Updated 2019 Nov 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482365
- Sun A, Crombez EA, Wong D. Arginase Deficiency. 2004 Oct 21 [Updated 2020 May 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1159
- Arginase deficiency. https://ghr.nlm.nih.gov/condition/arginase-deficiency
- Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: do their functions overlap? Mol. Genet. Metab. 2004 Apr;81 Suppl 1:S38-44.
- Therrell BL, Currier R, Lapidus D, Grimm M, Cederbaum SD. Newborn screening for hyperargininemia due to arginase 1 deficiency. Mol. Genet. Metab. 2017 Aug;121(4):308-313.
- Jain-Ghai S, Nagamani SC, Blaser S, Siriwardena K, Feigenbaum A. Arginase I deficiency: severe infantile presentation with hyperammonemia: more common than reported? Mol. Genet. Metab. 2011 Sep-Oct;104(1-2):107-11.
- Amayreh W, Meyer U, Das AM. Treatment of arginase deficiency revisited: guanidinoacetate as a therapeutic target and biomarker for therapeutic monitoring. Dev Med Child Neurol. 2014 Oct;56(10):1021-4.





{kind=link}