
Argininosuccinate lyase deficiency
Argininosuccinate lyase (ASL) deficiency also called argininosuccinic aciduria or argininosuccinic acid lyase deficiency, is an inherited disorder that causes ammonia to accumulate in the blood 1. Argininosuccinate lyase (ASL) enzyme is one of six enzymes that play a role in the breakdown and removal of nitrogen from your body, a process known as the urea cycle. Argininosuccinate lyase (ASL) enzyme cleaves argininosuccinic acid to produce arginine and fumarate in the fourth step of the urea cycle. The lack of argininosuccinate lyase enzyme results in excessive accumulation of nitrogen, in the form of ammonia (hyperammonemia), in the blood. Ammonia is formed when proteins are broken down in the body, is toxic if the levels become too high. Ammonia is a neurotoxin, which means that it damages or inhibits the function of neurons, the cells of the central nervous system. Excess ammonia travels to the central nervous system (CNS) through the blood, resulting in the symptoms and physical findings associated with the disorder. Affected infants may experience vomiting, refusal to eat, progressive lethargy, and coma.
Argininosuccinic acid lyase deficiency is inherited as an autosomal recessive trait.
Argininosuccinate lyase deficiency may present as a severe neonatal-onset form and a late-onset form:
- Severe neonatal-onset form. The clinical presentation of the severe neonatal-onset form, which is indistinguishable from that of other urea cycle disorders, is characterized by hyperammonemia within the first few days after birth that can manifest as increasing lethargy, somnolence, refusal to feed, vomiting, tachypnea, and respiratory alkalosis. Newborns typically appear healthy for the first 24 hours but within the next few days develop vomiting, lethargy, and refusal to accept feeds 2. Tachypnea and respiratory alkalosis are early findings. Failure to recognize and treat the defect in ureagenesis leads to worsening lethargy, seizures, coma, and even death. The findings of hepatomegaly and trichorrhexis nodosa (coarse and friable hair) at this early stage are the only clinical findings that may suggest the diagnosis of argininosuccinate lyase deficiency 2.
- Late-onset form. In contrast to the neonatal-onset form, the manifestations of the late-onset form range from episodic hyperammonemia, triggered by acute infection, stress, or non-compliance with dietary and/or medication recommendations, to cognitive impairment, behavioral abnormalities, and/or learning disabilities in the absence of any documented episodes of hyperammonemia 2.
Argininosuccinate lyase deficiency usually becomes evident in the first few days of life. An infant with argininosuccinic aciduria may be lacking in energy (lethargic) or unwilling to eat, and have a poorly controlled breathing rate or body temperature. Some babies with this disorder experience seizures or unusual body movements, or go into a coma. Complications from argininosuccinate lyase deficiency may include developmental delay and intellectual disability. Progressive liver damage, high blood pressure (hypertension), skin lesions, and brittle hair may also be seen.
Occasionally, individuals may inherit a mild form of the disorder. These individuals can have an accumulation of ammonia in the bloodstream only during periods of illness or other stress, or mild intellectual disability or learning disabilities with no evidence of elevated ammonia levels.
Whereas manifestations secondary to hyperammonemia are common to all urea cycle disorders, many individuals with argininosuccinate lyase deficiency deficiency can present with a complex clinical phenotype. The incidence of (1) neurocognitive deficiencies; (2) hepatitis, cirrhosis; (3) trichorrhexis nodosa; and (4) systemic hypertension are overrepresented in individuals with argininosuccinate lyase deficiency deficiency 3. These manifestations may be unrelated to the severity or duration of hyperammonemic episodes 4.
Argininosuccinate lyase deficiency is a rare disorder that occurs in approximately 1 in 70,000 to 218,000 newborns, with fewer than a thousand people in the United States 5. Males and females are affected in equal numbers. Most cases of argininosuccinate lyase deficiency are detected shortly after birth by newborn screening. Onset of symptoms usually occurs at birth, but may not be noticeable for days or weeks. In some children, onset of symptoms may not occur until later during infancy or childhood.
Treatment involves rapid control of hyperammonemia during metabolic decompensations and long-term management to help prevent episodes of hyperammonemia and long-term complications.
Figure 1. The urea cycle
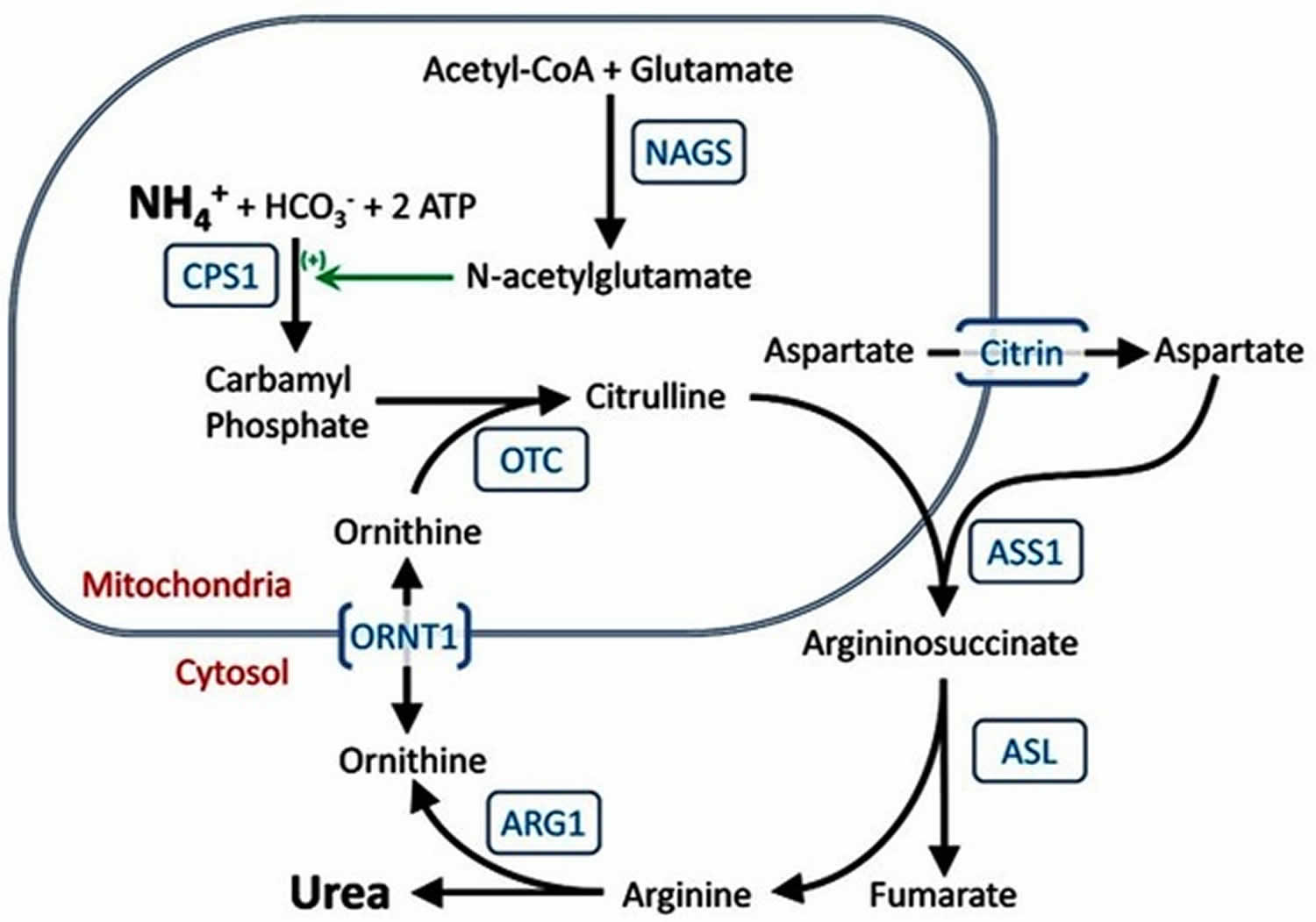
Argininosuccinate lyase deficiency causes
Mutations in the ASL gene cause argininosuccinate lyase deficiency. Argininosuccinate lyase deficiency belongs to a class of genetic diseases called urea cycle disorders because they are caused by problems with a process in the body called the urea cycle (Figure 1). The urea cycle is a sequence of reactions that occurs in liver cells. This cycle breaks down excess nitrogen, which is made when protein is used by the body, to make a compound called urea. Urea is removed from the body in urine. Breaking down excess nitrogen and excreting it as urea prevents it from accumulating in the body as ammonia.
The ASL gene provides instructions for making an enzyme called argininosuccinate lyase, which is needed for the fourth step of the urea cycle. The specific role of the argininosuccinate lyase enzyme is to start the reaction in which the amino acid arginine, a building block of proteins, is produced from argininosuccinate, the molecule that carries the waste nitrogen collected earlier in the urea cycle. The arginine is later broken down into urea, which is excreted, and ornithine, which restarts the urea cycle.
In people with argininosuccinate lyase deficiency, argininosuccinate lyase is dysfunctional or missing. As a result, the urea cycle cannot proceed normally, arginine is not produced, and nitrogen is not broken down efficiently. The excess nitrogen accumulates in the blood in the form of ammonia. This buildup of ammonia (hyperammonemia) damages the brain and other tissues and causes neurological problems and other signs and symptoms of argininosuccinate lyase deficiency. It is unclear how a lack of arginine contributes to the features of argininosuccinic acid lyase deficiency.
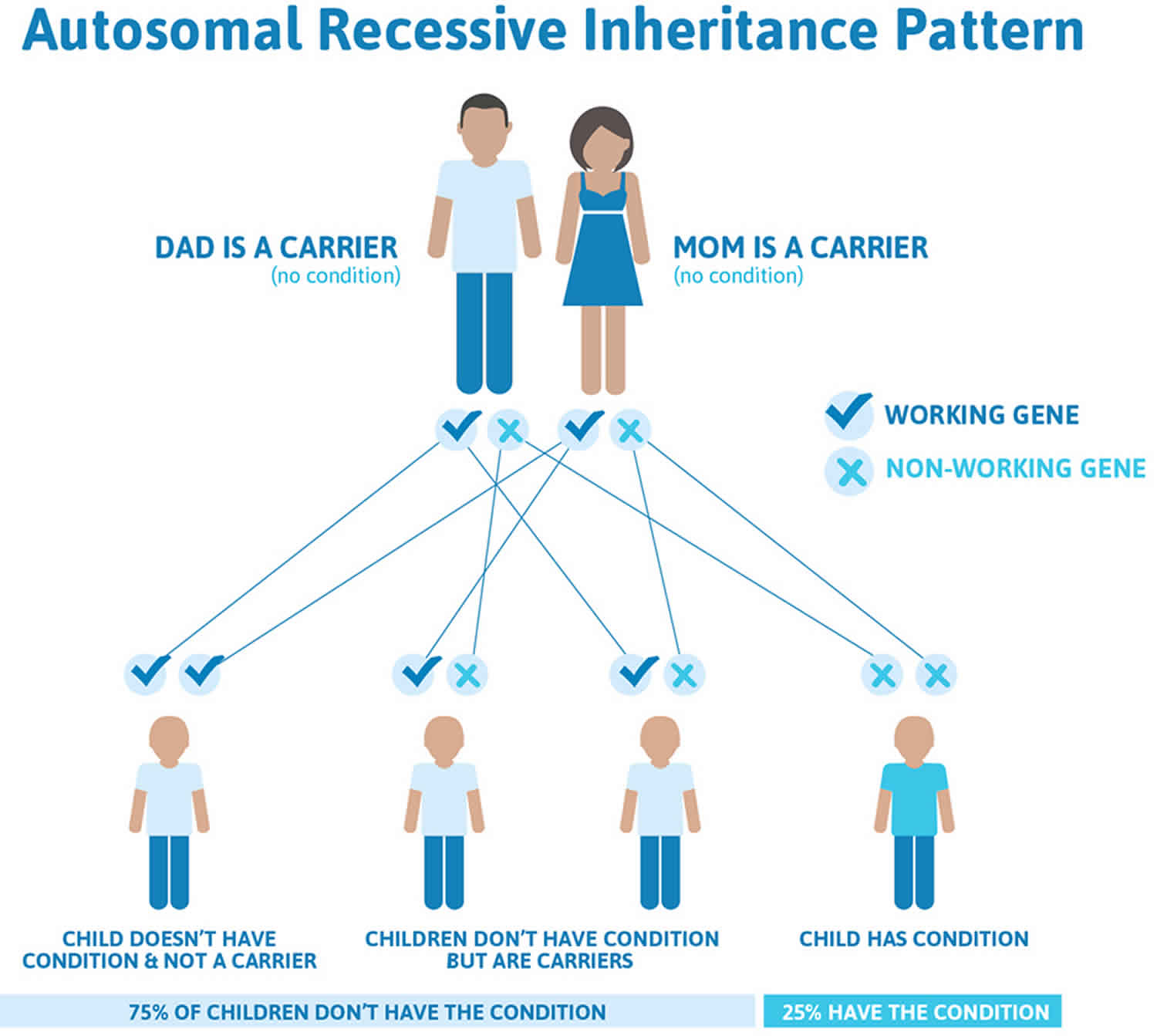
Argininosuccinate lyase deficiency inheritance pattern
Argininosuccinate lyase deficiency is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Argininosuccinate lyase deficiency autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Argininosuccinate lyase deficiency symptoms
The severity and specific symptoms of argininosuccinate lyase deficiency varies from one person to another 6. A severe form of the disorder, which is characterized by a complete or near complete lack of the ASL enzyme, occurs shortly after birth (neonatal period). A milder form of the disorder, which is characterized by partial lack of the ASL enzyme, affects some individuals later during infancy or childhood or even adulthood (late-onset form).
Symptoms are caused by the accumulation of ammonia in the blood. The severe form occurs within 24-72 hours after birth, usually following a protein feeding. This form is initially characterized by a refusal to eat, lethargy, lack of appetite, vomiting, and irritability. Affected infants may also experience seizures, breathing (respiratory) abnormalities, the accumulation of fluid in the brain (cerebral edema), and an abnormally large liver (hepatomegaly). Less commonly, some individuals develop progressive liver disease and dysfunction such as the buildup of scar tissue (fibrosis) and cirrhosis. In rare instances, chronic kidney (renal) disease has been reported. Abnormally rapid breathing (tachypnea) may be detected and sometimes is the first sign recognized of elevated ammonia in the blood. As affected individuals grow older, they may have coarse and brittle (friable) hair that breaks off easily and can leave patches of hair loss, a condition known as trichorrhexis nodosa.
In some instances, due to high levels of ammonia in the blood (hyperammonemic coma), the disorder may progress to coma. In such instances, argininosuccinate lyase deficiency may potentially result in neurological abnormalities including delays in reaching developmental milestones (developmental delays) and intellectual disability. The severity of such neurological abnormalities is more severe in infants who are in hyperammonemic coma for more than three days. If left untreated, the disorder will result in life-threatening complications. However, even individuals without significant hyperammonemia may develop neurological abnormalities suggesting alternative causes of injury.
In infants with partial enzyme deficiency, onset of the disorder may not occur until later during infancy or childhood (late onset form). Symptoms may include failure to grow and gain weight at the expected rate (failure to thrive), avoidance of protein from the diet, inability to coordinate voluntary movements (ataxia), lethargy, and vomiting. Affected infants and children may also have dry, brittle hair. Some individuals with the late onset form may not develop any symptoms (asymptomatic).
Individuals with argininosuccinate lyase deficiency may present with the following nonspecific supportive clinical features and preliminary laboratory findings that vary by age.
In the neonatal period
- Hyperammonemia can manifest as increasing lethargy, somnolence, refusal to feed, vomiting, tachypnea, and respiratory alkalosis.
- The presentation is typically indistinguishable from that of other proximal urea cycle disorders (i.e., carbamoyl-phosphate synthetase 1 deficiency, ornithine transcarbamylase deficiency, and citrullinemia type 1).
In individuals outside the neonatal period
- Episodic hyperammonemia that is triggered by acute infection, stress, or non-compliance with dietary restrictions or medications
- Liver involvement including hepatomegaly, elevated transaminases, liver fibrosis, or cirrhosis
- Neurocognitive deficits such as ADHD, developmental delay, learning disability, and seizures that may be independent of hyperammonemia
- Trichorrhexis nodosa consisting of coarse and brittle hair that breaks easily. See images.
- Hypertension that may occur in late childhood and adolescence, in the absence of secondary causes
- Hypokalemia of unknown etiology that may be chronic and secondary to excess urinary loss of potassium
Signs and symptoms of argininosuccinate lyase deficiency that appear to be unrelated to the severity or duration of hyperammonemic episodes 1:
- Neurocognitive deficiencies (attention-deficit/hyperactivity disorder, developmental delay, seizures, and learning disability)
- Liver disease (hepatitis, cirrhosis)
- Trichorrhexis nodosa (coarse brittle hair that breaks easily)
- Systemic hypertension
Infants with the mild form may alternate between periods of wellness and hyperammonemia. Episodes of hyperammonemia are usually triggered by acute infection, stress, certain medications, or non-compliance with the recommended dietary restrictions (e.g. high protein intake). Other individuals with the mild form may not have any documented episodes of hyperammonemia, but can still develop behavioral abnormalities such as attention deficit/hyperactivity disorder, cognitive impairment, and learning disabilities.
Both the severe and late-onset forms of argininosuccinate lyase deficiency can be associated with long-term complications including liver dysfunction, neurocognitive deficits such as cognitive impairment, seizures, brittle hair, and high blood pressure (hypertension). These long-term complications appear to be unrelated to the frequency, length or severity of episodes of hyperammonemia. Increasingly, high blood pressure has been diagnosed in children and adults with this condition. This may be due to an inability of the body to generate a chemical called nitric oxide.
Researchers have determined that argininosuccinate lyase deficiency is a more complex metabolic disorder than originally suspected. Affected individuals have developed some of the long-term complications described above (e.g. liver disease, hypertension, neurocognitive issues) despite not having any episodes of hyperammonemia and having an overall good metabolic profile. Researchers theorize that the deficient enzyme, argininosuccinate lyase, may have more roles in the body other than breaking down nitrogen (i.e. its role in the urea cycle) including the production of nitric oxide. More research is necessary to fully understand the complex, underlying mechanisms of argininosuccinate lyase deficiency.
Argininosuccinate lyase deficiency complications
Neurocognitive deficiencies. In a cross-sectional study of individuals with a urea cycle disorder, it was observed that persons with argininosuccinate lyase deficiency had a higher incidence of developmental delay and neurologic abnormalities than did individuals with ornithine transcarbamylase deficiency 7.
Individuals with argininosuccinate lyase deficiency also had an increased incidence of attention-deficit/hyperactivity disorder (ADHD), developmental delay (intellectual disability, behavioral abnormalities, and/or learning disability), and seizures compared to persons with all other urea cycle disorders 7. In a recent retrospective study, developmental delay and epilepsy were observed in 92% (48/52) and 42% (22/52) of individuals, respectively 8. Though neurocognitive deficits are common in argininosuccinate lyase deficiency, they are not universally present; many individuals with argininosuccinate lyase deficiency who are treated with protein restriction and supplemental arginine have normal cognition and development 4.
The increasing reliance on newborn screening programs for early diagnosis of argininosuccinate lyase deficiency allows the evaluation of early treatment on disease progression, especially in the late-onset form:
- Ficicioglu et al 4 reported the long-term outcome of 13 infants diagnosed between age four and six weeks by newborn screening programs. All had low argininosuccinate lyase enzyme activity; in spite of optimal therapy with protein restriction and arginine supplementation, four of 13 had learning disability, three had mild developmental delay, three had seizures, and six had an abnormal EEG including abnormal sharp irregular background activity, frequent bilateral paroxysms, and increased slow wave activity.
- In a separate cohort of 17 individuals with argininosuccinate lyase deficiency diagnosed by newborn screening in Austria, IQ was average or above average in 11 (65%), low average in five (29%), and in the mild intellectual disability range in one (6%). Four had an abnormal EEG without evidence of clinical seizures 9. The overall favorable outcomes in persons in this cohort may be attributable not only to early dietary and therapeutic interventions but also to the high proportion of persons with very mild disease.
Liver disease in individuals with argininosuccinate lyase deficiency also appears to be independent of the defect in ureagenesis. The spectrum of hepatic involvement ranges from hepatomegaly to elevations of liver enzymes to severe liver fibrosis 7. Liver involvement has been noted even in individuals treated with protein restriction and arginine supplementation who had not experienced significant hyperammonemia 9. In a recent retrospective study, hepatomegaly and elevated alanine aminotransferase (ALT) were observed in nearly half of individuals with argininosuccinate lyase deficiency 8. At present no biochemical or molecular features help predict liver dysfunction in people with argininosuccinate lyase deficiency. Given the potential direct toxicity of argininosuccinate on hepatocytes, lowering of the argininosuccinate levels in plasma (a reflection of its production by the liver) may have potential benefit 10.
Trichorrhexis nodosa is characterized by nodular swellings of the hair shaft accompanied by frayed fibers and loss of cuticle. About half of individuals with argininosuccinate lyase deficiency have an abnormality of the hair manifest as dull, brittle hair surrounded by areas of partial alopecia 11. Normal hair contains 10.5% arginine by weight; hair that is deficient in arginine as a result of argininosuccinate lyase deficiency is weak and tends to break. Thus, this clinical feature responds to arginine treatment.
Hypertension. Whereas there have only been anecdotal reports of hypertension in argininosuccinate lyase deficiency, preclinical data and systematic analysis of blood pressures from one controlled clinical trial have shown that argininosuccinate lyase deficiency can directly result in endothelial dysfunction and hypertension 3. Usually no secondary causes of hypertension are detected, suggesting that this finding is related to the tissue-autonomous loss of ASL in the vascular endothelium.
Electrolyte imbalances. Some individuals develop electrolyte imbalances such as hypokalemia. The hypokalemia is observed even in individuals who are not treated with sodium phenylbutyrate. The etiology is unclear; increased renal wasting has been suggested.
Argininosuccinate lyase deficiency diagnosis
A diagnosis of a urea cycle disorder, such as argininosuccinate lyase deficiency, should be considered in any newborn that has an undiagnosed illness characterized by vomiting, progressive lethargy, and irritability. All 50 states in the U.S. include argininosuccinate lyase deficiency in newborn screening programs.
A diagnosis of argininosuccinate lyase deficiency can be made through a detailed patient/family history, identification of characteristic findings, and a variety of specialized tests. Blood tests may reveal excessive amounts of ammonia in the blood, which is the main criterion for a diagnosis of urea cycles disorders including argininosuccinate lyase deficiency. Blood tests may also reveal high levels of an amino acid called citrulline. The typical range of citrulline at presentation is 100-300 µmol/L 2. The typical plasma levels of argininosuccinic acid are between 5 and 110 µmol/L 4. However, high levels of ammonia or citrulline in the blood may characterize other disorders such as the organic acidemias, congenital lactic acidosis, and fatty acid oxidation disorders and are also present in other urea cycle disorders.
A diagnosis can be confirmed by identifying elevated levels of argininosuccinic acid in blood or urine samples. A diagnosis can also be confirmed by molecular genetic testing, which detects the gene alteration that causes the disorder.
Plasma ammonia concentration
- In the severe forms of argininosuccinate lyase deficiency, the initial plasma ammonia concentration (before treatment) may be greater than 1,000 µmol/L, though typically elevations are in the ranges of few hundred µmol/L.
- In the milder neonatal and late-onset forms of argininosuccinate lyase deficiency, the elevations of plasma ammonia concentration may be less pronounced but above the upper limits of normal for age (see Table 1).
Table 1. Upper limits of normal plasma ammonia concentration by age
| Age | Upper Limits of Normal Ammonia Concentration (µmol/L) 1 |
|---|---|
| 0-7 days | 94 |
| 8-30 days | 80 |
| 1-12 months | 47 |
| 1-15 years | 48 |
| >16 years | 26 |
Footnote: 1. The values depicted are only representative of the normal ranges; the normal reference ranges of individual laboratories should be used for clinical interpretation.
Table 2. Age-related plasma amino acid concentrations in argininosuccinate lyase deficiency
| Metabolite | Normal Plasma Levels Age <2 Years (µmol/L) 1 | Normal Plasma Levels Age 2-18 Years (µmol/L) 1 | In argininosuccinate lyase deficiency |
|---|---|---|---|
| Citrulline | 2-41 | 6-38 | Elevated |
| Argininosuccinic acid | 0-1 | 0-1 | Elevated 2 |
| Arginine | 42-132 | 18-127 | Low to normal |
| Glycine | 104-344 | 92-346 | Normal to high |
| Glutamine | 238-842 | 266-746 | Normal to high |
| Alanine | 148-420 | 103-528 | Normal to high |
Footnotes:
1. The values depicted are only representative of the normal ranges; the normal references of individual laboratories should be used for clinical interpretation.
2. The argininosuccinate chromatographic peak may co-elute with leucine or isoleucine, resulting in an apparent increase in one of these two amino acids. The anhydrides that elute later in the run allow for the correct identification of argininosuccinate.
Urinary analysis
- Orotic acid excretion is typically normal (0.3-2.8 mmol/mol of creatinine); however, orotic aciduria may be observed 12.
- Argininosuccinic acid is significantly elevated. Urinary concentration of argininosuccinate is typically greater than 10,000 µmol/g of creatinine on urine amino acid analysis (normal range 0-1 µmol/L) 4..
Argininosuccinate lyase deficiency treatment
Treatment may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, geneticists, dieticians, and physicians who are familiar with metabolic disorders may need to work together to ensure a comprehensive approach to treatment. Occupational, speech language, and physical therapists may be needed to treat children with developmental disabilities. Genetic counseling is recommended for affected individuals and their families.
The treatment of argininosuccinate lyase deficiency is aimed at preventing excessive ammonia from being formed or from removing excessive ammonia during a hyperammonemic episode. Long-term therapy combines dietary restrictions and the stimulation of alternative methods of converting and excreting nitrogen from the body (alternative pathways therapy).
Dietary restrictions in individuals with argininosuccinate lyase deficiency are aimed at limiting the amount of protein intake to avoid the development of excess ammonia. However, enough protein must be taken in by an affected infant to ensure proper growth. Infants with argininosuccinate lyase deficiency are placed a low protein, high calorie diet supplemented by essential amino acids. A combination of a high biological value natural protein such as breast milk or cow’s milk formulate, an essential amino acid formula (e.g., UCD-1 Ross, or Cyclinex, Mead Johnson), and a calorie supplement without protein is often used (e.g., MJ80056, Mead Johnson).
Individuals with argininosuccinate lyase deficiency benefit from treatment with arginine, which helps to promote the excretion of nitrogen. Arginine supplementation has shown benefits in improving or reversing changes to the hair, but its impact on the long-term, chronic complications of the disorder are not fully understood. The dose of arginine is often higher than is used in other forms of urea cycle disorder and it is effective in decreasing ammonia in emergent situations of elevated ammonia. However, chronic treatment with high doses of arginine may contribute to liver disease as it produces higher levels of argininosuccinic acid. Therefore, in individuals with liver disease, lower doses should be considered for long term treatment. In this situation, other medications like alternative pathway therapies may be needed. Multiple vitamins and calcium supplements may also be used in the treatment of argininosuccinate lyase deficiency. Finally, because of decreased production of nitric oxide in patients with argininosuccinate lyase deficiency, the addition of low protein foods rich in nitrite may be helpful.
Prompt treatment is necessary when individuals have extremely high ammonia levels (severe hyperammonemic episode). Prompt treatment can avoid hyperammonemic coma and associated neurological symptoms. However, in some individuals, especially those with complete enzyme deficiency, prompt treatment will not prevent recurrent episodes of hyperammonemia and the potential development of serious complications.
In some instances, despite early treatment and good metabolic control, affected individuals may develop certain symptoms such as neurocognitive deficiencies, behavior issues such as ADHD, developmental disability and seizures.
In addition to dietary restrictions and supplements, individuals with argininosuccinate lyase deficiency are treated by medications that stimulate the removal of nitrogen from the body. These medications provide an alternative method to the urea cycle in converting and removing nitrogen waste. This is known as alternative pathway therapy or nitrogen scavenging therapy. This includes sodium benzoate, sodium phenylbutyrate, and glycerol triphenylbutyrate.
In 2013, the U.S. Food and Drug Administration (FDA) approved Ravicti (glycerol phenylbutyrate) for the chronic management of urea cycle disorders including argininosuccinate lyase deficiency in affected individuals age 2 years and older. Ravicti is a liquid therapy that helps to remove ammonia from the body. Ravicti is used in individuals who cannot management the disorder through a low-protein diet and dietary supplements alone.
In 1996, the FDA approved Buphenyl (sodium phenylbutyrate) for chronic management of urea cycle disorders including argininosuccinate lyase deficiency. Buphenyl is a powder therapy that helps to remove ammonia from the body. A generic form of Buphenyl is also now available.
Sodium benzoate is a powder that is not FDA approved for treatment urea cycle disorders, but it has been used in chronic treatment of urea cycle disorders. It is not believed to be as effective as Buphenyl or Ravicti based on theoretical considerations, though this has never been tested in patients.
In 2005, the FDA approved the use Ammonul (sodium benzoate and sodium phenylacetate) as an intravenous, rescue therapy for the prevention and treatment of hyperammonemia and associated disease of the brain (encephalopathy) in individuals with urea cycle disorders.
Aggressive treatment is needed in hyperammonemic episodes that have progressed to vomiting and increased lethargy. Affected individuals may be hospitalized and protein may be completely eliminated from the diet for 24 hours. Affected individuals may also receive treatment with intravenous administration of arginine and a combination of sodium benzoate and sodium phenylacetate. Non-protein calories may be also provided as glucose.
In individuals where there is no improvement or where hyperammonemic coma develops, the removal of wastes by filtering an affected individual’s blood through a machine (hemodialysis) may be necessary. Hemodialysis is also used to treat infants, children, and adults who are first diagnosed with argininosuccinate lyase deficiency during hyperammonemic coma.
In some individuals, a liver transplant may be recommended. This is an option of last resort for specific individuals who have progressive liver disease, experience recurrent medical crises and hospitalizations despite therapy, or who have a poor quality of life.
Acute hyperammonemic treatment
During acute hyperammonemic episodes severe enough to cause neurologic symptoms, the treatment includes the following (Table 3).
Table 3. Acute inpatient treatment in individuals with argininosuccinate lyase deficiency
| Manifestation/ Concern | Treatment | Consideration/Other |
|---|---|---|
| Acute hyperammonemic episodes | Discontinue oral protein intake. | |
| Supplement oral intake w/IV lipids, glucose, & insulin if needed (w/close monitoring of blood glucose) to promote anabolism. | ||
| IV nitrogen-scavenging therapy. A loading dose of 600 mg/kg L-arginine-hydrochloride & 250 mg/kg each of sodium benzoate & sodium phenylacetate in 25-35 mL/kg of 10% dextrose solution given intravenously over a 90-min period is recommended, followed by a sustained IV infusion of 600 mg/kg L-arginine-hydrochloride & 250 mg/kg each of sodium benzoate & sodium phenylacetate over a 24-hr period. | When available, plasma concentrations of ammonia-scavenging drugs should be monitored to avoid toxicity. In the absence of drug levels, a serum anion gap of >15 mEq/L & an anion gap that has risen >6 mEq/L could indicate drug accumulation & ↑risk for toxicity. | |
| Failure to decrease ammonia levels w/medical therapy | Prompt institution of hemodialysis |
|
Footnote: Inpatient emergency treatment should: (a) take place at the closest medical facility equipped to treat individuals with metabolic disorders, (b) be started without delay, and (c) be supervised by physicians and specialist dieticians at the responsible metabolic center, who should be contacted without delay.
Abbreviations: HCL = hydrochloride; IV = intravenous
Long-term management
Dietary restriction of protein and dietary supplementation with arginine are the mainstays of long-term management as detailed in Table 4.
Table 4. Routine daily treatment in individuals with argininosuccinate lyase deficiency
| Principle/ Manifestation | Treatment | Consideration/Other |
|---|---|---|
| Dietary restriction of protein | Lifelong dietary management is necessary & requires the services of a metabolic nutritionist. 1 |
|
| Arginine base supplementation | The doses of arginine base routinely recommended are 400-700 mg/kg/day in persons weighing ˂20 kg & 8.8-15.4 g/m2/day in those weighing >20 kg. The authors prefer to use a lower dose of arginine whenever possible, in the range of 100-250 mg/kg/day. |
|
| Oral nitrogen-scavenging therapy (an alternative pathway therapy in which sodium benzoate & phenyl butyrate stimulate the excretion of nitrogen in the form of hippuric acid & phenylacety-lglutamine, respectively) | The typical dose ranges 2 for the medications:
| Individuals who have had frequent metabolic decompensations or episodes of ↑ ammonia despite being on a protein-restricted diet & arginine base supplementation are candidates for oral nitrogen-scavenging therapy. |
| Orthotopic liver transplantation (OLT) | Recommended only in those w/recurrent hyperammonemia or metabolic decompensations that are resistant to conventional medical therapy, or in those who develop cirrhosis w/associated metabolic decompensations [Author, personal observations] | Orthotopic liver transplantation does not correct the arginine deficiency or elevation of argininosuccinic acid at the tissue level, two abnormalities thought to account for the long-term complications of argininosuccinate lyase deficiency. |
| Hypertension |
| |
| Hypokalemia | Electrolyte (potassium) supplementation is appropriate when indicated. | |
| Neurocognitive delay | Special educational services & therapies as needed |
Footnotes:
- Some of the correlations between compliance with the prescribed diet and outcome are contradictory. Although in some patients dietary therapy along with arginine supplementation have been shown to reverse the abnormalities of hair, to improve cognitive outcome, and to reverse abnormalities on EEG 4, in many dietary therapy has not been shown to influence the outcome of liver disease or cognitive impairment 9.
- The dose ranges depicted are those typically used in individuals with argininosuccinate lyase deficiency. The safety and efficacy of phenylbutyrate doses >20 g/day are not known. The dose of glycerol phenylbutyrate depicted is the recommended initial dose in phenylbutyrate-naïve patients. When switching from sodium phenylbutyrate, the total daily dosage of glycerol phenylbutyrate (mL) = total daily dosage of sodium phenylbutyrate (g) x 0.86. The maximal daily dose for benzoate is 12 grams. When prescribing doses in the upper ranges of the recommended dosing, toxicity should be monitored.
RDA = recommended daily allowance
Agents and circumstances to avoid
Avoid the following:
- Excess protein intake
- Large boluses of protein or amino acids
- Less than recommended intake of protein
- Prolonged fasting or starvation
- Obvious exposure to communicable diseases
- Valproic acid
- Intravenous steroids
- Hepatotoxic drugs in individuals with hepatic involvement
Surveillance
After diagnosis of argininosuccinate lyase deficiency, steps can be taken to anticipate the onset of a hyperammonemic episode. Affected individuals should receive periodic blood tests to determine the levels of ammonia in the blood. Detection of elevated levels of ammonia may allow treatment before clinical symptoms appear. Monitoring for complications such as high blood pressure, liver inflammation and fibrosis, and developmental delay should be closely monitored from the time of diagnosis.
Table 5. Recommended surveillance for individuals with argininosuccinate lyase deficiency
| Manifestation/ Concern | Evaluation | Frequency/Comment |
|---|---|---|
| Management of the disorder | Follow up in a metabolic clinic w/a qualified metabolic dietician & clinical biochemical geneticist | Laboratory & clinical monitoring frequency should depend on metabolic status of the individual. In general:
Monitor following changes in medical or dietary prescriptions. |
| Abnormal amino acid levels | Analysis of plasma amino acids to identify deficiency of essential amino acids as well as impending hyperammonemia 1 | |
| Hypertension | Measurement of blood pressure using the appropriate-sized cuff & plotting the centile values for age & stature | At each clinic visit |
| Abnormal liver function | Liver function tests (ALT, AST) | Every 6-12 months as required |
| Abnormal electrolytes | Serum electrolyte analysis | Every 1-2 yrs as required |
Footnote: ALT = alanine aminotransferase; AST = aspartate aminotransferase
1. Early signs of impending hyperammonemic episodes in older individuals include mood changes, headache, lethargy, nausea, vomiting, refusal to feed, ankle clonus, and elevated plasma concentrations of glutamine, alanine, and glycine. Plasma glutamine concentration may rise 48 hours in advance of increases in plasma ammonia concentration in such individuals.
Argininosuccinate lyase deficiency prognosis
Argininosuccinate lyase deficiency prognosis is guarded. Although intellectual impairment is the rule, even among patients who receive excellent and timely treatment, some patients with argininosuccinate lyase deficiency reportedly develop normally 13.
References- Nagamani SCS, Erez A, Lee B. Argininosuccinate Lyase Deficiency. 2011 Feb 3 [Updated 2019 Mar 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK51784
- Brusilow S, Horwich A. Urea cycle enzymes. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. Chapter 85. New York: McGraw-Hill; 2001:1909-63.
- Kho J, Tian X, Wong WT, Bertin T, Jiang MM, Chen S, Jin Z, Shchelochkov OA, Burrage LC, Reddy AK, Jiang H, Abo-Zahrah R, Ma S, Zhang P, Bissig KD, Kim JJ, Devaraj S, Rodney GG, Erez A, Bryan NS, Nagamani SCS, Lee BH. Argininosuccinate lyase deficiency causes an endothelial-dependent form of hypertension. Am J Hum Genet. 2018;103:276–87.
- Ficicioglu C, Mandell R, Shih VE. Argininosuccinate lyase deficiency: longterm outcome of 13 patients detected by newborn screening. Mol Genet Metab. 2009;98:273–7.
- Argininosuccinic aciduria. https://ghr.nlm.nih.gov/condition/argininosuccinic-aciduria
- Argininosuccinic Aciduria. https://rarediseases.org/rare-diseases/argininosuccinic-aciduria/
- Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, Kerr DS, Diaz GA, Seashore MR, Lee HS, McCarter RJ, Krischer JP, Batshaw ML, et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397–402.
- Baruteau J, Jameson E, Morris AA, Chakrapani A, Santra S, Vijay S, Kocadag H, Beesley CE, Grunewald S, Murphy E, Cleary M, Mundy H, Abulhoul L, Broomfield A, Lachmann R, Rahman Y, Robinson PH, MacPherson L, Foster K, Chong WK, Ridout DA, Bounford KM, Waddington SN, Mills PB, Gissen P, Davison JE. Expanding the phenotype in argininosuccinic aciduria: need for new therapies. J Inherit Metab Dis. 2017;40:357–68.
- Mercimek-Mahmutoglu S, Moeslinger D, Häberle J, Engel K, Herle M, Strobl MW, Scheibenreiter S, Muehl A, Stöckler-Ipsiroglu S. Long-term outcome of patients with argininosuccinate lyase deficiency diagnosed by newborn screening in Austria. Mol Genet Metab. 2010;100:24–8.
- Nagamani SC, Shchelochkov OA, Mullins MA, Carter S, Lanpher BC, Sun Q, Kleppe S, Erez A, O’Brian Smith E, Marini JC, Lee B, et al. A randomized controlled trial to evaluate the effects of high-dose versus low-dose of arginine therapy on hepatic function tests in argininosuccinic aciduria. Mol Genet Metab. 2012c;107:315–21.
- Fichtel JC, Richards JA, Davis LS. Trichorrhexis nodosa secondary to argininosuccinicaciduria. Pediatr Dermatol. 2007;24:25–7.
- Brosnan ME, Brosnan JT. Orotic acid excretion and arginine metabolism. J Nutr. 2007;137:1656S–61S.
- Argininosuccinate Lyase (ASL) Deficiency. https://emedicine.medscape.com/article/950752-overview





{kind=link}