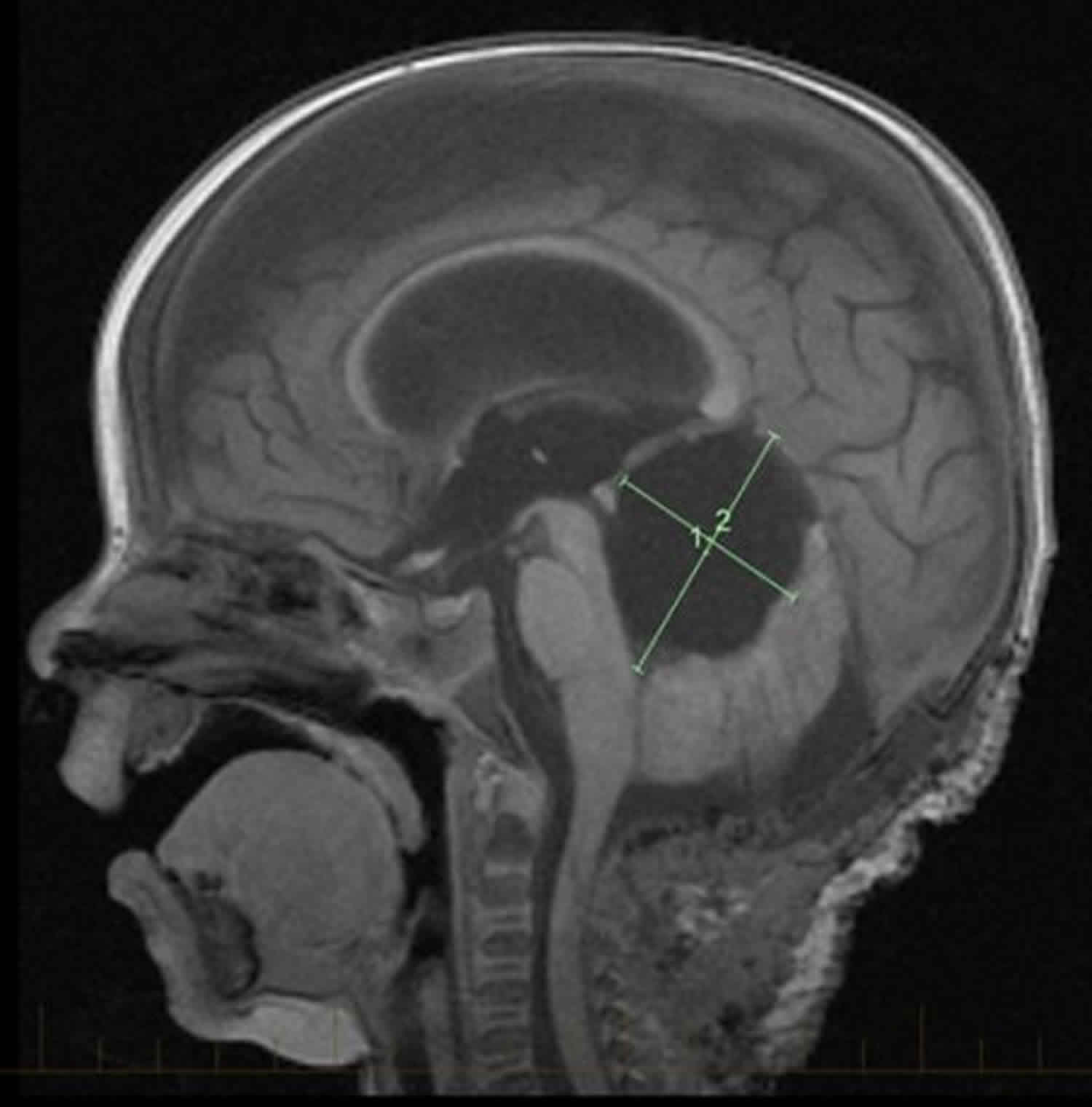
ATRT tumor
ATRT is short for atypical teratoid rhabdoid tumor, is a very rare fast-growing and aggressive embryonal tumor of the brain and spinal cord 1. ATRT cancer is very rare, accounting for about 1-2% of central nervous system (CNS) tumors in children. ATRT tumor most often occurs in young children aged three years and younger, although it can occur in older children and adults. Approximately 50% of embryonal tumors in infants under 1 year of age are ATRTs. There are about 75 new cases of ATRT each year in the United States.
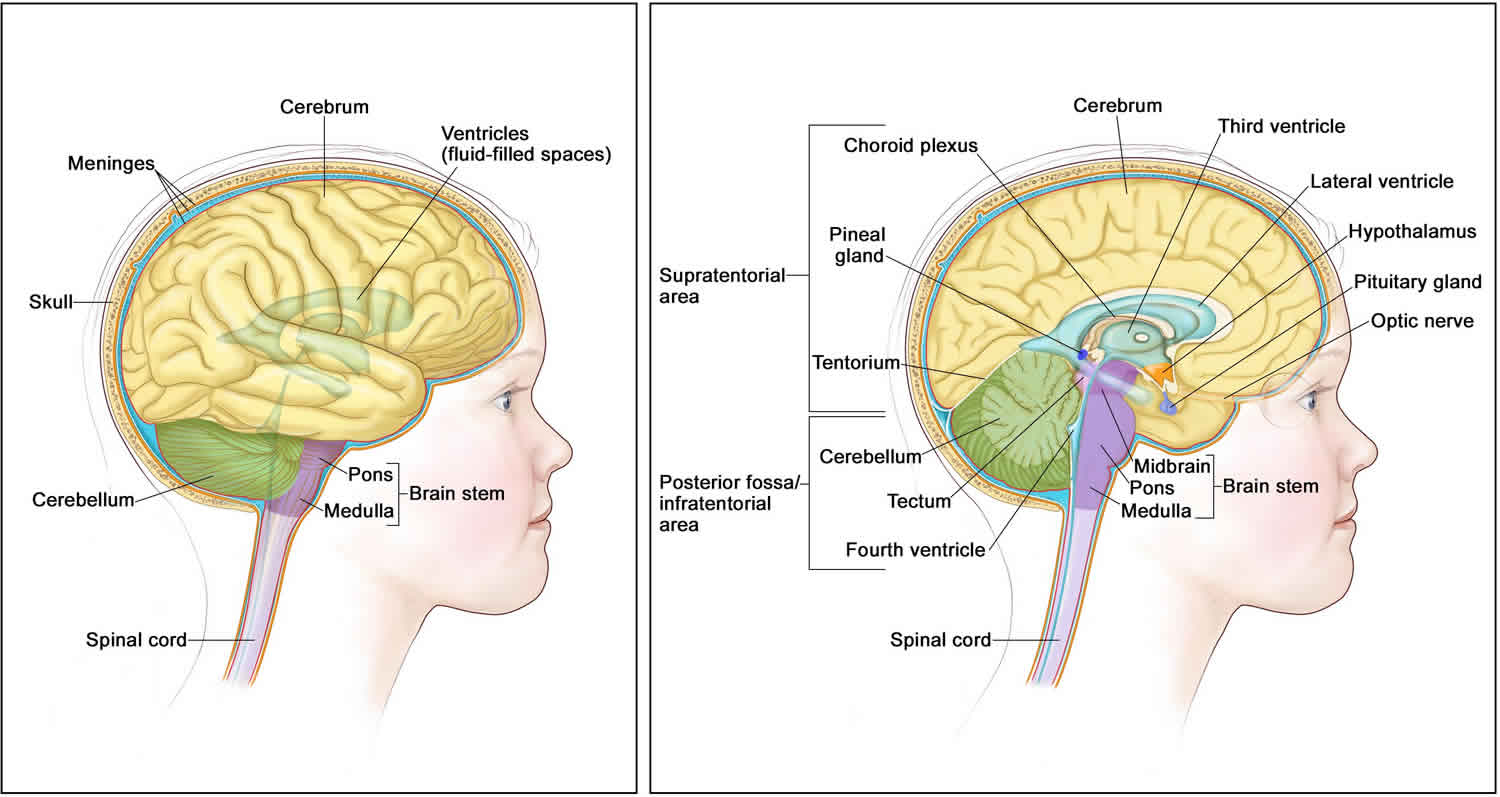
About half of ATRT tumors form in the cerebellum or brain stem. The cerebellum is the part of the brain that controls movement, balance, and posture. The brain stem controls breathing, heart rate, and the nerves and muscles used in seeing, hearing, walking, talking, and eating. ATRT tumor may also be found in other parts of the central nervous system (brain and spinal cord).
The exact incidence of childhood central nervous system (CNS) ATRT is difficult to determine because the tumor has only been recognized since 1996 2.
- In two North American prospective studies performed by the Children’s Cancer Group and the Pediatric Oncology Group for children aged 3 years or younger at diagnosis, retrospective review disclosed that approximately 10% of children with brain tumors had ATRTs 3.
- A Taiwanese study found that ATRTs account for 26% of primitive or embryonal tumors in children younger than 3 years 4.
- The Austrian Brain Tumor Registry (recruitment period, 1996–2006) confirmed that ATRTs represented the sixth most common malignant brain tumor among 311 newly diagnosed children (6.1%), with a peak incidence during the first 2 years of life 5.
The incidence in older patients is unknown. However, in the Central Nervous System Atypical Teratoid Rhabdoid Tumor Registry (ATRT Registry), 12 of the 42 patients (29%) were older than 36 months at the time of diagnosis 6.
ATRT treatment usually involves a combination of therapies which may include surgery, chemotherapy, and radiation therapy. Even with current treatments, ATRT cancer is a very difficult cancer to cure.
Treatment depends on the following:
- The age of the child.
- How much cancer remains after surgery to remove the tumor.
Results from the following procedure are also used to plan treatment:
- Ultrasound exam: A procedure in which high-energy sound waves (ultrasound) are bounced off internal tissues or organs, such as the kidney, and make echoes. The echoes form a picture of body tissues called a sonogram. The picture can be printed to be looked at later. This procedure is done to check for tumors that may also have formed in the kidney.
Figure 1. Brain anatomy
ATRT tumor causes
Certain genetic changes may increase the risk of atypical teratoid rhabdoid tumor.
Anything that increases the risk of getting a disease is called a risk factor. Having a risk factor does not mean that you will get cancer; not having risk factors doesn’t mean that you will not get cancer. Talk with your child’s doctor if you think your child may be at risk.
About 95% of ATRT tumor cases are related to a gene that does not function properly. ATRT tumor may be linked to changes in the tumor suppressor genes SMARCB1 (also known as INI1, SNF5, and BAF47 gene). In less than 5% of cases, ATRT tumors are due to a defect in another tumor suppressor gene called SMARCA4. Genes of this type make a protein that helps control cell growth. When the gene does not work, the protein is not made, and tumor growth is not controlled. Changes in the DNA of tumor suppressor genes like SMARCB1 or SMARCA4 may lead to cancer.
Changes in the SMARCB1 or SMARCA4 genes may be inherited (passed on from parents to offspring) or it may only occur in the cells of the tumor. When this gene change is inherited, tumors may form in two parts of the body at the same time (for example, in the brain and the kidney). Most of the time, the gene mutation is only in the cells of the tumor. However, all patients with ATRT, genetic counseling (a discussion with a trained professional about inherited diseases and a possible need for gene testing) may be recommended.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
ATRT tumor symptoms
Symptoms of ATRT vary widely based on the child’s age and tumor location. Because atypical teratoid rhabdoid tumor is fast growing, signs and symptoms may develop quickly and get worse over a period of days or weeks. Signs and symptoms may be caused by ATRT or by other conditions. Check with your child’s doctor if your child has any of the following:
- Headache, often worse in the morning and/or improves after vomiting
- Nausea and vomiting
- Changes in activity levels, lethargy
- Fatigue or unusual sleepiness or change in activity level
- Loss of balance, coordination or problems walking
- Asymmetric eye or face movements
- Change in behavior, irritability
- Increased head size in infants
- Increased fullness of the fontanel (“soft spot” at the top of the skull)
ATRT tumor diagnosis
Tests that examine the brain and spinal cord are used to detect (find) atypical teratoid rhabdoid tumor.
The following tests and procedures may be used:
- Physical exam and history: An exam of the body to check general signs of health, including checking for signs of disease, such as lumps or anything else that seems unusual. A history of the patient’s health habits and past illnesses and treatments will also be taken.
- Neurological exam: A series of questions and tests to check the brain, spinal cord, and nerve function. The exam checks a person’s mental status, coordination, and ability to walk normally, and how well the muscles, senses, and reflexes work. This may also be called a neuro exam or a neurologic exam.
- MRI (magnetic resonance imaging): A procedure that uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the brain and spinal cord. This procedure is also called nuclear magnetic resonance imaging (NMRI).
- Lumbar puncture: A procedure used to collect cerebrospinal fluid (CSF) from the spinal column. This is done by placing a needle between two bones in the spine and into the CSF around the spinal cord and removing a sample of fluid. The sample of CSF is checked under a microscope for signs of tumor cells. The sample may also be checked for the amounts of protein and glucose. A higher than normal amount of protein or lower than normal amount of glucose may be a sign of a tumor. This procedure is also called an LP or spinal tap.
- SMARCB1 and SMARCA4 gene testing: A laboratory test in which a sample of blood or tissue is tested for certain changes in the SMARCB1 and SMARCA4 genes.
- A biopsy is performed to diagnose ATRT. In a biopsy, a small sample of the tumor is removed during surgery using a needle. A pathologist looks at the tissue sample under a microscope to identify the specific type of tumor. The cells of ATRT look different from healthy cells. Cellular markers in the tissue are used to examine specific proteins in the cells. A specific antibody stain is used to check for the SMARCB1 or SMARCA4 protein in the cancer cells. ATRT cells lack this protein, which confirms the diagnosis of the tumor. If cancer cells are found, the doctor may remove as much tumor as safely possible during the same surgery. It is often difficult to completely remove ATRT because of where the tumor is in the brain and because it may already have spread at the time of diagnosis.
ATRT stages
There is no standard staging system for ATRT. Tumors are classified as newly diagnosed or recurrent. ATRT is an aggressive cancer. Approximately 15-30% of patients have spread of disease to the meninges or cerebrospinal fluid (CSF) at diagnosis. This spread of disease to the meninges is called leptomeningeal metastases.
ATRT tumor treatment
There are different types of treatment for patients with central nervous system atypical teratoid rhabdoid tumor. Treatment depends on several factors including the size and location of the tumor and the child’s age.
ATRT is a very aggressive cancer, and most patients will receive multiple types of treatments. ATRT treatment may include surgery, chemotherapy, and radiation. Treatment for ATRT is usually within a clinical trial. A treatment clinical trial is a research study meant to help improve current treatments or obtain information on new treatments for patients with cancer.
Children with atypical teratoid rhabdoid tumor should have their treatment planned by a team of health care providers who are experts in treating cancer in children.
Treatment will be overseen by a pediatric oncologist, a doctor who specializes in treating children with cancer. The pediatric oncologist works with other pediatric health care providers who are experts in treating children with central nervous system cancer and who specialize in certain areas of medicine. These may include the following specialists:
- Pediatrician.
- Pediatric neurosurgeon.
- Radiation oncologist.
- Neurologist.
- Pediatric nurse specialist.
- Rehabilitation specialist.
- Psychologist.
- Social worker.
- Geneticist or genetic counselor.
Four types of treatment are used:
Surgery
Surgery is used to both diagnose and treat central nervous system atypical teratoid rhabdoid tumor. If ATRT is confirmed with a biopsy, the surgeon will remove as much of the tumor as possible. Atypical teratoid rhabdoid tumors are often difficult to remove completely because of their location in the brain and how quickly they spread.
After the doctor removes all the cancer that can be seen at the time of the surgery, most patients will be given chemotherapy and possibly radiation therapy after surgery to kill any cancer cells that are left. Treatment given after the surgery, to lower the risk that the cancer will come back, is called adjuvant therapy.
Chemotherapy
Chemotherapy is a cancer treatment that uses drugs to stop the growth of cancer cells, either by killing the cells or by stopping them from dividing.
- When chemotherapy is placed directly into the cerebrospinal fluid, an organ, or a body cavity such as the abdomen, the drugs mainly affect tumor cells in those areas (regional chemotherapy). Regular doses of anticancer drugs given by mouth or vein to treat brain and spinal cord tumors cannot cross the blood-brain barrier and reach the tumor. Anticancer drugs injected into the cerebrospinal fluid are able to reach the tumor. This is called intrathecal chemotherapy.
- When chemotherapy is taken by mouth or injected into a vein or muscle, the drugs enter the bloodstream and can reach tumor cells throughout the body (systemic chemotherapy). High doses of some anticancer drugs given into a vein can cross the blood-brain barrier and reach the tumor.
Radiation therapy
Radiation therapy is a cancer treatment that uses high-energy x-rays or other types of radiation to kill cancer cells or keep them from growing. There are two types of radiation therapy:
- External radiation therapy uses a machine outside the body to send radiation toward the cancer.
- Internal radiation therapy uses a radioactive substance sealed in needles, seeds, wires, or catheters that are placed directly into or near the cancer.
The way the radiation therapy is given depends on the type of tumor being treated and whether it has spread. External radiation therapy may be given to the brain and spinal cord.
Because radiation therapy can affect growth and brain development in young children, especially children who are three years old or younger, the dose of radiation therapy may be lower than in older children.
High-dose chemotherapy with stem cell transplant
High doses of chemotherapy are given to kill cancer cells. Healthy cells, including blood-forming cells, are also destroyed by the cancer treatment. Stem cell transplant is a treatment to replace the blood-forming cells. Stem cells (immature blood cells) are removed from the blood or bone marrow of the patient or a donor and are frozen and stored. After the patient completes chemotherapy, the stored stem cells are thawed and given back to the patient through an infusion. These reinfused stem cells grow into (and restore) the body’s blood cells.
Targeted therapy
Children with ATRT are eligible for ongoing clinical trials using targeted therapy. Targeted therapy is a type of treatment that uses drugs or other substances to attack specific cancer cells. Targeted therapies include the aurora kinase A inhibitor, alisertib, and the EZH2 inhibitor, tazemetostat. Targeted therapies usually cause less harm to normal cells than chemotherapy or radiation therapy do. Targeted therapy is being studied in the treatment of recurrent childhood central nervous system atypical teratoid rhabdoid tumor.
Immunotherapy is also being studied in the treatment of ATRT.
For some patients, taking part in a clinical trial may be the best treatment choice. Clinical trials are part of the cancer research process. Clinical trials are done to find out if new cancer treatments are safe and effective or better than the standard treatment.
Many of today’s standard treatments for cancer are based on earlier clinical trials. Patients who take part in a clinical trial may receive the standard treatment or be among the first to receive a new treatment.
Patients who take part in clinical trials also help improve the way cancer will be treated in the future. Even when clinical trials do not lead to effective new treatments, they often answer important questions and help move research forward.
Some clinical trials only include patients who have not yet received treatment. Other trials test treatments for patients whose cancer has not gotten better. There are also clinical trials that test new ways to stop cancer from recurring (coming back) or reduce the side effects of cancer treatment.
Supportive therapy
Supportive care for ATRT patients includes appropriate rehabilitation and neurological consultation. Steroid and anti-seizure medications may be needed. Additional support may be needed to address issues in learning, developmental milestones, and coping with cancer.
Follow-up tests
Some of the tests that were done to diagnose ATRT cancer may be repeated. Some tests will be repeated in order to see how well the treatment is working. Decisions about whether to continue, change, or stop treatment may be based on the results of these tests.
Some of the tests will continue to be done from time to time after treatment has ended. The results of these tests can show if your child’s condition has changed or if the cancer has recurred (come back). These tests are sometimes called follow-up tests or check-ups.
Life after ATRT
Children with ATRT and germline SMARCB1 or SMARCA4 alterations have rhabdoid tumor predisposition syndrome. They and other family members should receive genetic testing and counseling since they are at an increased risk of developing other tumors. These children need additional monitoring including periodic ultrasound exams of the kidney to monitor for the development of kidney tumors.
Children treated for ATRT should be monitored for adverse effects of treatment. Late effects may include neurocognitive and endocrine problems due to radiation. Chemotherapy and radiation therapy also increase risk for second cancers. Families should talk to their doctors about risks related to the specific treatments children received.
Balancing quality of life with cancer-directed therapy is important. Families should talk to their care team about what problems to expect and ways to help manage them. Including palliative care or quality of life services can help families manage symptoms, navigate difficult conversations, and make decisions that align family wishes with goals of care.
ATRT survival rate
ATRTs are fast-growing and difficult to treat. Prognosis is usually poor, but advances in therapy have helped some children.
Certain factors affect prognosis (chance of recovery) and treatment options.
The prognosis (chance of recovery) and treatment options depend on the following:
- Whether there are certain inherited gene changes.
- Age of the child. Children under 3 years old have a poorer prognosis.
- How much of the tumor remains after surgery. Complete resection of the tumor improves chance of survival.
- Whether the cancer has spread to other parts of the central nervous system (brain and spinal cord) or to the kidney at the time of diagnosis.
The 5-year survival rate for children with ATRT is approximately 50%. However, this varies widely depending upon the age at diagnosis and the presence of metastases. Children less than 3-years of age with metastatic disease have the worst prognosis with less than 10% chance of long term cure.
Most published data on outcomes of patients with ATRT are from small series and are retrospective in nature. Initial retrospective studies reported an average survival from diagnosis of only about 12 months 7. In a retrospective report 7, 2-year overall survival was better for patients who underwent a gross-total resection than for those who had a subtotal resection. However, in this study, the effect of radiation therapy on survival was less clear.
There are reports of long-term survivors 8. Notably, improved survival has been reported for those receiving intensive multimodal therapy 9.
- Children aged 3 years and older with ATRT who received postoperative craniospinal irradiation and high-dose, alkylator-based chemotherapy had improved survival compared with patients younger than 3 years. In this report, the incidence of leptomeningeal metastases was also higher in the infant group of patients 10.
- In one prospective study of 25 children with ATRT who received intensive multimodal therapy, including radiation and intrathecal chemotherapy, the reported 2-year progression-free survival rate was 53%, and the overall survival rate was 70% 11.
- Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment (PDQ®)–Patient Version. https://www.cancer.gov/types/brain/patient/child-cns-atrt-treatment-pdq
- Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85 (1): 56-65, 1996.
- Packer RJ, Biegel JA, Blaney S, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24 (5): 337-42, 2002 Jun-Jul.
- Ho DM, Hsu CY, Wong TT, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99 (5): 482-8, 2000.
- Woehrer A, Slavc I, Waldhoer T, et al.: Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer 116 (24): 5725-32, 2010.
- Hilden JM, Meerbaum S, Burger P, et al.: Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22 (14): 2877-84, 2004.
- Lafay-Cousin L, Hawkins C, Carret AS, et al.: Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 48 (3): 353-9, 2012.
- Olson TA, Bayar E, Kosnik E, et al.: Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17 (1): 71-5, 1995.
- Bartelheim K, Nemes K, Seeringer A, et al.: Improved 6-year overall survival in AT/RT – results of the registry study Rhabdoid 2007. Cancer Med 5 (8): 1765-75, 2016.
- Tekautz TM, Fuller CE, Blaney S, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23 (7): 1491-9, 2005.
- Chi SN, Zimmerman MA, Yao X, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27 (3): 385-9, 2009.





{kind=link}