
What is Bartter syndrome
Bartter syndrome is a group of very similar but rare inherited kidney disorders that cause an imbalance of potassium, sodium, chloride, and related molecules in the body 1. Bartter syndrome is caused by impairment in the sodium-potassium-chloride cotransporter (NKCC2) or the apical potassium channel (ROMK) affect the transport of sodium, potassium, and chloride in the thick ascending limb of the loop of Henle 2. This results in increased distal delivery of these ions, where only some sodium is reabsorbed, and potassium is secreted.
Bartter syndrome is caused by a tubular defect in the kidney’s ability to reabsorb potassium and chloride. This makes the kidneys remove too much potassium and chloride from the body. Failure to reabsorb chloride results in failure to reabsorb sodium and leads to excessive sodium and chloride (salt) delivery to the distal tubules, leading to excessive salt and water loss from the body, resulting in hypokalemic alkalosis, hypercalciuria/nephrocalcinosis, increased levels of plasma renin and aldosterone, low blood pressure and vascular resistance to angiotensin 2 in some patients 3.
In some cases, Bartter syndrome becomes apparent before birth. Bartter syndrome can cause polyhydramnios, which is an increased volume of fluid surrounding the fetus (amniotic fluid). Polyhydramnios increases the risk of premature birth.
Beginning in infancy, affected individuals often fail to grow and gain weight at the expected rate (failure to thrive). They lose excess amounts of salt (sodium chloride) in their urine, which leads to dehydration, constipation, and increased urine production (polyuria). In addition, large amounts of calcium are lost through the urine (hypercalciuria), which can cause weakening of the bones (osteopenia). Some of the calcium is deposited in the kidneys as they are concentrating urine, leading to hardening of the kidney tissue (nephrocalcinosis). Bartter syndrome is also characterized by low levels of potassium in the blood (hypokalemia), which can result in muscle weakness, cramping, and fatigue. Rarely, affected children develop hearing loss caused by abnormalities in the inner ear (sensorineural deafness).
The exact prevalence of Bartter syndrome is unknown, although it likely affects about 1 per 1,000,000 people worldwide. Bartter syndrome appears to be more common in Costa Rica and Kuwait than in other populations.
Bartter syndrome is caused by mutations in any one of at least 5 genes and is usually inherited in an autosomal recessive manner 3. The different types of Bartter syndrome are classified according to the age of onset, severity, and the specific gene that causes the condition 1. Treatment depends on the type of the syndrome present but chiefly focuses on restoring and maintaining the proper balance of fluids and electrolytes in the body 4.
Types of Bartter’s syndrome:
- Bartter syndrome type 1 (Antenatal Bartter syndrome; Loop disorder type 1). Type 1 Bartter syndrome results from mutations in the sodium chloride/potassium chloride cotransporter gene (NKCC2) (SLC12A1 gene).
- Bartter syndrome type 2 (Antenatal Bartter syndrome; Loop disorder type 2). Type 2 Bartter syndrome results from mutations in the ROMK gene.
- Bartter syndrome type 3 (Classic Bartter syndrome; distal convoluted tube (DCT) disorder type 2). Type 3 Bartter syndrome results from mutations in the chloride channel gene (CLC-Kb), which encodes the CLC-Kb chloride channel involved in NaCl reabsorption in the renal tubule 5
- Bartter syndrome type 4A (Loop-distal convoluted tube (DCT) disorder type 1). This is a type of neonatal Bartter syndrome associated with sensorineural deafness and has been shown to be caused by mutations in the BSND gene 6. BSND encodes barttin, an essential beta subunit that is required for the trafficking of the chloride channel ClC-K (ClC-Ka and ClC-Kb) to the plasma membrane in the TALH and the marginal cells in the scala media of the inner ear that secrete potassium ion ̶ rich endolymph 7. Thus, loss-of-function mutations in barttin cause Bartter syndrome with sensorineural deafness. In contrast to other Bartter types, the underlying genetic defect in type IV is not directly in an ion-transporting protein. The defect instead indirectly interferes with the barttin-dependent insertion in the plasma membrane of chloride channel subunits ClC-Ka and ClC-Kb 8.
- Bartter syndrome type 4B (Loop-distal convoluted tube (DCT) disorder type 2). Type 4B Bartter syndrome is a recently renamed form. It is associated with sensorineural deafness but is not caused by mutations in the BSND gene.
- Type 5 Bartter syndrome also called Gitelman syndrome (Distal convoluted tube (DCT) disorder type 1). Type 5 Bartter syndrome results from mutations in extracellular calcium ion-sensing receptor and in the SLC12A3 genes that encode the chloride channel subunits, ClC-Ka and ClC-Kb.
- Most recently, an international team of researchers has identified an X-linked disorder characterized by polyhydramnios with prematurity and a severe but transient form of antenatal Bartter syndrome 9. The disorder results from mutations in MAGED2, a gene on the X chromosome that encodes melanoma-associated antigen D2 (MAGE-D2), which is essential for fetal renal salt reabsorption, amniotic fluid homeostasis, and the maintenance of pregnancy. In their study of 13 infants, four died perinatally and 11 survived; in the survivors, all symptoms disappeared spontaneously during follow-up.
Two major forms of Bartter syndrome are distinguished by their age of onset and severity. One form begins before birth (antenatal) and is often life-threatening. The other form, often called the classical form, begins in early childhood and tends to be less severe. Once the genetic causes of Bartter syndrome were identified, researchers also split the disorder into different types based on the genes involved. Types 1, 2, and 4 have the features of antenatal Bartter syndrome. Because type 4 is also associated with hearing loss, it is sometimes called antenatal Bartter syndrome with sensorineural deafness. Type 3 usually has the features of classical Bartter syndrome. An antenatal variant of Bartter syndrome presents with severe hypokalemia, metabolic alkalosis, and profound systemic manifestations. Bartter syndrome type 3 and 5 usually present later in life and have mild symptoms 10.
Bartter syndrome can be caused secondary to aminoglycoside use 10. Hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalcemia commonly are seen with an aminoglycoside-induced Bartter-like syndrome 10.
There is no cure for Bartter’s syndrome.
The treatments consist of supplements to replace what is lost and medications to prevent urinary wasting of potassium and magnesium. Bartter syndrome is treated by eating foods rich in potassium or taking potassium chloride supplements. Many people also need salt (sodium chloride) and magnesium supplements. Medicine such as potassium-sparing diuretics may be needed that blocks the kidney’s ability to get rid of potassium. In younger children growth hormone (GH) may be used to prevent the short stature and prostaglandin inhibitors to decrease the elevated prostaglandin levels. High doses of nonsteroidal anti-inflammatory drugs (NSAIDs) may also be used (e.g., indomethacin). In high-stress situations such as illness or trauma, blood electrolyte levels can change rapidly, which may require immediate intravenous treatment. Genetic counseling may benefit affected individuals and their families.
Is Bartter’s syndrome curable?
Bartter syndrome cannot be cured because it is an inherited disorder.
What is the long term outlook for people Bartter syndrome?
Currently there is no cure for Bartter syndrome, but treatments are available. Severity of symptoms (and associated complications) vary from person to person. People with Bartter syndrome must take medications consistently, as prescribed, throughout their lifetime. They also must be careful to maintain an adequate fluid and electrolyte balance. With treatment, prognosis in many cases is good 11. However, life expectancy and quality of life may be affected by complications such as growth delays, developmental problems, kidney failure and multiple hospitalizations 11.
Is genetic testing available for Bartter syndrome?
The Genetic Testing Registry (https://www.ncbi.nlm.nih.gov/gtr/all/conditions/?term=bartter%20syndrome) provides information about the genetic tests for this condition. The intended audience for the Genetic Testing Registry (https://www.ncbi.nlm.nih.gov/gtr/all/conditions/?term=bartter%20syndrome) is health care providers and researchers. Patients and consumers with specific questions about a genetic test should contact a health care provider or a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counsellors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
- The National Cancer Institute provides a Cancer Genetics Services Directory (https://www.cancer.gov/about-cancer/causes-prevention/genetics/directory), which lists professionals who provide services related to cancer genetics. You can search by type of cancer or syndrome, location, and/or provider name.
If you have a health condition that has not been diagnosed, you may be interested in the Undiagnosed Diseases Network (https://undiagnosed.hms.harvard.edu/). They have information about how to apply for this multicenter research study.
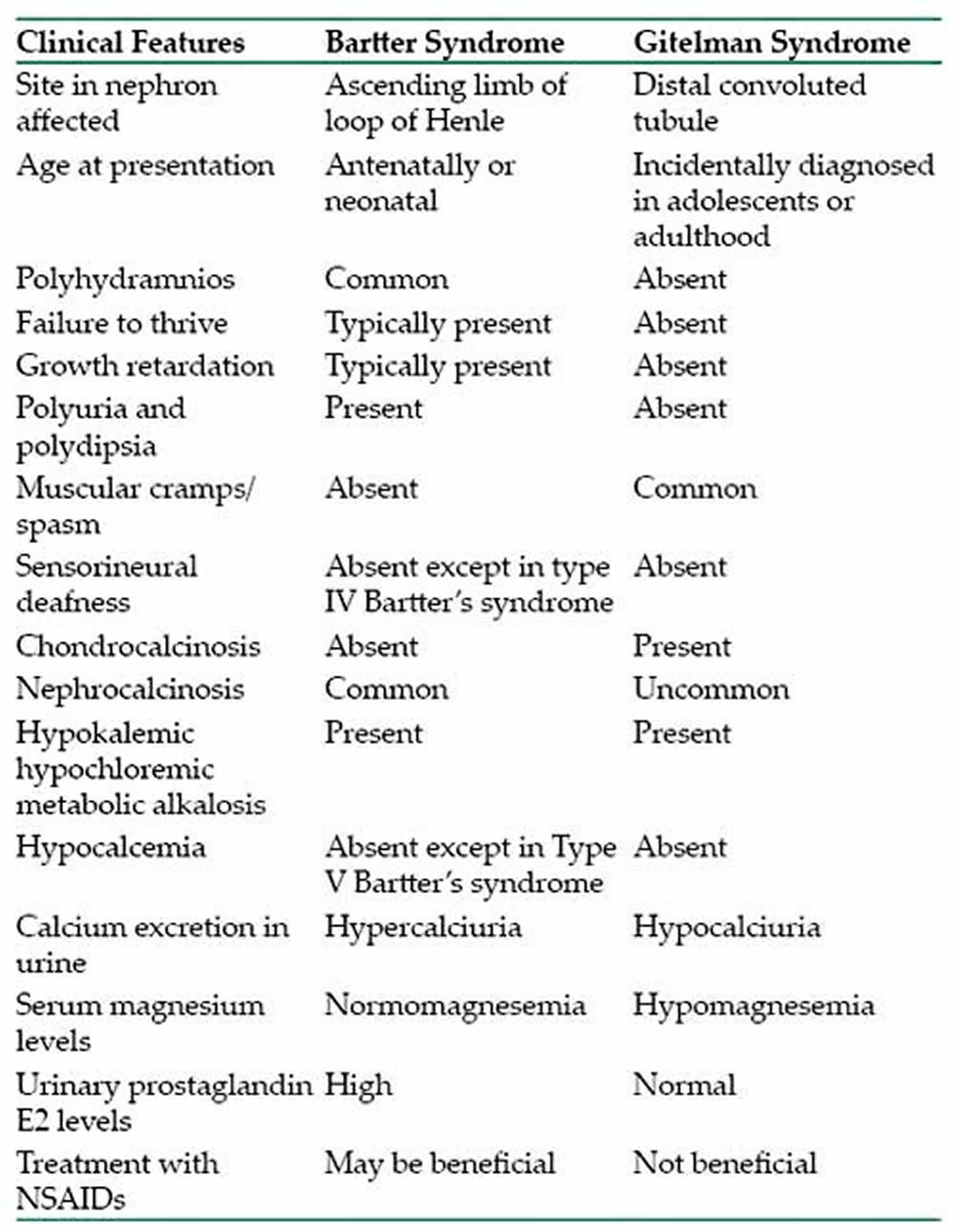
What conditions are similar to Bartter syndrome, also causing low potassium levels?
The condition that most resembles Bartter syndrome in individuals with the characteristic signs and symptoms is another genetic disorder called Gitelman syndrome, which is also sometimes called the hypocalciuric variant of Bartter syndrome (Bartter syndrome is typically characterized by hypercalciuria). In addition, individuals who chronically take certain types of diuretics, or individuals who have self-induced vomiting (such as bulimia), can have laboratory test results that are very similar to those seen in individuals with Bartter syndrome. When these circumstances cause the feautures of Bartter syndrome, it is sometimes referred to as pseudo-Bartter syndrome. There is also a large group of hormonal disorders that may present with hypokalemic alkalosis, however, unlike Bartter syndrome, these disorders are characterized by low renin levels and high blood pressure 12. Celiac disease is also part of the differential diagnosis 3.
General causes of potassium loss leading to low potassium levels in the blood (hypokalemia) may include chronic kidney failure, diabetic ketoacidosis, diarrhea, excessive sweating, excessive use of laxatives, prescription diuretic (water or fluid pills) use, primary aldosteronism, and vomiting.
Genetic conditions that predispose affected individuals to dehydration and failure to thrive (which occur in individuals with Bartter syndrome) may include nephrogenic diabetes insipidus, cystic fibrosis, pseudohypoaldosteronism, and congenital adrenal hyperplasia. However, laboratory test results usually make differentiating these disorders from Bartter syndrome easy 12.
Gitelman syndrome
Gitelman syndrome, also known as familial hypokalemia-hypomagnesemia, is a rare genetic disorder in which there is a specific defect in kidney function 13. Sometimes known as a variant of Bartter syndrome, Gitelman syndrome can show significant overlap with Bartter syndrome type 3; in specific cases, it is extremely difficult to distinguish between these disorders. Some researchers believe it is better to consider the Bartter syndrome and Gitelman syndrome as a spectrum of disease rather than distinct disorders. These disorders may be broadly classified as renal tubulopathies (because certain small tubes within the kidneys are affected), salt-wasting disorders (because affected individuals excrete excess amounts of salt), salt-losing tubulopathies or channelopathies (because the ion channels in the kidneys are affected). Gitelman syndrome causes metabolic abnormalities resembling treatment with high dosage of thiazide diuretics while Bartter syndrome resembles treatment with high dosage of loop diuretics.
This defect impairs the kidney’s ability to reabsorb salt and causes changes in various electrolyte concentrations as well as contraction of extracellular fluid volume (thus causing symptoms of dehydration). The electrolytes affected are primarily mineral ions, specifically potassium, calcium, magnesium, sodium, and chloride. Fundamentally, like Bartter’s syndrome, Gitelman syndrome is a salt wasting nephropathy. The symptoms and severity of the disorder can vary greatly from one person to another and can range from mild to severe. For unknown reasons, the onset of symptoms is frequently delayed until the second decade of life. Symptoms and severity can even vary greatly among members of the same family. Common symptoms can include episodes of fatigue, muscle weakness, and muscle cramps sometimes accompanied by gastrointestinal problems such as abdominal pain, nausea and vomiting. Some individuals may need to urinate frequently and will pass a large volume of urine (polyuria). This symptom is the result of failure to fully concentrate urine in the face of dehydration. Most cases of Gitelman syndrome are caused by mutations in the SLC12A3 gene and are inherited in an autosomal recessive manner.
Gitelman syndrome affects males and females in equally. The disorder occurs in approximately 1 in 40,000 Caucasian individuals. However, many cases of these disorders may go undiagnosed or misdiagnosed, making it difficult to determine the true frequency of Gitelman syndrome in the general population. The prevalence of individuals with one mutated copy of a gene (known as heterozygotes or carriers of the disease) is approximately 1% of European populations. These heterozygotes may enjoy a benefit of a small degree of salt wasting: they have lower blood pressures than the general population.
Most medical sources will use specific terminology to describe the electrolyte imbalances that characterize Gitelman syndrome. These terms refer to findings on laboratory tests rather than specific symptoms. Such terms include low levels of potassium in the blood (hypokalemia), low levels of chloride in the blood (hypochloremia), excess alkaline levels in the body (metabolic alkalosis), low levels of magnesium in the blood (hypomagnesemia), low levels of calcium in the urine (hypocalciuria), high levels of renin in the blood (hyperreninemia), and high levels of aldosterone in the blood (hyperaldosteronemia). The latter two laboratory findings are appropriate regulatory responses to dehydration caused by salt wasting kidney.
disease.
Gitelman syndrome causes
Most cases of Gitelman syndrome are caused by mutations in the SLC12A3 gene. In a minority of cases, mutations in the CLCNKB gene cause the disorder. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, absent, or not inserted in a tubule membrane properly. Depending upon the functions of the particular protein, this can affect many organ systems of the body. In the case of Gitelman syndrome, defective protein structure causes failure to reclaim filtered sodium and chloride (channelopathy). More severe salt wasting is caused by a defect in a different channel in Bartter syndrome.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
The SLC12A3 gene that causes the majority of cases of Gitelman syndrome produces (encodes) a protein known as thiazide-sensitive NaCl cotransporter, which helps to transport salts through ion channels in the kidney. Ion channels, which are pores in cell membranes, regulate the movement of electrically-charged particles called ions, which include electrolytes such as potassium and sodium ions, in certain structures of the kidneys. Mutations in this gene result in abnormal functioning of the NaCl cotransporter protein that transports electrolytes through the ion channels. This abnormal functioning or channel inception in the tubular membrane prevents sodium and chloride (salt) from being reabsorbed (reclaimed) from the distal renal tubule. This causes salt and water wastage (negative balance) and results in volume depletion (dehydration). The kidney attempts to attenuate dehydration by activating the renin angiotensin aldosterone system. Hypokalemia is the adverse consequence of renin angiotensin aldosterone system activation. Because salt balance can never be fully achieved; the hypokalemia in Gitelman syndrome can only rarely be corrected.
The human kidney filters 180 liters of serum each day through selective filtration in glomeruli. All but 1-1.5 liters of this glomerular filtrate is selectively reclaimed) by renal tubules including the distal convoluted tubule (which functions abnormally in Gitelman syndrome) and the thick ascending limb (which functions abnormally in Bartter syndrome). Both abnormalities cause salt wasting and, in turn, symptoms related to dehydration as well as those due to secondary electrolyte disturbances (hypokalemia and hypomagnesemia.)
Gitelman syndrome signs and symptoms
Gitelman syndrome usually becomes apparent anywhere from late childhood (usually over the age of six) to early adulthood. The disorder is highly variable, even among individuals in the same family. Some people do not develop any symptoms (asymptomatic), while others can develop chronic issues that can impact their quality of life.
Muscle weakness, spasms, and cramps may occur and generally are more common in Gitelman syndrome than the related Bartter syndrome. Affected individuals may experiences episodes of fatigue, dizziness, fainting (due to low blood pressure), muscle weakness, muscle aches, cramps and spasms. Affected individuals may also experience a specific form of cramping spasms called tetany. Tetany is marked by cramping spasms of certain muscles, particularly those of the hands and feet, arms, legs and/or face. Tetany may be provoked by hyperventilation during periods of anxiety.
Symptomatic episodes may also be accompanied by abdominal pain, vomiting, diarrhea or constipation, and fever. Vomiting or diarrhea in a patient with Gitelman syndrome may lead to the misdiagnosis of eating disorder or cathartic abuse as the cause of hypokalemia. Falsely accusing a patient with Gitelman or Bartter syndrome of these behaviors can cause loss of trust as well as adverse psychological and emotional consequences. Measurement of urinary chloride will help differentiate Gitelman syndrome (high urinary chloride) from hypokalemia resulting from GI fluid losses. (urine chloride < 10 meQ/L). Seizures may also occur and in some people may be the initial reason they seek medical assistance. A loss of sensation or feeling of the face (facial paresthesia) characterized by numbness or tingling is common. Less often, tingling or numbness may affect the hands. The severity of fatigue can vary widely. Some individuals are severely fatigued to the point where it interferes with daily activities; other individuals never report fatigue as a specific symptom.
Affected individuals may or may not experience excessive thirst (polydipsia) and a frequent need to urinate (polyuria) including the excessive need to urinate at night (nocturia). When these symptoms do occur they are usually mild. Blood pressure can be abnormally low (hypotension) in comparison to the general population. Affected individuals often crave salt or high-salt foods. Salt craving frequently begins in childhood and is helpful in making a correct diagnosis.
Some affected adults develop chondrocalcinosis, a condition characterized by the accumulation of calcium in the joints. Its development is thought to be related to hypomagnesemia. Affected joints may be swollen, tender, reddened, and warm to the touch. In some individuals, chondrocalcinosis and its complications are the only symptoms that develop.
Gitelman syndrome is generally considered to be a milder variant of Bartter syndrome, with symptoms often overlapping with Bartter syndrome type 3 (classic Bartter syndrome). Renal salt wasting is more severe and begins earlier in life in Bartter syndrome than in Gitelman syndrome. However, researchers have determined that in rare cases more severe complications can occur in the newborn (neonatal) period. In these cases, affected infants experience severe hypokalemia and hypomagnesemia, which can be associated with an increased need to urinate and passage of large amounts of urine (polyuria), diminished muscle tone (hypotonia), muscle spasms, growth delays and a failure to grow and gain weight as would be expected based on age and gender (failure to thrive). Earlier-onset, more severe cases have occurred in greater frequency in male infants than female infants.
In affected individuals who experience significant electrolyte imbalances, irregular heartbeats (cardiac arrhythmias) may develop. Although rare, if untreated, these cardiac arrhythmias can potentially progress to cause sudden cardiac arrest and potentially sudden death. These cardiac issues result from a prolonged QT interval. The QT-interval is measured on the electrocardiogram and, if prolonged, indicates that the heart muscle is taking longer than usual to recharge between beats.
Some affected individuals may develop the breakdown of muscle tissue causing the release of toxic content of muscle cells into the body fluids (rhabdomyolysis). Rhabdomyolysis is a serious condition that can potentially damage the kidneys. Additional symptoms have been reported in the medical literature but are quite rare. These symptoms include blurred vision, vertigo, and an impaired ability to coordinate voluntary movements (ataxia). A study from Yale indicated that Gitelman and Bartter syndrome have significant impact to quality of life 14.
In a study of a series of individuals with Gitelman syndrome 15, it was determined that affected individuals can develop abnormally high blood pressure (hypertension) later during life (median 55 years of age). This is counterintuitive in a salt-wasting disorder that can cause low blood pressure earlier in life. The exact reason for the development of hypertension is unknown, but may be related to prolonged exposure to renin and aldosterone levels and often occurs in the presence of traditional risk factors for hypertension. Some women have experienced severe potassium wasting during pregnancy and have required increased potassium and magnesium supplementation.
Gitelman syndrome diagnosis
A diagnosis of Gitelman syndrome is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. A diagnosis may be suspected after other more common causes of hypokalemia and metabolic acidosis are ruled out.
Clinical testing and workup
Laboratory tests that are used to diagnose Gitelman syndrome include blood tests to determine serum electrolyte levels, specifically low serum concentrations of magnesium and potassium and/or elevated serum concentrations of renin, and aldosterone. Urine electrolyte measurement seeks to determine the presence inappropriately high urine potassium in the face of hypokalemia. Low urine chloride should always suggest GI losses from vomiting and/or diarrhea. Low urinary calcium is comparable with a diagnosis of Gitelman syndrome. Hypertension in a hypokalemic patient who is not taking diuretics should always suggest primary hyperaldosteronism, not Gitelman or Bartter syndromes.
Molecular genetic testing can confirm a diagnosis of Gitelman syndrome. Genetic testing can detect mutations in the specific genes known to cause the disorder, but is available only as a diagnostic service at specialized laboratories. Generally, genetic testing is not needed to make a diagnosis.
Gitelman syndrome treatment
The treatment of Gitelman syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians or general internists, kidney specialists (nephrologists or pediatric nephrologists), cardiologists, social workers and other healthcare professionals may need to systematically and comprehensively plan individual’s treatment. Genetic counseling may be of benefit for affected individuals and their families. Because this is a rare disease, even well trained private practice or academic nephrologists may have little experience diagnosing or treating this disease.
Individuals who do not develop symptoms (asymptomatic) often do not require treatment, but it is recommended that they receive outpatient monitoring one or twice a year. They should be aware that they will be prone to rapidly become dehydrated should they experience vomiting or diarrhea from GI illness. They may require saline and intravenous potassium supplementation during these illnesses. All individuals with Gitelman syndrome are encouraged to follow a high-sodium chloride added diet. Dietary potassium should also be high. Dried fruit is an excellent source of supplemental potassium. Such a diet can help reduce exposure to potassium chloride supplements which irritate the stomach lining. These patents should never be treated with ACE inhibitors or angiotensin receptor blockers (ARBs).
There is no cure for Gitelman syndrome. The mainstay of treatment for affected individuals is a high salt diet with oral potassium and magnesium supplements. Potassium rich foods such as dried fruit are helpful. Magnesium supplements in single large doses cause diarrhea and should be avoided. Magnesium supplements should be taken in small frequent (4-6 times/ day) in order to avoid magnesium associated diarrhea which may worsen hypokalemia and symptoms of volume depletion. For many individuals, lifelong daily supplementation with magnesium is recommended. In some cases, during severe muscle cramps, magnesium has been given intravenously. In general, the goal of therapy should always be attenuation of symptoms rather than normalization of electrolyte abnormalities. Because of the risk of infection and thrombosis, central catheters should be discouraged.
Some affected individuals may receive medications known as potassium-sparing diuretics such as spironolactone, eplerenone or amiloride. These drugs are mild diuretics that spare potassium excretion. While these agents improve hypokalemia, they rarely normalize serum potassium concentrations. The goal of therapy is to improve symptoms not to normalize laboratory abnormalities. When chondrocalcinosis causes symptoms, supplementation with magnesium, pain medications and/or nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen may be beneficial.
A specific nonsteroidal anti-inflammatory drug (NSAID) known as indomethacin has been used to treat some infants and children with Gitelman syndrome. This drug is commonly used to treat individuals with Bartter syndrome, but is being used more often in Gitelman syndrome, particularly to treat growth deficiency in severe, early-onset forms of the disorder.
Affected individuals may be encouraged to undergo a cardiac workup to screen for risk factors for cardiac arrhythmias. Individuals with a prolonged QT interval should avoid drugs that prolong the QT interval. For a list of such drugs, contact the Sudden Arrhythmia Death Syndromes Foundation (https://www.sads.org/).
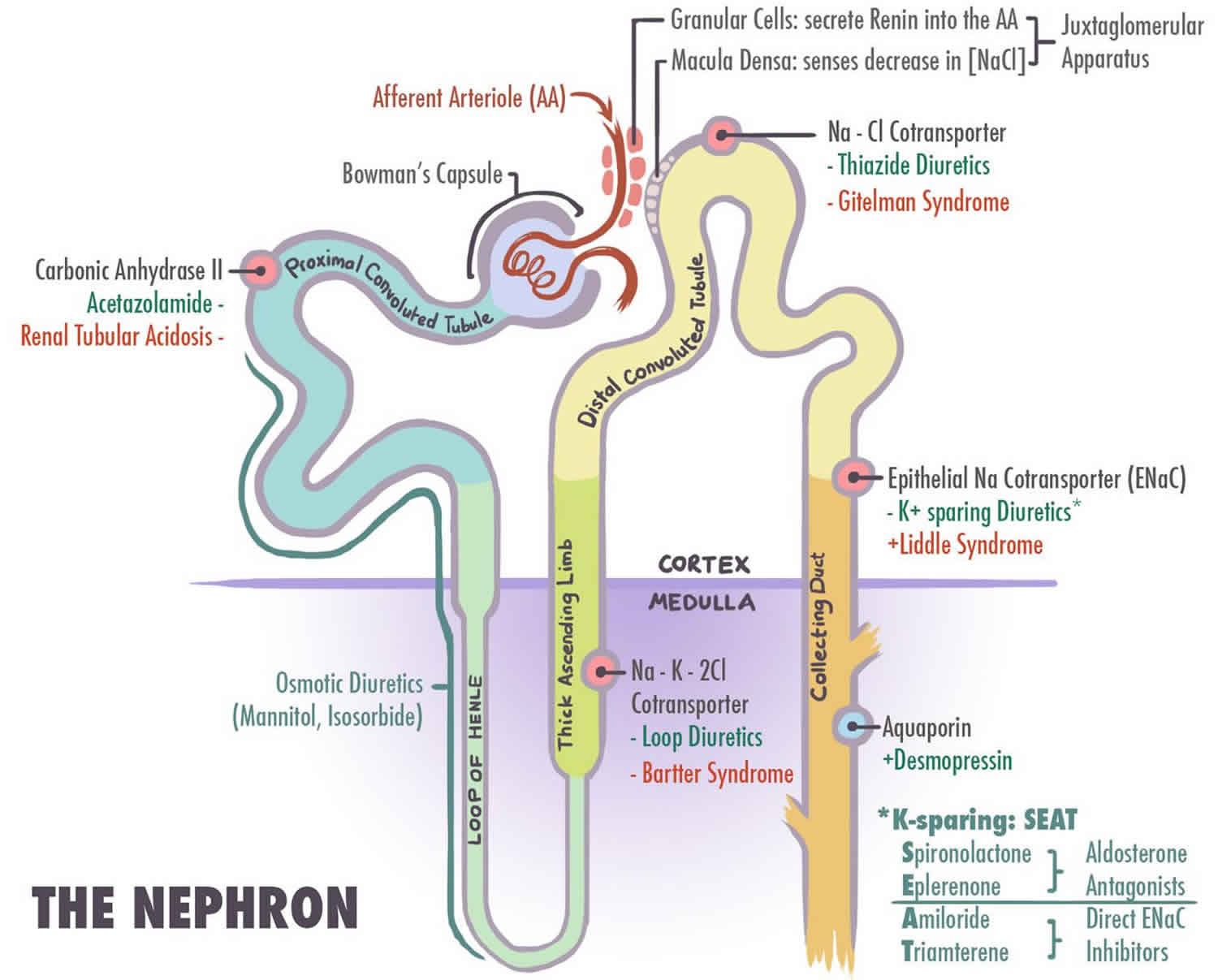
Bartter syndrome pathophysiology
Bartter syndrome is a renal tubular salt-wasting disorder in which the kidneys cannot reabsorb sodium and chloride in the thick ascending limb of the loop of Henle. This leads to increased distal delivery of salt and excessive salt and water loss from the body. Salt and water depletion due to an inability to conserve sodium in the thick ascending limb of the loop of Henle or the distal convoluted tubule (DCT) leads to activation of the renin-angiotensin-aldosterone system (RAAS) and subsequent secondary hyperaldosteronism. This helps the kidneys retain sodium distal to the site of the mutation, but at the expense of losing potassium. Long-term renin-angiotensin-aldosterone system (RAAS) stimulation causes hyperplasia of juxtaglomerular apparatus and hence increased renin levels.
Figure 1. Normal transport mechanisms in the thick ascending limb of the loop of Henle
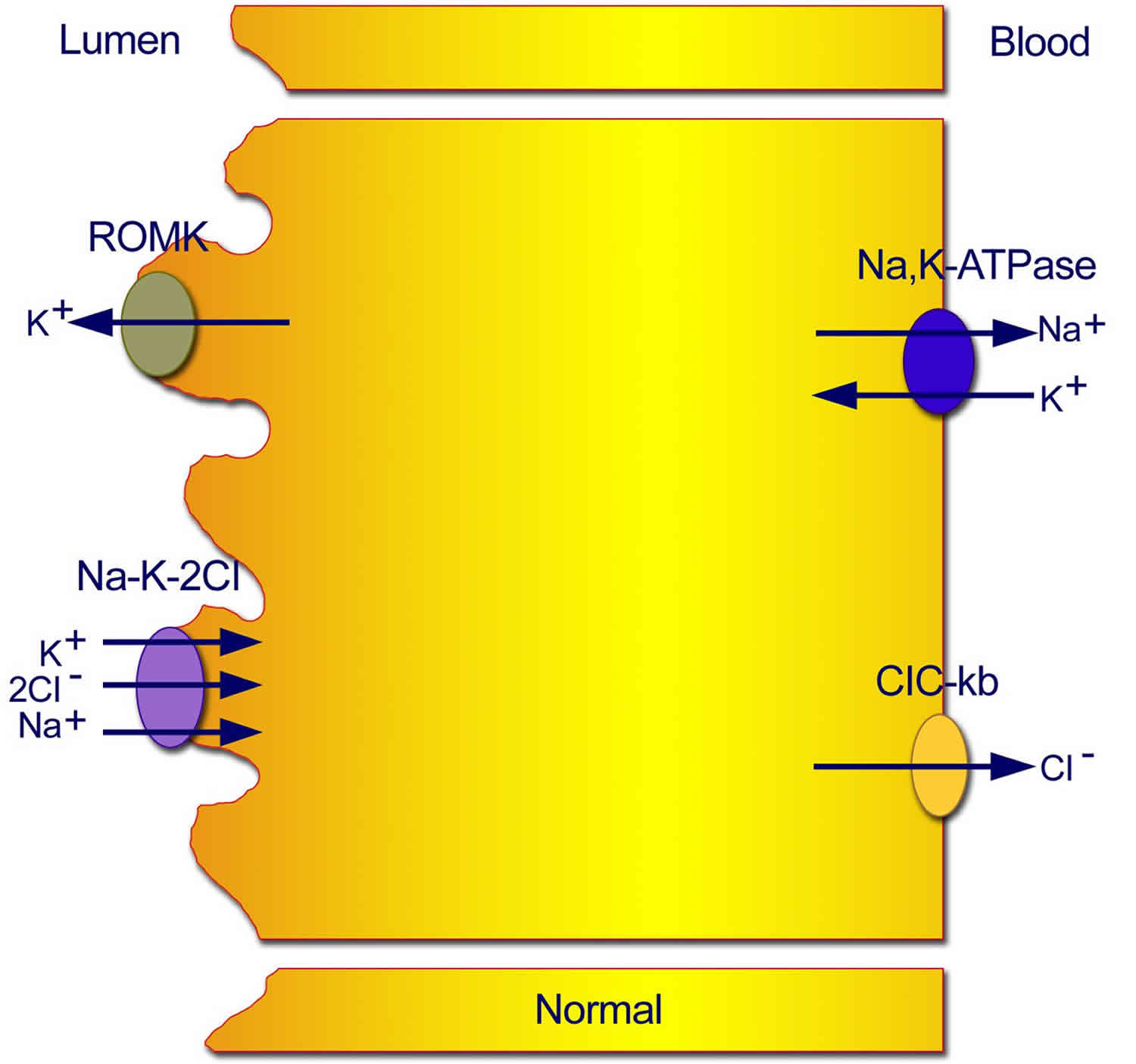
Footnote: Normal transport mechanisms in the thick ascending limb of the loop of Henle. Reabsorption of sodium chloride is achieved with the sodium chloride/potassium chloride cotransporter, which is driven by the low intracellular concentrations of sodium, chloride, and potassium. Low concentrations are maintained by the basolateral sodium pump (sodium-potassium adenosine triphosphatase), the basolateral chloride channel (ClC-kb), and the apical potassium channel (ROMK).
Figure 2. Type 1 Neonatal Bartter syndrome
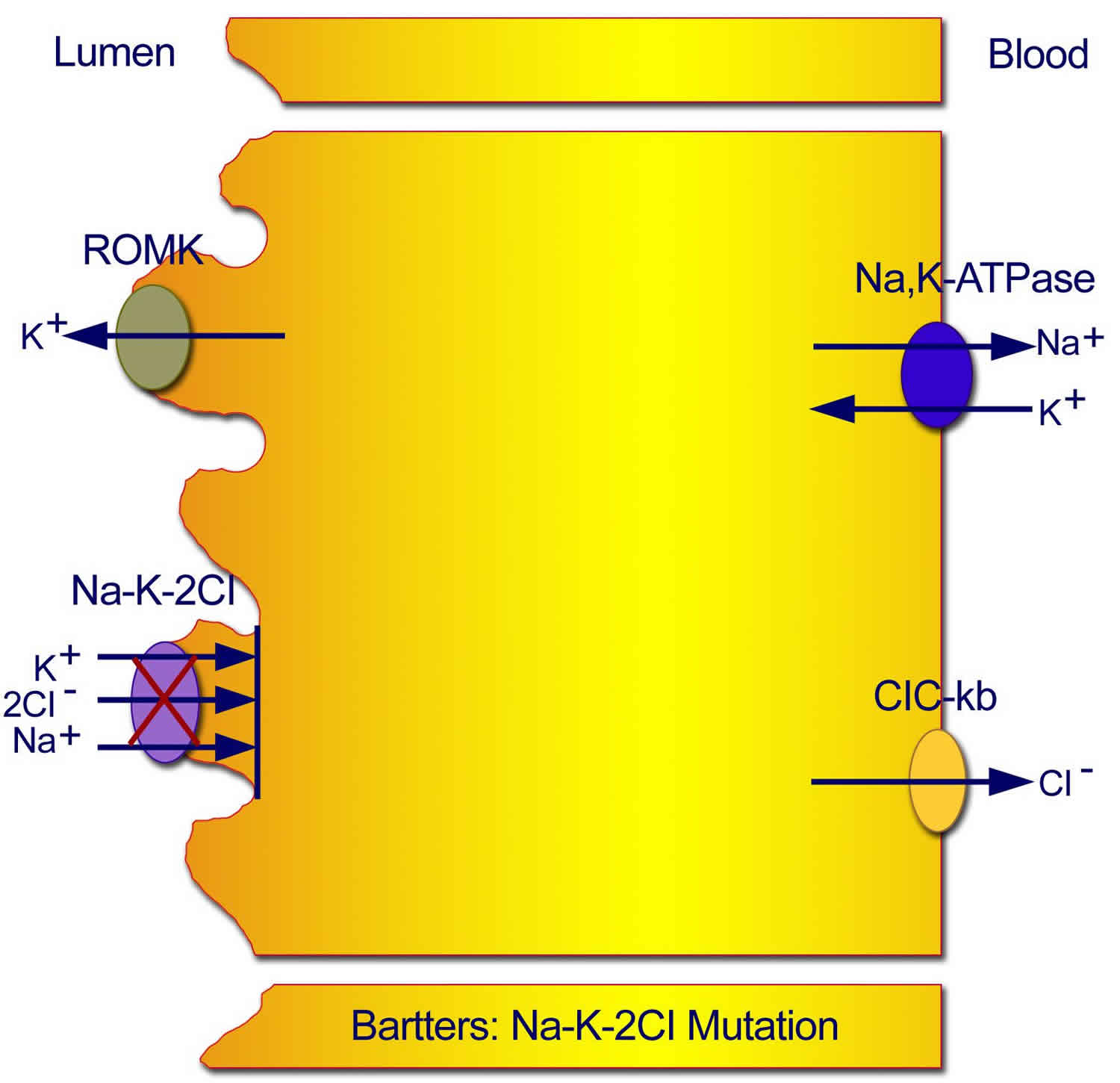
Footnote: Mutations in the sodium chloride/potassium chloride cotransporter gene result in defective reabsorption of sodium, chloride, and potassium.
Figure 3. Type 2 neonatal Bartter syndrome
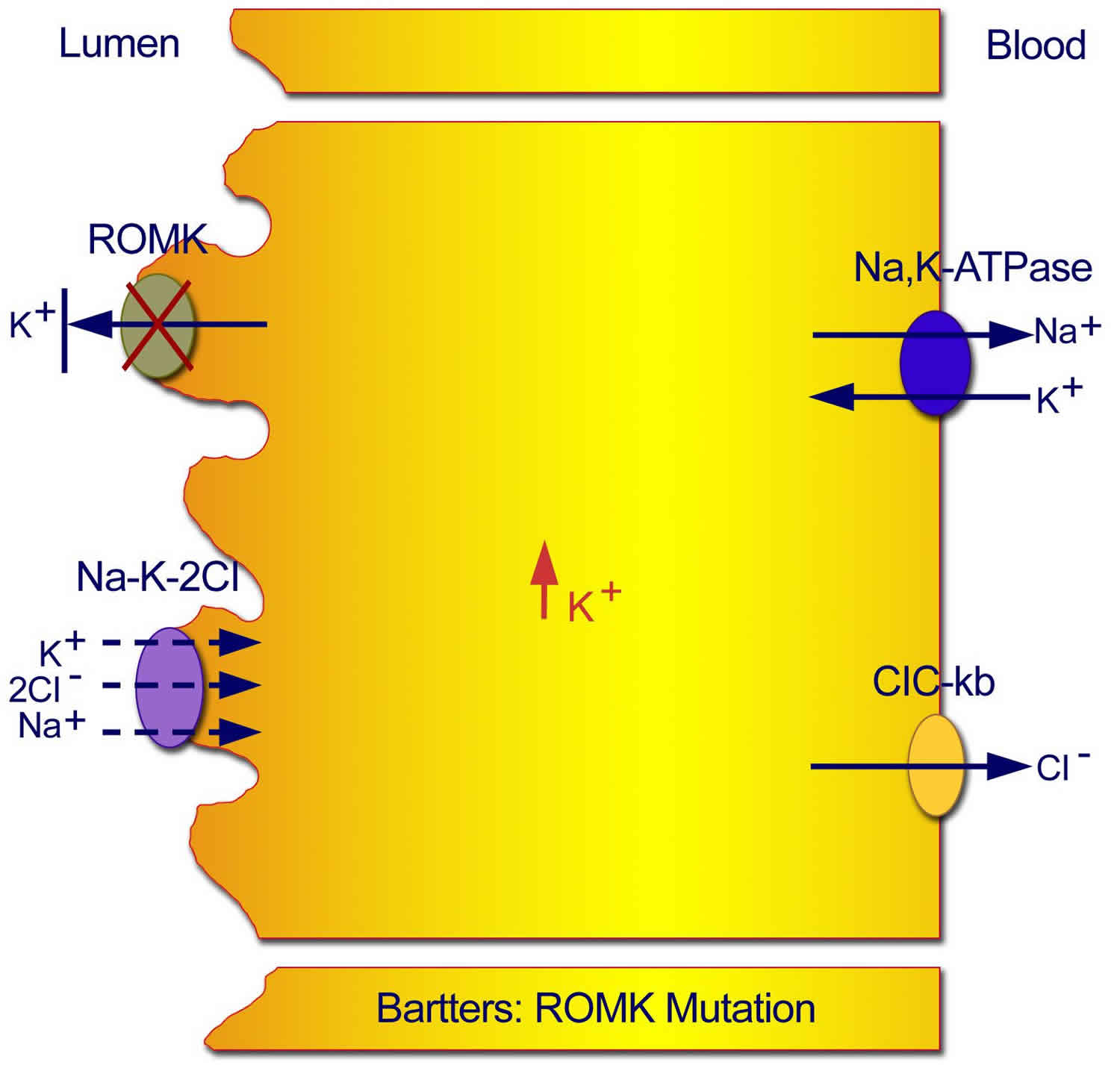
Footnote: Mutations in the ROMK gene result in an inability to recycle potassium from the cell back into the tubular lumen, with resultant inhibition of the sodium chloride/potassium chloride cotransporter.
Figure 4. Classic Bartter syndrome (Type 3 Bartter syndrome)
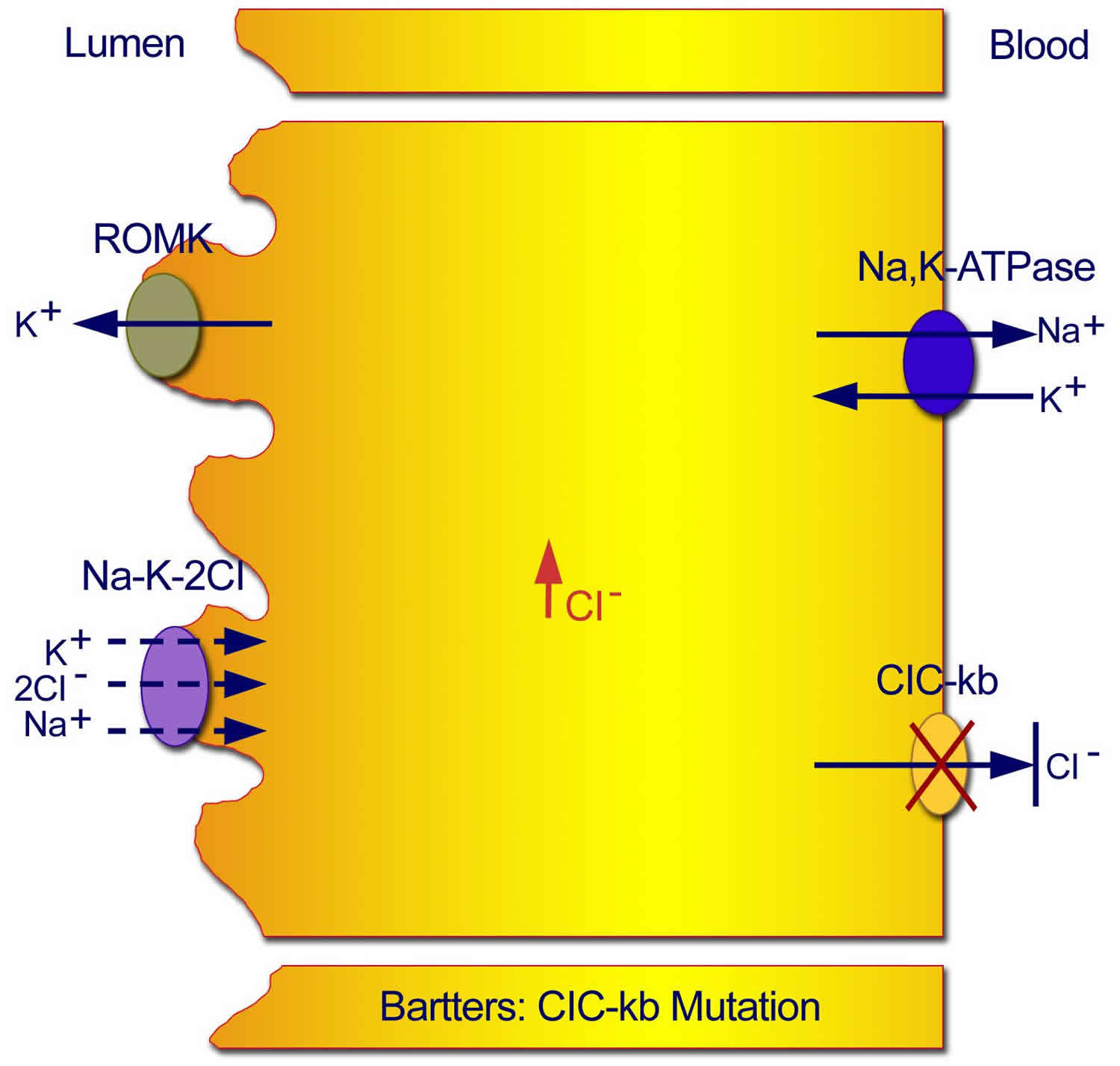
Footnote: Mutations in the CLC-kb chloride channel lead to an inability of chloride to exit the cell, with resultant inhibition of the sodium chloride/potassium chloride cotransporter.
Excessive distal delivery of sodium results in enhanced distal convoluted tubule sodium reabsorption and exchange with the negatively charged potassium or hydrogen ion and leads to increased loss of potassium in urine and increased hydrogen (H+) secretion. Decreased chloride reabsorption leads to a decreased exchange of bicarbonate for chloride, thus increased bicarbonate retention and hypokalemia result in metabolic alkalosis.
Urinary concentrating and diluting abilities are compromised in Bartter syndrome. Impaired urinary concentrating ability is secondary to defective sodium absorption in the loop of Henle. Under normal circumstances, salt absorption in the loop of Henle in the presence of normal ADH (antidiuretic hormone) is the main driving force for maintaining concentration gradient in medulla needed for concentrated urine formation. Other implicated factors include polyuria, hypokalemia, and elevated prostaglandin E2 levels. The defective sodium chloride transport in the loop of Henle associated with Bartter syndrome leads to the impaired electrochemical gradient, which is necessary for calcium and magnesium reabsorption, leading to increased urinary loss of calcium and magnesium.
Nephrocalcinosis commonly is seen in patients with Bartter syndrome. Likely explanation is secondary to excess calcium wasting in urine. Chloride transporters malfunction in the thick ascending limb of Loop of Henle, resulting in malabsorption of calcium in the thick ascending limb of Loop of Henle. Under normal conditions, calcium and magnesium are absorbed paracellularly under the influence of positive charge in lumen due to reabsorption of negatively charged chloride ions.
Bartter syndrome causes
Bartter syndrome can be caused by mutations in at least five genes. Mutations in the SLC12A1 gene cause type 1. Type 2 results from mutations in the KCNJ1 gene. Mutations in the CLCNKB gene are responsible for type 3. Type 4 can result from mutations in the BSND gene or from a combination of mutations in the CLCNKA and CLCNKB genes.
The genes associated with Bartter syndrome play important roles in normal kidney function. The proteins produced from these genes are involved in the kidneys’ reabsorption of salt. Mutations in any of the five genes impair the kidneys’ ability to reabsorb salt, leading to the loss of excess salt in the urine (salt wasting). Abnormalities of salt transport also affect the reabsorption of other charged atoms (ions), including potassium and calcium. The resulting imbalance of ions in the body leads to the major features of Bartter syndrome.
In some people with Bartter syndrome, the genetic cause of the disorder is unknown. Researchers are searching for additional genes that may be associated with this condition.
Inheritance pattern
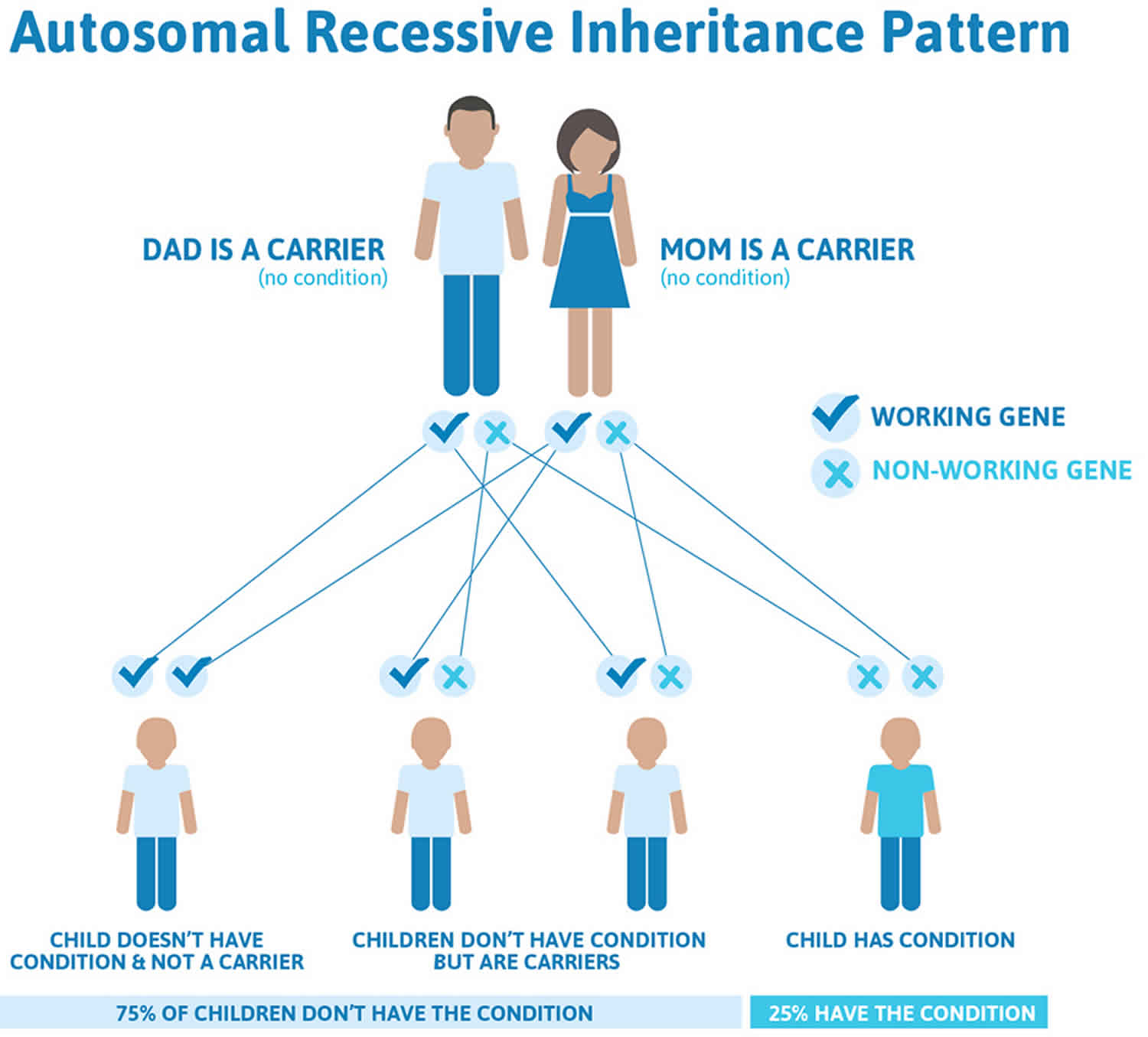
Bartter syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Both parents carry at least 1 gene for the disorder. Statistically, only 1 of 4 siblings will be completely healthy. Whether carrying 1 gene for this abnormality leads to long-term problems late in life if 1 kidney is removed is unknown.
Figure 5. Bartter syndrome autosomal recessive inheritance pattern
Bartter syndrome symptoms
The signs and symptoms associated with Bartter syndrome can vary depending on the form of Bartter syndrome an affected individual has 16. The antenatal forms (beginning before birth) can be life-threatening, while the classical form, beginning in early childhood, tends to be less severe. The age of onset, severity and specific symptoms associated with Bartter syndrome can vary greatly from one person to another, even among individuals who have the same subtype. Some individuals may have mild cases; others may experience severe, potentially life-threatening complications, at birth.
Bartter syndrome usually occurs in childhood. Bartter syndrome symptoms include:
- Constipation
- Rate of weight gain is much lower than that of other children of similar age and gender (growth failure)
- Needing to urinate more often than usual (urinary frequency or polyuria)
- Low blood pressure
- Kidney stones
- Muscle cramping and weakness
- Fatigue
- Polydipsia (Increased Thirst)
- Nocturia (Waking up at night to urinate)
- Generalized weakness
- Salt Cravings
- Dehydration
- Mental confusion
- Vomiting
- Muscle weakness
- Muscle spasms
- Tetany
- Failure to thrive
- Short stature (if untreated)
- Tetany, muscle spasms, Chvostek’s sign and Trousseau’s sign may be seen in hypokalemia, hypocalcemia, and hypomagnesemia patients.
The antenatal forms of Bartter syndrome (types 1, 2 and 4) may first be characterized by abnormally high levels of amniotic fluid surrounding the affected fetus (polyhydramnios); premature delivery; and possibly life-threatening salt (sodium-chloride) loss. Affected newborns may experience excessive urination (polyuria) and life-threatening episodes of fever and dehydration. Vomiting and diarrhea may also occur. Some affected infants have distinctive facial features (triangular face, prominent forehead, large eyes, protruding ears, and drooping mouth), failure to thrive, delayed growth, and/or intellectual disability. Individuals with type 4 may also have sensorineural deafness (hearing loss caused by abnormalities in the inner ear).
Classical Bartter syndrome typically becomes apparent in childhood and is characterized by muscle weakness, cramping, spasms, and fatigue. Excessive thirst (polydipsia), excessive urination and the need to urinate at night (nocturia) may also be present. This can lead to dehydration. Some children crave salt. Additional symptoms include constipation, vomiting, elevated body temperature, growth delay and developmental delay.
Some individuals with Bartter syndrome have significant electrolyte imbalances which can lead to irregular heartbeats (cardiac arrhythmias). This can increase the risk for sudden cardiac arrest. Another complication is excessive levels of calcium in the kidneys (nephrocalcinosis). This can lead to blood in the urine, vomiting, and fever. Over time, this calcium buildup can affect kidney function.
Sensorineural deafness
Sensorineural deafness associated with Bartter syndrome type 4 is due to defects in the barttin subunit of the ClC-Ka and CIC-Kb channels 7.
Nephrocalcinosis
A review of 61 cases of Bartter syndrome reported 29 with nephrocalcinosis, a condition that is often associated with hypercalciuria.
Bartter syndrome prognosis
Currently there is no cure for Bartter syndrome, but treatments are available. Severity of symptoms (and associated complications) vary from person to person. People with Bartter syndrome must take medications consistently, as prescribed, throughout their lifetime. They also must be careful to maintain an adequate fluid and electrolyte balance 16. With treatment, prognosis in many cases is good 17. However, life expectancy and quality of life may be affected by complications such as growth delays, developmental problems, kidney failure and multiple hospitalizations 17. Infants who have severe growth failure may grow normally with treatment. Over time, some people with the condition will develop kidney failure due to interstitial fibrosis 17.
Renal failure is fairly uncommon in Bartter syndrome. In a review of 63 patients, 5 developed progressive renal disease requiring dialysis or transplantation. In 2 reports of patients who underwent biopsies before developing end-stage renal disease, 1 patient had interstitial nephritis, and the other had mesangial and interstitial fibrosis. One report relates the case of a patient developing reversible acute renal failure from rhabdomyolysis due to hypokalemia.
Short stature/growth retardation
Nearly all patients with Bartter syndrome have growth retardation. In a review of 66 patients, 62 had growth retardation, often severe (below the fifth percentile for age). Treatment with potassium, indomethacin, and growth hormone (GH) has been effective.
Additional complications
Other complications in Bartter syndrome include the following:
- Cardiac arrhythmia and sudden death – May result from electrolyte imbalances
- Failure to thrive and developmental delay – Common in untreated patients
- Significant decrease in bone mineral density – Has been documented in patients with either the neonatal or classic form
Bartter syndrome diagnosis
A diagnosis of one of the Bartter syndromes is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests.
Bartter syndrome usually is seen in children and adolescents who also have stunted growth and complaints of polyuria, polydipsia, cramps, vomiting, dehydration, constipation, growth delays, and failure to thrive. A family history of nephrocalcinosis and detailed personal history ruling out the possibility of surreptitious vomiting and diuretic abuse should be practiced before making the diagnosis. Patients usually are emaciated with prominent forehead, large eyes, strabismus, protruding ears, sensorineural deafness, and drooping mouth. Normal or low blood pressures usually are recorded. Long-standing cases may present with elevated blood pressures.
Offspring with antenatal Bartter syndrome present with polyhydramnios secondary to intrauterine polyuria and usually are delivered prematurely. Fever, sensorineural deafness, profound polyuria, vomiting, and diarrhea leading to dehydration are common observations after birth.
Clinical Testing and Workup
Diagnosis is made by pertinent findings in the history and physical exam, potentiated with specific laboratory abnormalities.
Bartter syndrome is usually suspected when a blood test finds a low level of potassium in the blood. Unlike other forms of kidney disease, this condition does not cause high blood pressure. There is a tendency toward low blood pressure.
Laboratory tests may show:
- High levels of potassium, calcium, and chloride in the urine
- High levels of the hormones renin and aldosterone in the blood
- Low blood chloride
- Metabolic alkalosis
- Low serum potassium levels
- Normal-low serum magnesium levels (20% have decreased magnesium levels)
- Increased Prostaglandin E2 excretion
- Normal-high urinary magnesium excretion
- Normal – Low Blood Pressure
- Increased plasma angiotensin 2
- Nephrocalcinosis
These same signs and symptoms can also occur in people who take too many diuretics (water pills) or laxatives. Urine tests can be done to rule out other causes.
Bartter syndrome is associated with electrolyte and acid-base abnormalities, including hypokalemia and metabolic alkalosis in almost all cases. Other abnormalities include increased serum renin and aldosterone levels with decreased magnesium and phosphate levels in few patients. Urine electrolytes show elevated sodium, potassium, and prostaglandin E2 excretion. Elevated 24-hour urine calcium excretion helps exclude Gitelman syndrome, which is associated with low calcium excretion. Spot urine chloride concentration helps differentiate from surreptitious vomiting, where it is less than 25 meq/L. Usually, urine chloride is elevated (greater than 35 meq/L) in Barrter syndrome.
Polyhydramnios and intrauterine growth retardation are seen on ultrasound with the antenatal subtypes of Bartter syndrome. Amniotic fluid chloride levels may be elevated.
Abdominal radiographs, intravenous pyelograms (IVPs), renal ultrasonograms, or spiral CT scans can be done to document nephrocalcinosis. Genetic testing can be considered to rule out specific mutations.
Bartter syndrome treatment
The treatment of the Bartter syndromes is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians or general internists, kidney specialists (nephrologists or pediatric nephrologists), social workers, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment. Genetic counseling may be of benefit for affected individuals and their families. Psychosocial support for the entire family is essential as well.
There is no cure for Bartter syndromes, which require lifelong administration of certain supplements and medications. The mainstay of treatment is restoring the proper balance of fluids and electrolytes in the body. This includes potassium chloride supplementation to help correct electrolyte imbalances. Potassium chloride supplementation is preferred to salt supplementation because of the corresponding chloride deficiencies. Some infants with severe, life-threatening loop disorders (antenatal Bartter syndromes) may require salt and water replacement via a central vein catheter. Because aggressive fluid replacement may worsen polyuria, treatment with a drug that prevents the production of prostaglandin 2 such as indomethacin.
Indomethacin is a nonsteroidal anti-inflammatory drug (NSAID). This drug reduces prostaglandin levels in the body, thereby reducing excess urine production and the need for potassium supplements. Indomethacin has generally been effective and well tolerated in individuals with Bartter syndromes.
Some affected individuals may receive medications known as potassium-sparing diuretics such as spironolactone or amiloride. These drugs increase the excretion of water in urine, but retain potassium preventing potassium (hypokalemia). They have not always proven effective in treating hypokalemia and they may worsen the loss of salt in the body (renal salt wasting).
Drugs that inhibit or block the renin-aldosterone-angiotensin system (RAAS inhibitors) have been used to treat individuals with Bartter syndromes in addition to other therapies (adjunct therapy). Renin-aldosterone-angiotensin system inhibitors include aldosterone antagonists, angiotensin 2 receptor blockers (ARBs), and angiotensin-converting enzyme (ACE) inhibitors. These drugs can prevent the secretion of aldosterone from the adrenal glands and counteract the effects of renin on the kidneys, thereby reducing potassium loss in the distal tubules. The body conserves potassium, and less oral potassium supplementation is needed. Use of these drugs needs to be monitored because they may lower blood pressure, which may already be low in individuals with Bartter syndromes, and can potentially impact kidney and cardiovascular function.
Growth hormone (GH) therapy has been successful for the treatment of growth retardation and short stature potentially associated with Bartter syndrome. Although not well studied, at least 1 report describes a patient with low growth hormone (GH) levels and Gitelman syndrome who was below the third percentile for height and whose growth rate improved 4-fold during growth hormone (GH) treatment. Dose depends on brand used. Somatropin (up to 0.3 mg/kg weekly subcutaneously) and somatropin (rDNA origin, 0.1 mg/kg daily subcutaneously) have been used.
In some cases, calcium and/or magnesium supplementation is required to treat muscle spasms or tetany.
Adequate salt and water intake is necessary. Affected individuals have a large appetite for salt due to salt cravings. Affected individuals may be encouraged to eat foods that are high in potassium.
Cochlear implants can be used to treat deafness associated with Bartter syndromes type 4A and 4B.
In stressful situations, blood electrolytes can change rapidly, require prompt intravenous treatment. Stressful situations can include surgical procedures, trauma, and the presence of another type of disease or infection (intercurrent disease).
Diet and Activity
Diet
Adequate salt and water intake is necessary to prevent hypovolemia, and adequate potassium intake is essential to replace urinary potassium losses. Patients should consume foods and drinks that contain high levels of potassium (e.g., tomatoes, bananas, orange juice).
With growth retardation, adequate overall nutritional balance (protein-calorie intake) is important. Whether other dietary supplements (eg, citrate, magnesium, vitamins) are helpful is not clear.
Activity
No restriction on general activity is required, but precautions against dehydration should be taken. Patients should avoid strenuous exercise avoided because of the danger of dehydration and functional cardiac abnormalities secondary to potassium imbalance.
Kidney transplant
Bartter and Gitelman syndromes, by themselves, do not lead to chronic renal insufficiency; however, in patients with these syndromes who develop end-stage renal disease for other reasons, transplants from living relatives are an option and result in normal urinary handling of sodium, potassium, calcium, and magnesium 18.
Reports of kidney transplants from living relatives in end-stage renal disease patients with Bartter syndrome suggest that many endocrinologic abnormalities in Bartter syndrome improve or normalize after transplantation 18.
Because the genetic abnormality in Bartter syndrome may be found only in the kidneys (which is certain in Na-K-Cl cotransporter but may not be the case for some of the other mutations), transplantation corrects the problem by replacing unhealthy kidneys with normal ones.
References- Bartter syndrome. https://ghr.nlm.nih.gov/condition/bartter-syndrome
- Bokhari SRA, Mansur A. Bartter Syndrome. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK442019/
- Bartter syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=112
- Bartter’s Syndrome. https://rarediseases.org/rare-diseases/bartters-syndrome/
- Seys E, Andrini O, Keck M, Mansour-Hendili L, Courand PY, et al. Clinical and Genetic Spectrum of Bartter Syndrome Type 3. J Am Soc Nephrol. 2017 Aug. 28 (8):2540-2552.
- Kitanaka S, Sato U, Maruyama K, Igarashi T. A compound heterozygous mutation in the BSND gene detected in Bartter syndrome type IV. Pediatr Nephrol. 2006 Feb. 21(2):190-3.
- Janssen AG, Scholl U, Domeyer C, et al. Disease-causing dysfunctions of barttin in Bartter syndrome type IV. J Am Soc Nephrol. 2009 Jan. 20(1):145-53.
- Krämer BK, Bergler T, Stoelcker B, Waldegger S. Mechanisms of Disease: the kidney-specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat Clin Pract Nephrol. 2008 Jan. 4(1):38-46.
- Laghmani K, Beck BB, Yang SS, Seaayfan E, Wenzel A, et al. Polyhydramnios, Transient Antenatal Bartter’s Syndrome, and MAGED2 Mutations. N Engl J Med. 2016 May 12. 374 (19):1853-63.
- Bokhari SRA, Mansur A. Bartter Syndrome. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK442019
- Bartter Syndrome. https://emedicine.medscape.com/article/238670-overview
- Colussi G. Bartter syndrome. https://www.orpha.net/data/patho/Pro/en/Bartter-FRenPro259.pdf
- Gitelman syndrome. https://rarediseases.org/rare-diseases/gitelman-syndrome/
- Cruz AJ, Castro A. Gitelman or Bartter type 3 syndrome? A case of distal convoluted tubulopathy caused by CLCNKB gene mutation. BMJ Case Rep. 2013;2013:bcr2012007929. Published 2013 Jan 22. doi:10.1136/bcr-2012-007929 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3604527/
- Berry MR, Robinson C, Karet Frankl FE. Unexpected clinical sequelae of Gitelman syndrome: hypertension in adulthood is common and females have higher potassium requirements. Nephrol Dial Transplant. 2013;28(6):1533-42. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3685308/
- Bartter syndrome. https://rarediseases.org/rare-diseases/bartters-syndrome/
- Bartter syndrome. https://emedicine.medscape.com/article/238670-overview
- Bartter Syndrome Treatment & Management. https://emedicine.medscape.com/article/238670-treatment#showall





{kind=link}