
Biotin deficiency
Biotin deficiency also called vitamin B7 deficiency is rare 1, and severe biotin deficiency in healthy individuals eating a normal mixed diet has never been reported 2.
Although clinically overt biotin deficiency or vitamin B7 deficiency is very rare, the human requirement for dietary biotin has been demonstrated in three different situations: prolonged intravenous feeding (parenteral nutrition) without biotin supplementation, infants fed an elemental formula devoid of biotin, and consumption of raw egg white for a prolonged period (many weeks to years) 3. Raw egg white contains avidin; this antimicrobial protein binds biotin with an affinity and specificity that is almost unique as a reversible binding. Because native avidin is resistant to mammalian and microbial digestion, avidin prevents biotin absorption. Cooking egg white denatures avidin, rendering it susceptible to digestion and therefore unable to block the absorption of dietary biotin 4.
The signs and symptoms of biotin deficiency typically appear gradually and can include thinning hair with progression to loss of all hair on the body (alopecia); scaly, red rash around body openings (eyes, nose, mouth, and perineum) also called seborrheic dermatitis; conjunctivitis (pink eye); ketolactic acidosis (which occurs when lactate production exceeds lactate clearance) and aciduria (abnormal amounts of acid in urine); seizures; skin infection; brittle nails; neurological findings (e.g., depression, lethargy, hallucinations, and paresthesias of the extremities) in adults; and hypotonia, lethargy, and developmental delay in infants 5. The rash and unusual distribution of facial fat in people with biotin deficiency is known as “biotin deficiency facies” 2.
The neurological and psychological symptoms can occur with only mild biotin deficiencies while dermatitis, conjunctivitis, and hair loss generally occur only when biotin deficiency becomes more severe 6. Individuals with hereditary disorders of biotin deficiency additionally have evidence of impaired immune system function, including increased susceptibility to bacterial and fungal infections 7.
Regardless of the cause, biotin deficiency or vitamin B7 deficiency can usually be addressed directly with oral biotin supplementation.
Figure 1. Biotin deficiency hair loss
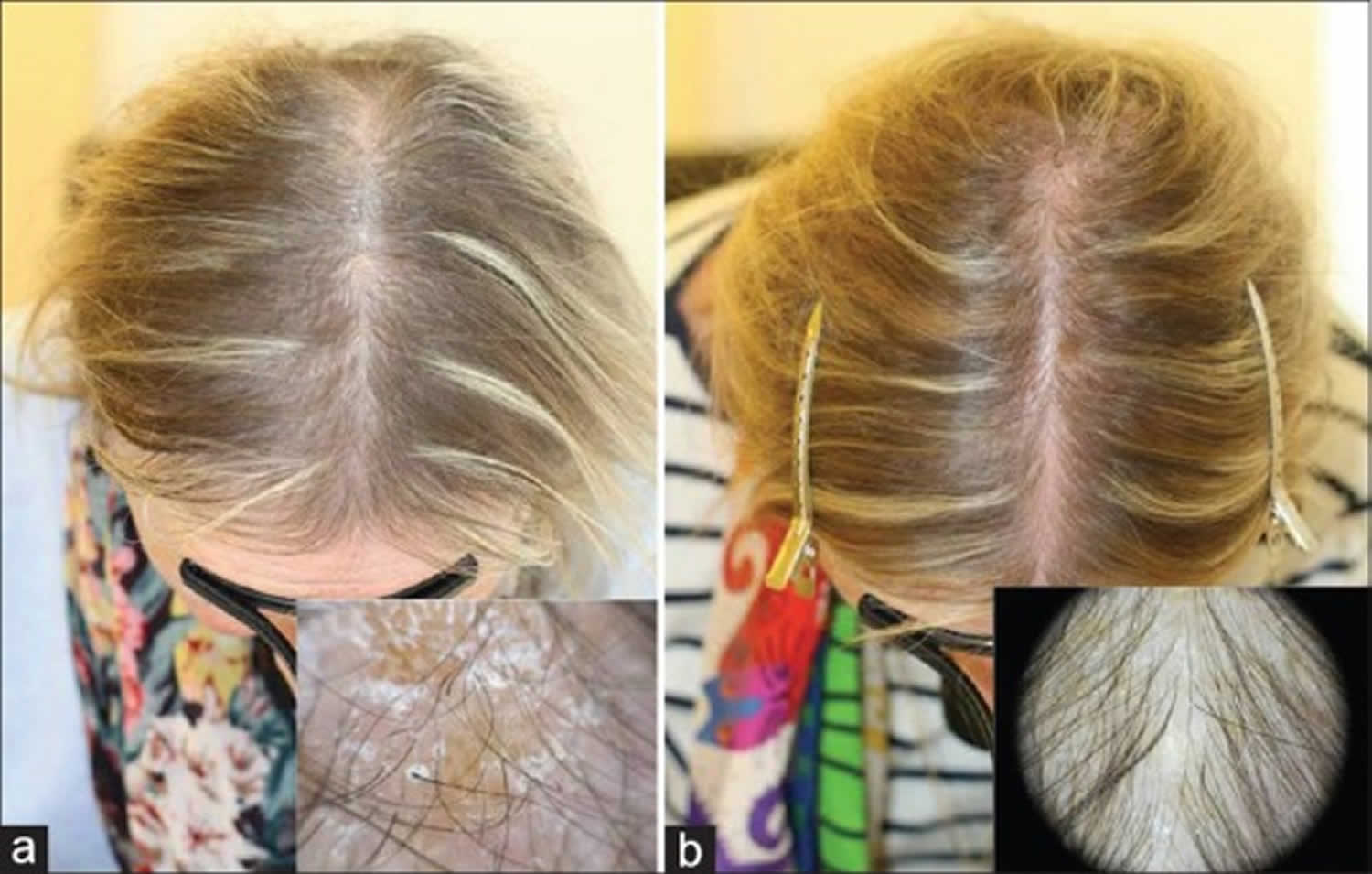
Footnotes: Hair loss (alopecia) and seborrheic-like dermatitis in a patient with biotin deficiency (a) before, and (b) after treatment with 5 mg oral biotin for 3 months.
[Source 6 ]Figure 2. Biotin deficiency in an infant fed with amino acid formula
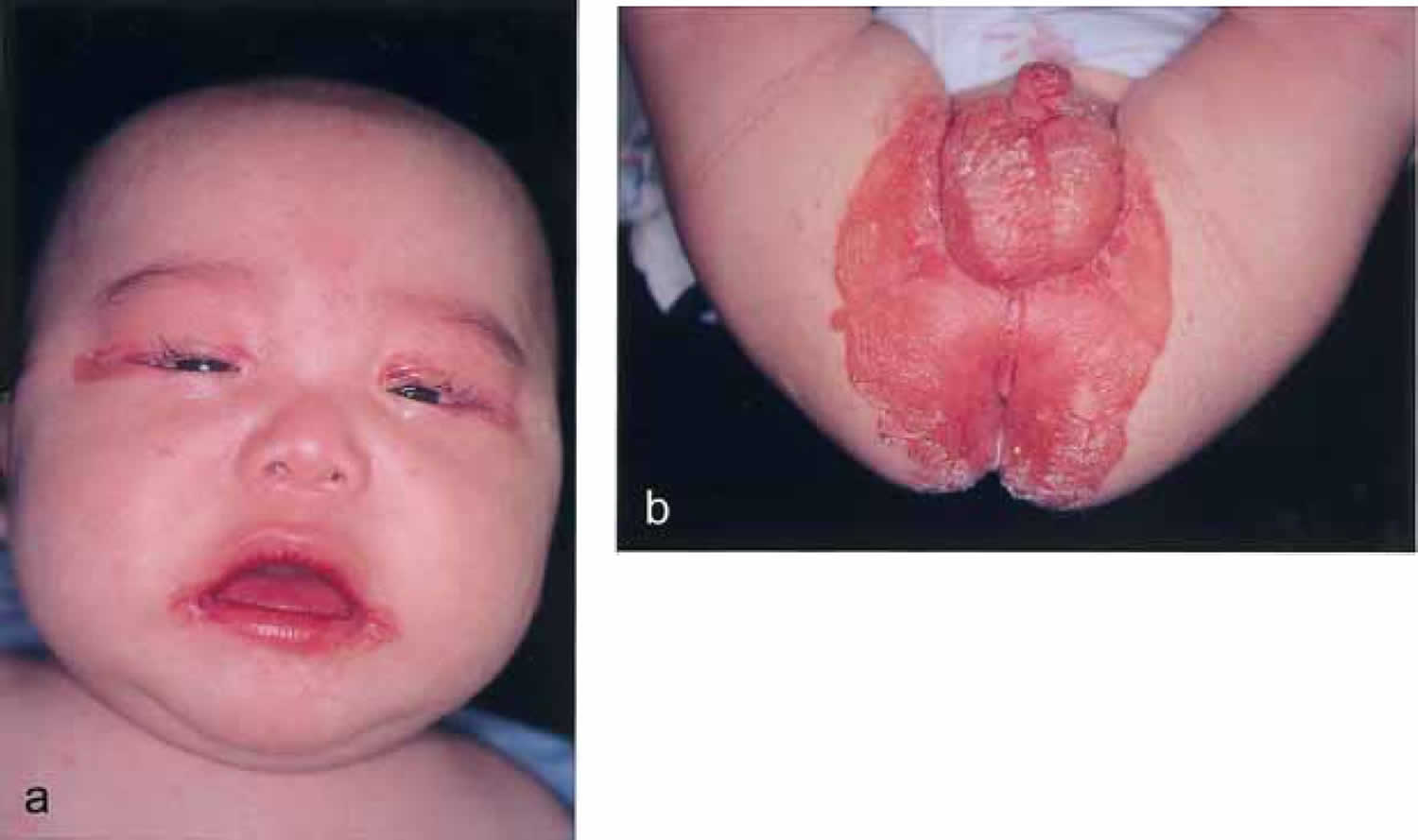
Footnotes: Biotin deficiency is rarely encountered in an infant on weaning from breast and formula feeding. Biotin deficiency is characterized by hair loss (alopecia) and scaly, erythematous dermatitis distributed around the body orifices. A 5-month-old Japanese infant with typical skin lesions who had been diagnosed as a neonate with dyspepsia and fed only an amino acid formula. Serum and urine levels of biotin were below the normal range, but zinc and biotinidase were within normal range. Urinary excretion of 3-methylcrotonylglycine, 3-hydroxyisovaleric acid, and methylcitric acid was significantly elevated. Daily oral supplementation with 1 mg of biotin resulted in dramatic improvement of the periorificial dermatitis and hair growth together with a complete disappearance of the organic aciduria.
[Source 8 ]Figure 3. Biotinidase deficiency
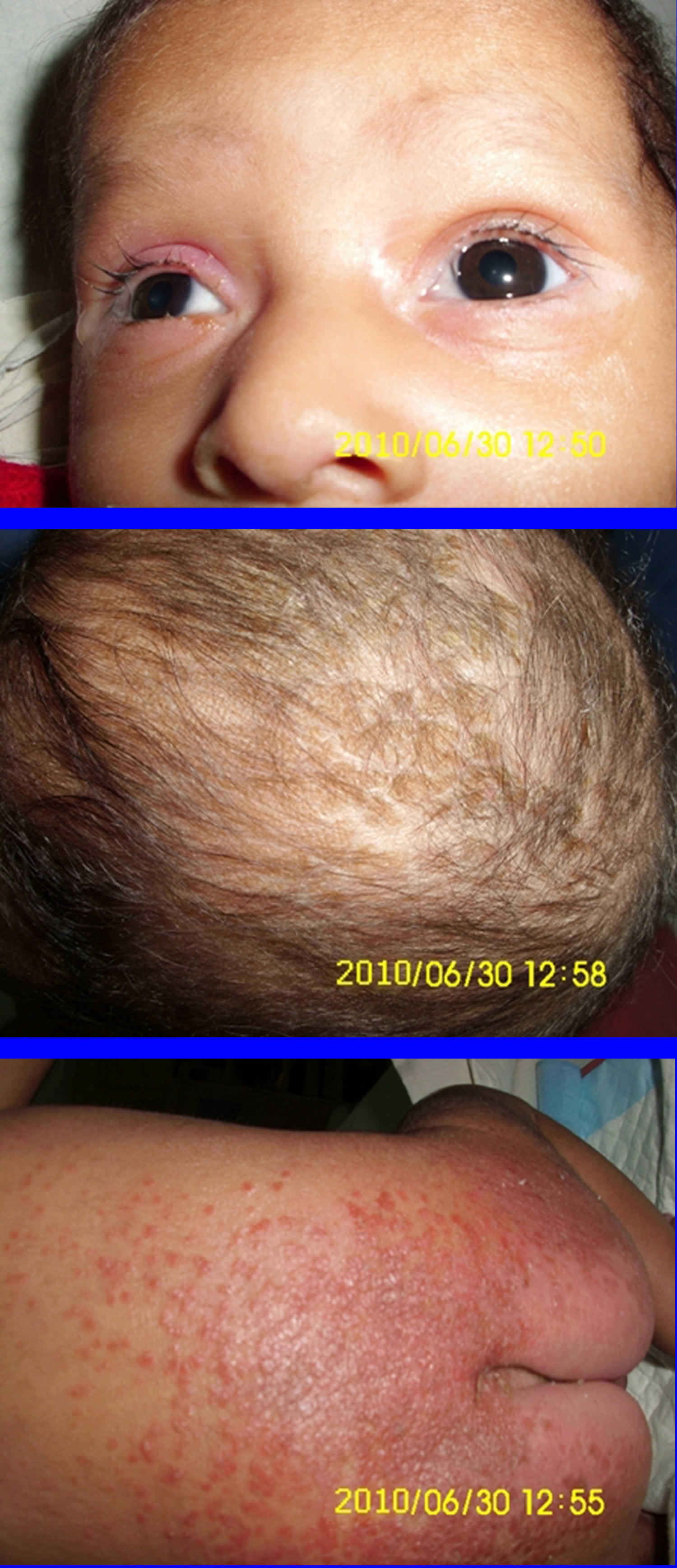
Footnotes: Biotinidase deficiency is a rare autosomal recessive inherited disorder in which the body is unable to recycle biotin, leading to biotin deficiency despite normal biotin intake from foods such as liver, egg yolks, and milk 9. Biotinidase deficiency is caused by genetic changes (mutations) in the BTD gene. The BTD gene provides instructions for making an enzyme called biotinidase 10. The biotinidase enzyme recycles biotin. Biotinidase removes biotin that is bound to proteins in food, leaving the vitamin in its free (unbound) state. Free biotin is needed by enzymes called biotin-dependent carboxylases to break down fats, proteins, and carbohydrates. Because several of these enzymes are impaired in biotinidase deficiency, the condition is considered a form of multiple carboxylase deficiency 9. Biotinidase deficiency in a 58-day-old male infant with seizures for 2 weeks prior to presentation. At admission, the child had multiple episodes of generalized tonic and myoclonic seizures not associated with fever. He also had respiratory distress and stridor. The skin showed erythematous maculopapular rash in the perioral and perianal regions. He had alopecia (hair loss), scanty eyebrows, blepharitis, conjunctivitis, balanitis and seborrheic dermatitis involving the scalp. Hair over the scalp was hypopigmented. On neurological examination, the child was convulsing intermittently. Hypertonia was noted in all the four limbs with exaggerated deep tendon reflexes. Fundus examination revealed papilloedema. Rest of the systemic examination was normal. Within 24 hour of hospitalization, the possibility of biotinidase deficiency was entertained and oral biotin was started empirically at a dose of 10 mg twice a day, after obtaining a blood sample for biotinidase assay. There was total cessation of seizures and considerable improvement in the rash by 48 hour. The hypertonia in the upper limbs resolved within 72 hours though it persisted in the lower limbs, even at discharge, albeit much less than before. The balanitis and blepharitis also resolved by 72 hours. Serum biotinidase levels showed a profound deficiency. The child was then started on regular biotin supplementation and was discharged after 5 days. Anticonvulsants were successfully stopped without recurrence of seizures. The child is currently 10 months old, healthy, with adequate weight gain and normal developmental milestones. At follow-up, there are no sequelae except for a mild persisting lower limb hypertonia.
[Source 11 ]Figure 4. Holocarboxylase synthetase deficiency in baby
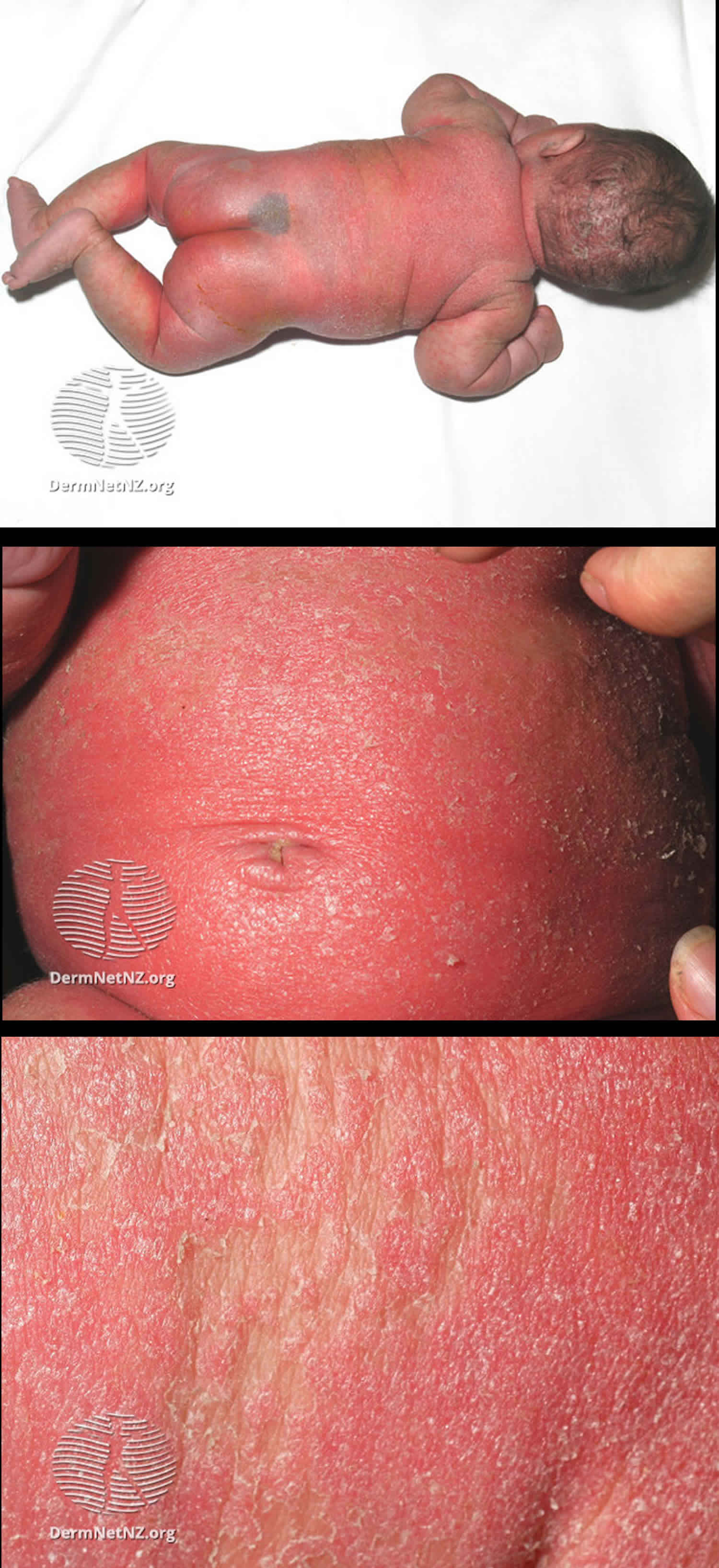
Figure 5. Holocarboxylase synthetase deficiency in adult
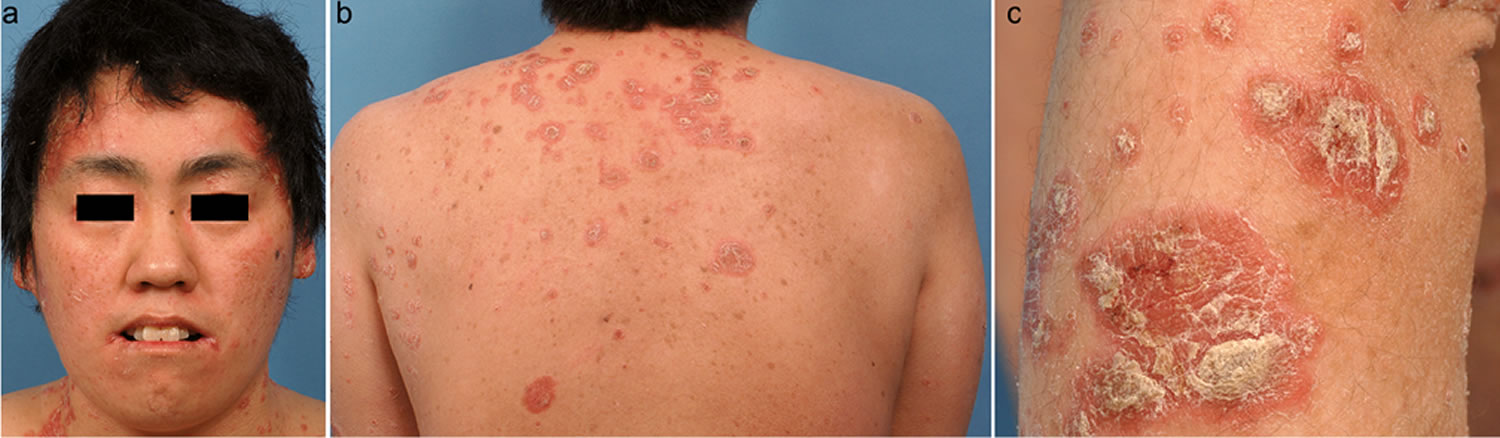
Footnotes: A 34-year-old woman presented with a skin eruption covering the whole body that had persisted since infancy. She had been born to healthy, unrelated Japanese parents. The patient had developed tachypnea and myoclonic seizures from the second day of life. Investigation confirmed severe metabolic acidosis and ketosis. The results of urinary organic acid analysis and fibroblast carboxylase tests were consistent with a diagnosis of holocarboxylase synthetase deficiency. Erythematous plaques with pustules and scales on (a) the face, (b) the abdomen and (c) the back.
[Source 13 ]What is Biotin?
Biotin also known as vitamin B7 or vitamin H, is a water soluble vitamin and is naturally present in liver, soy, beans and egg yolks. Raw egg whites, however, contain the protein avidin that binds to biotin and reduces its availability. Eating 2 or more uncooked egg whites daily for several months has caused biotin deficiency that is serious enough to produce symptoms. Biotin acts as a carrier of carbon dioxide and plays a role in carboxylase enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) that catalyze critical steps fatty acid metabolism, gluconeogenesis (the formation of glucose from sources other than carbohydrates, such as pyruvate, lactate, glycerol, and the glucogenic amino acids), and amino acids 5. Biotin also plays key roles in histone modifications, gene regulation (by modifying the activity of transcription factors), and cell signaling 14.
The recommended daily dietary allowance for biotin has not been formally established, but the amounts needed are small and biotin is found in many foods and is produced by intestinal bacteria. An adequate intake for biotin has been estimated as 30 micrograms (mcg) daily. Thus, most diets provide adequate amounts of biotin and its deficiency is rare. Although there are no nationally representative estimates of biotin intakes in the United States, the average biotin intake from foods in other western populations is about 35–70 mcg/day, indicating that most people in these countries consume adequate amounts of biotin and biotin deficiency is rare 5, 15.
Most biotin in foods is bound to protein, although some dietary biotin is in the free form 16. Gastrointestinal proteases and peptidases break down the protein-bound forms of ingested biotin into biocytin and biotin-oligopeptides, which undergo further processing by biotinidase, an enzyme, in the intestinal lumen to release free biotin 16. The free biotin is then absorbed in the small intestine, and most biotin is stored in the liver 15.
Biotin is available generically in many over-the-counter forms in doses of 5 to 10 mg and is included in most multivitamin preparations, usually in concentrations of 30 to 300 mcg. Biotin is typically added to parenteral nutrition and the doses for deficiencies is in the range of 10 mg daily.
Biotin deficiency has occurred in humans on parenteral nutrition (intravenous administration of nutrition). However, biotin deficiency is very rare in the United States. The signs and symptoms of biotin deficiency typically appear gradually and can include thinning hair with progression to loss of all hair on the body (alopecia); a scaly red rash around the eyes, nose, mouth, and anal area (seborrheic dermatitis); pinkeye (conjunctivitis); lactic acidosis (which occurs when lactate production exceeds lactate clearance) and aciduria (abnormal amounts of acid in urine); skin infection; brittle nails; nervous system disorders (e.g., depression, lethargy, seizures, hallucinations, ataxia and numbness and tingling [paresthesias] of the extremities) in adults; and hypotonia (weak muscle tone), lethargy, sluggishness and developmental delay in infants 5. The characteristic facial rash, together with unusual facial fat distribution in people with biotin deficiency is known as “biotin deficiency facies” 2. Individuals with hereditary disorders of biotin metabolism (inborn metabolic disorders) resulting in functional biotin deficiency often have similar physical findings, as well as seizures and evidence of impaired immune system function and increased susceptibility to bacterial and fungal infections 17, 18.
A limited number of reliable indicators of biotin status is available 19. In healthy adults, the concentration of biotin is 133–329 pmol/L in serum and 18–127 nmol/24 hours in urine 5. Abnormally low urinary excretion of biotin is an indicator of biotin deficiency, as is abnormally high excretion of 3-hydroxyisovaleric acid (higher than 3.3 mmol/mol creatinine) or 3-hydroxyisovalerylcarnitine (higher than 0.06 mmol/mol creatinine) resulting from reduced activity of methylcrotonyl-CoA carboxylase 19. The most reliable individual markers of biotin status, including deficiency and sufficiency, are biotinylated methylcrotonyl-CoA carboxylase and propionyl-CoA carboxylase in white blood cells 19. Oral administration of large doses of biotin increases serum concentrations of biotin and its metabolites 20. However, serum concentrations of biotin and its catabolites are not good indicators of marginal biotin deficiency because they do not decrease sufficiently in people with marginal biotin deficiency for these changes to be detectable with existing tests 15.

What happens if I don’t get enough biotin?
Biotin deficiency is very rare in the United States. Biotin deficiency can cause thinning hair and loss of body hair; a rash around the eyes, nose, mouth, and anal area; pinkeye; high levels of acid in the blood and urine; seizures; skin infection; brittle nails; and nervous system disorders. Symptoms of biotin deficiency in infants include weak muscle tone, sluggishness, and delayed development.
What does biotin do?
Biotin helps turn the carbohydrates, fats, and proteins in the food you eat into the energy you need. Biotin functions as a coenzyme; involved in carboxylation, transcarboxylation, and decarboxylation reactions of gluconeogenesis, lipogenesis, fatty acid synthesis, propionate metabolism, and the catabolism of leucine. Biotin acts as a carrier of carbon dioxide and functions as a covalently bound cofactor required for the biological activity of the five known mammalian biotin-dependent carboxylases enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) that catalyze critical steps fatty acid metabolism, gluconeogenesis, and amino acids 5. For acetyl-CoA carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2), biotin serves as a cofactor responsible for transfer of bicarbonate to acetyl-CoA, converting it to malonyl-CoA for fatty acid synthesis. Pyruvate carboxylase (PC) participates in gluconeogenesis. Methylcrotonyl-CoA carboxylase (MCC) catalyzes a step in leucine metabolism. Propionyl-CoA carboxylase (PCC) catalyzes a step in the metabolism of propionyl-CoA 21, 22. Metabolic degradation of the biotinylated carboxylases leads to the formation of biocytin. This compound is further degraded by biotinidase to release biotin, which is then reutilized by holocarboxylase synthetase 22.
Biotin also plays key roles in histone modifications, gene regulation (by modifying the activity of transcription factors), and cell signaling 14.
Enzyme cofactor
Biotin is an essential cofactor to five known mammalian biotin-dependent carboxylases enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) in intermediary metabolism and a key regulator of gene expression 23.
Five mammalian carboxylases catalyze essential metabolic reactions 23:
- Both acetyl-Coenzyme A (CoA) carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2) catalyze the conversion of acetyl-CoA to malonyl-CoA using bicarbonate and ATP; however, the two enzymes have different roles in metabolism and different intracellular locations. ACC1 is located in the cytosol, and the malonyl CoA generated by ACC1 is a rate-limiting substrate for the synthesis of fatty acids (Figures 6 and 7). Acetyl-Coenzyme A (CoA) carboxylase 1 (ACC1) is found in all tissues and is particularly active in lipogenic tissues (i.e., liver, white adipose tissue, and mammary gland), heart, and pancreatic islets. Acetyl-CoA carboxylase 2 (ACC2) is located on the outer mitochondrial membrane, and the malonyl CoA generated via ACC2 inhibits CPT1, an enzyme that regulates malonyl-CoA entry into the inner mitochondria, thereby regulating fatty acid oxidation (Figure 3). ACC2 is especially abundant in skeletal muscle and heart 24.
- Pyruvate carboxylase (PC) is a critical enzyme in gluconeogenesis (the formation of glucose from sources other than carbohydrates, such as pyruvate, lactate, glycerol, and the glucogenic amino acids). Pyruvate carboxylase (PC) catalyzes the ATP-dependent incorporation of bicarbonate into pyruvate, producing oxaloacetate; hence, pyruvate carboxylase is anaplerotic for the citric acid cycle (Figure 8). Oxaloacetate can then be converted to phosphoenolpyruvate and eventually to glucose.
- Methylcrotonyl-CoA carboxylase (MCC) catalyzes an essential step in the catabolism of leucine, an essential branched-chain amino acid (BCAA). Methylcrotonyl-CoA carboxylase (MCC) enzyme catalyzes the production of 3-methylglutaconyl-CoA from methylcrotonyl-CoA (Figure 9).
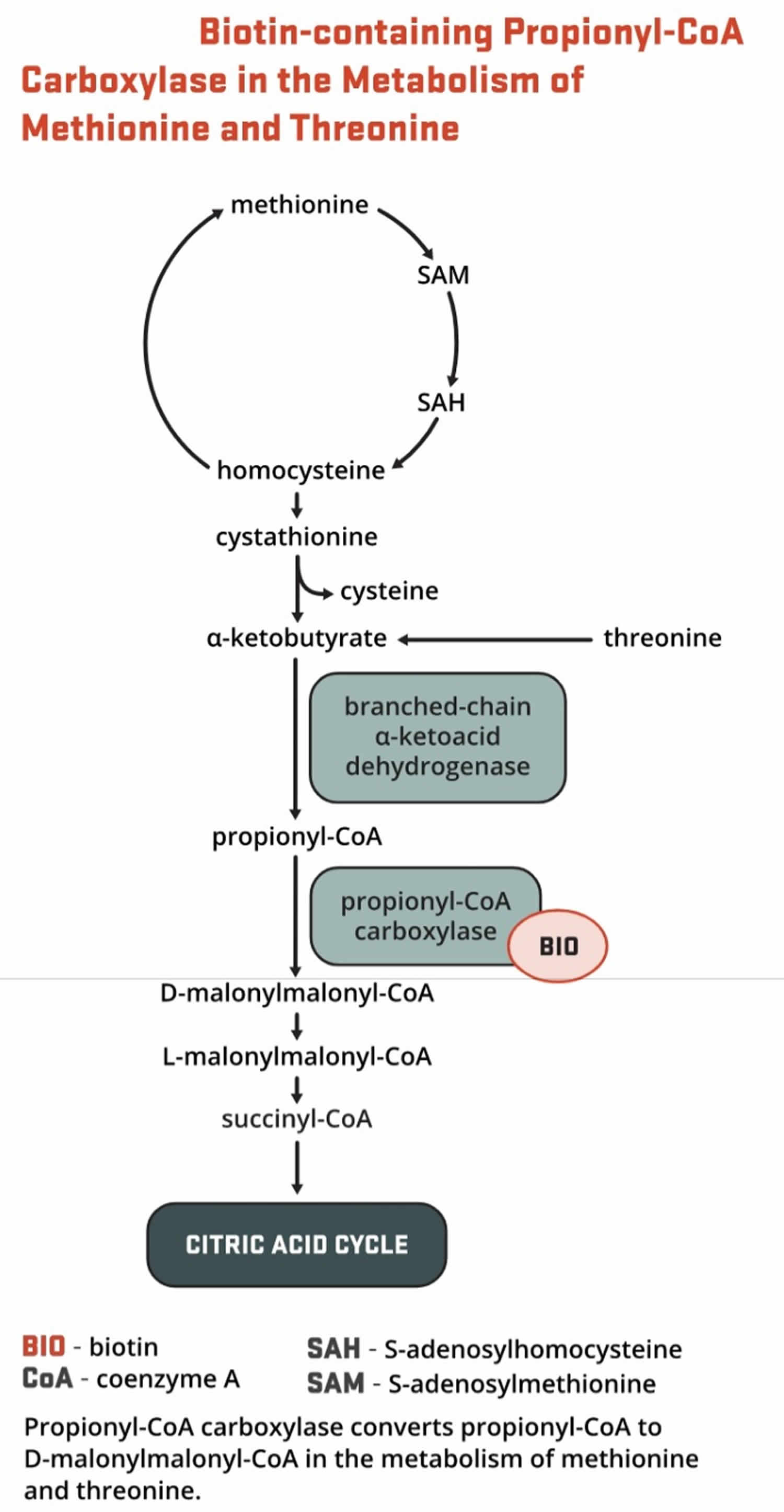
- Propionyl-CoA carboxylase (PCC) produces D-malonylmalonyl-CoA from propionyl-CoA, a by-product in the beta-oxidation of fatty acids with an odd number of carbon atoms (Figure 9). The conversion of propionyl-CoA to D-malonylmalonyl-CoA is also required in the catabolic pathways of two branched-chain amino acids (isoleucine and valine) and the side chain of cholesterol (Figure 9) and of the amino acids methionine and threonine (Figure 10).
Figure 6. Roles of the 5 biotin-dependent carboxylases
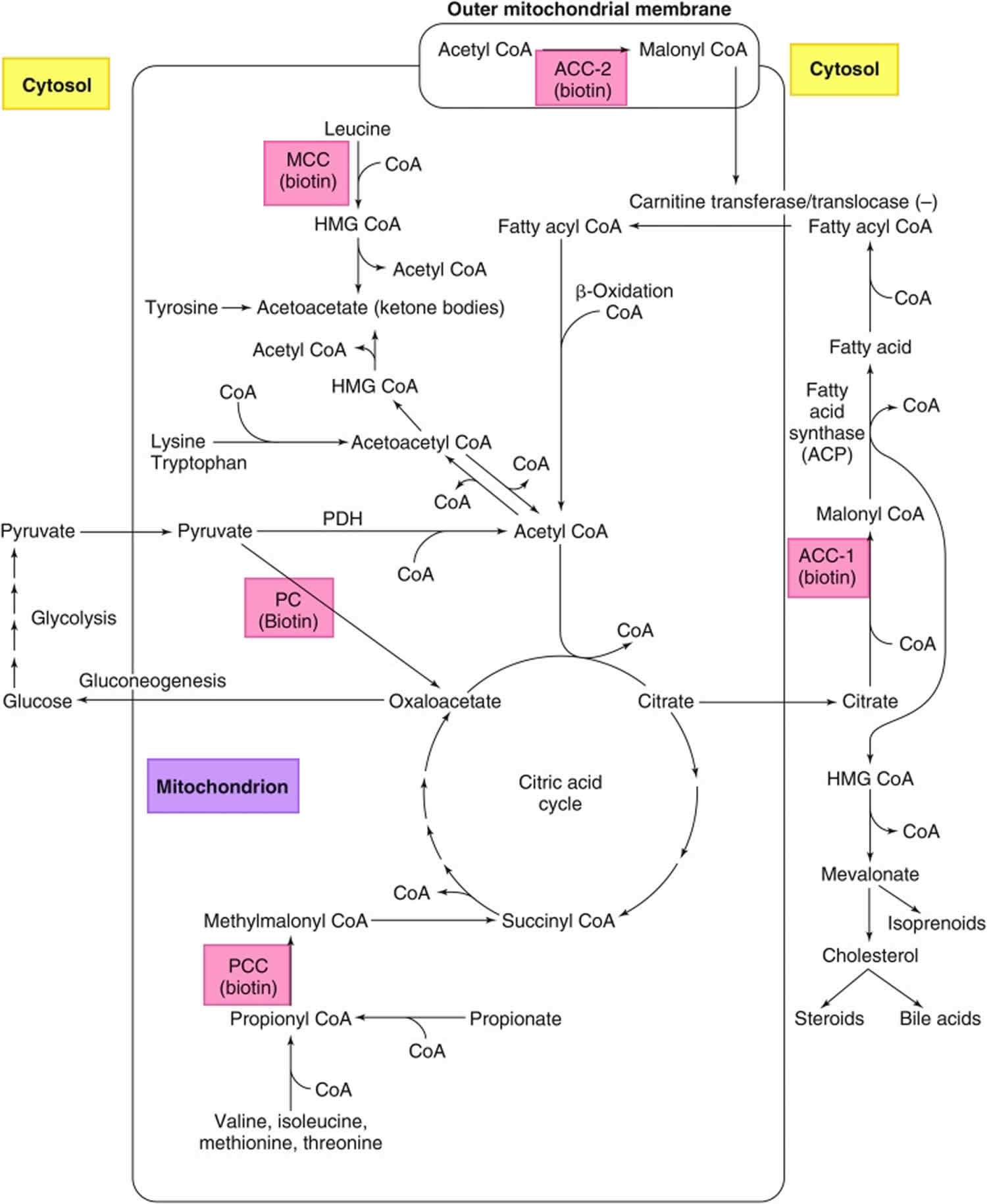
Footnotes: Roles of the 5 biotin-dependent carboxylases of coenzyme A (CoA) and acyl carrier protein (ACP) within the cell. Shown is an overview of the metabolic pathways of acetyl-CoA carboxylase 1 (ACC1) (cytosolic) and acetyl-CoA carboxylase 2 (ACC2) (outer mitochondrial membrane) and the 3 mitochondrial carboxylases propionyl-CoA carboxylase (PCC), methylcrotonyl-CoA carboxylase (MCC) and pyruvate carboxylase (PC).
Abbreviations: ACC1 = acetyl-CoA carboxylase 1; ACC2 = acetyl-CoA carboxylase 2; ACP = acyl carrier protein; HMG = 3-hydroxy-3-methylglutaryl; MCC = methylcrotonyl-CoA carboxylase; PC = pyruvate carboxylase; PCC = propionyl-CoA carboxylase; PDH = pyruvate dehydrogenase.
[Source 25 ]Figure 7. Biotin function as enzyme cofactor
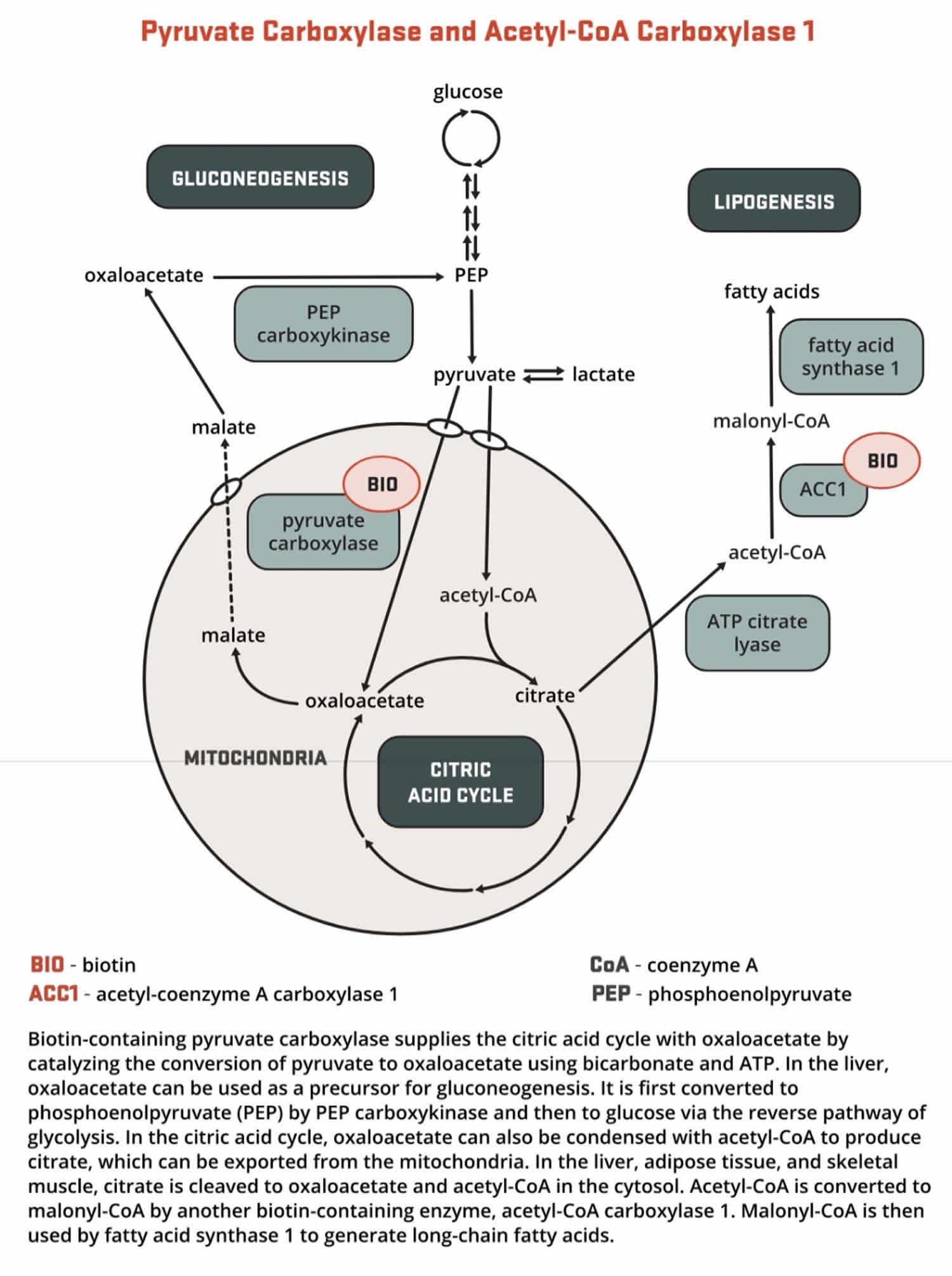
Figure 8. Biotin as enzyme cofactor in gluconeogenesis
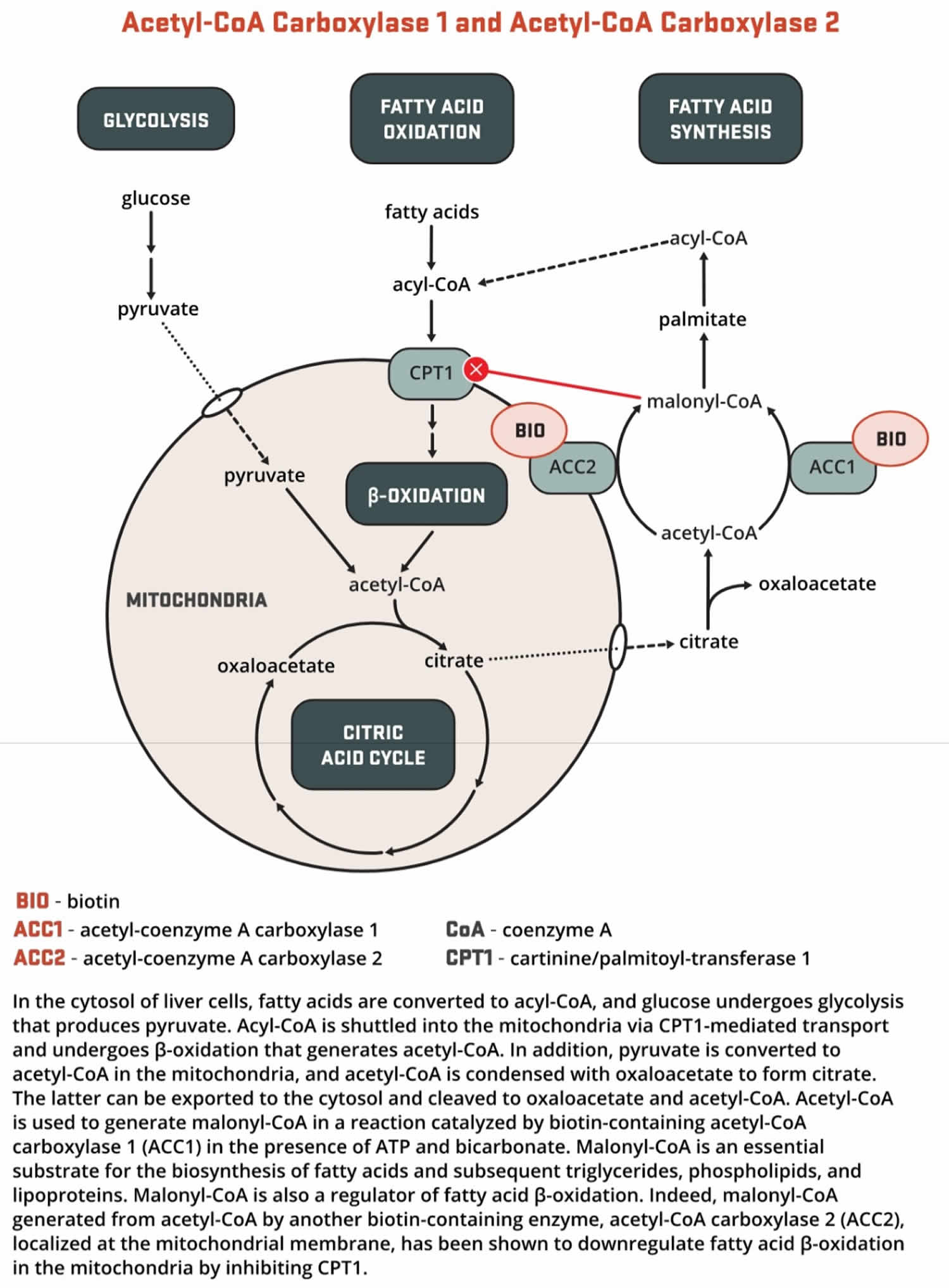
Figure 9. Biotin as enzyme cofactor in fatty acids, amino acids and cholesterol metabolism
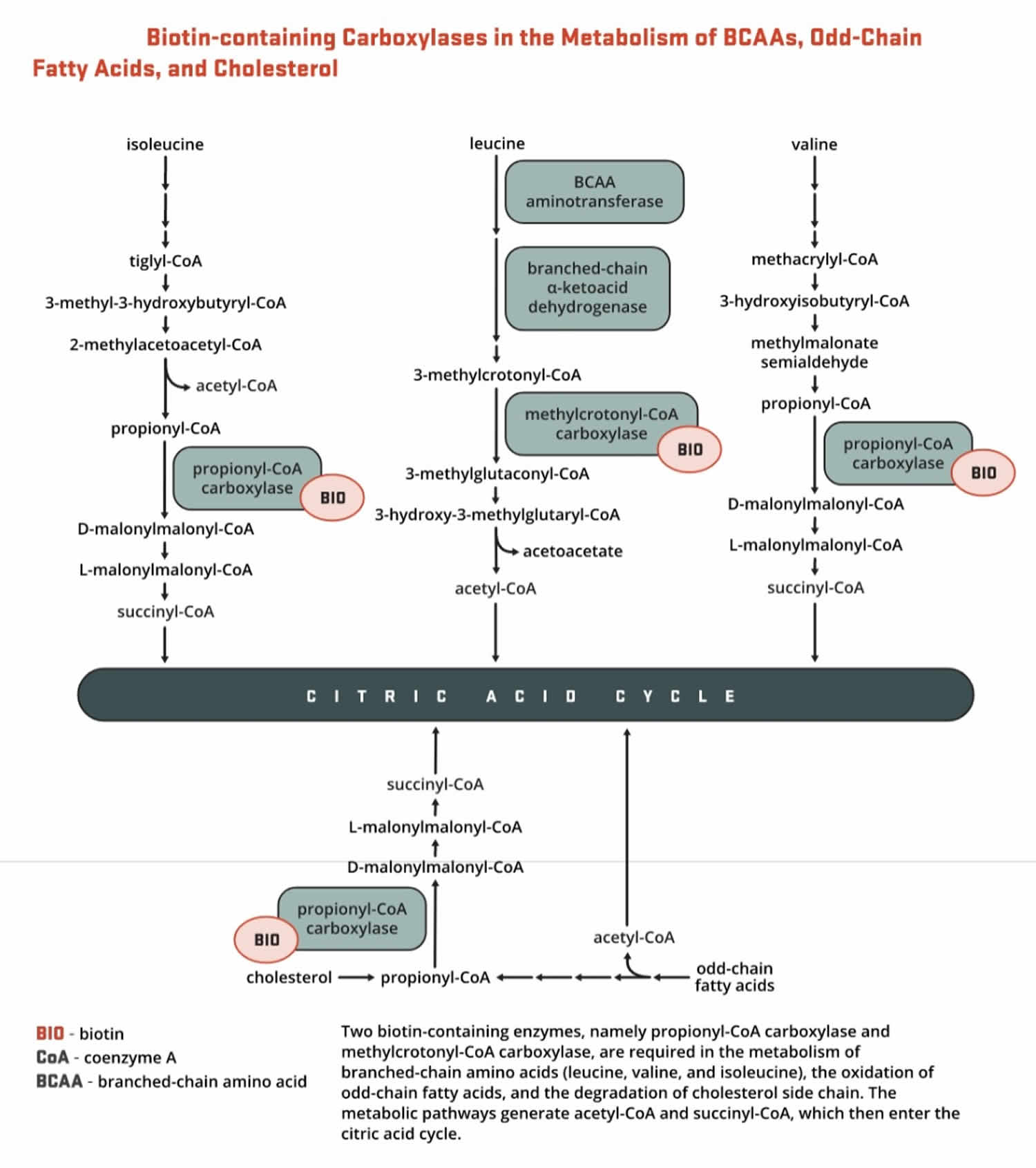
Figure 10. Biotin as enzyme cofactor in amino acids methionine and threonine metabolism
Regulation of chromatin structure and gene expression
In cells that have a nucleus, DNA is packaged into compact structures to form nucleosomes — fundamental units of chromatin. Each nucleosome is composed of 147 base pairs of DNA wrapped around eight histones (paired histones: H2A, H2B, H3, and H4) 23. The H1 linker histone is located at the outer surface of each nucleosome and serves as an anchor to fix the DNA around the histone core. The compact packaging of chromatin must be relaxed for DNA replication and transcription 23. Chemical modifications of DNA and histones affect the folding of chromatin, increasing or reducing DNA accessibility to factors involved in replication and transcription. DNA methylation and a number of chemical modifications within the N-terminal tail of core histones modify their electric charge and structure, thereby changing chromatin conformation and transcriptional activity of genes 23.
The modifications of histone tails (“marks”), including acetylation, methylation, phosphorylation, ubiquitination, SUMOylation, ADP-ribosylation, carbonylation, deimination, hydroxylation, and biotinylation, have various regulatory functions 23. Several sites of biotinylation have been identified in histones H2A, H3, and H4 4. Amongst them, histone H4 biotinylation at lysine (K) 12 (noted H4K12bio) appears to be enriched in heterochromatin, a tightly condensed chromatin associated with repeat regions in (peri)centromeres and telomeres. H4 biotinylation appears to be enriched in transposable elements known as long terminal repeats 26. These biotinylation marks also co-localize with well-known gene repression marks like methylated lysine 9 in histone H3 (H3K9me) in transcriptionally competent chromatin 27. For example, H4K12bio can be found at the promoter of the gene SLC5A6 that codes for the transporter mediating biotin uptake into cells, the human sodium-dependent multivitamin transporter (hSMVT). When biotin is abundant, HCS can biotinylate histones H4 in the SLC5A6 promoter, which down regulates hSMVT synthesis and reduces biotin uptake. Conversely, in biotin-deficient cells, biotinylation marks in the SLC5A6 promoter are removed increasing gene expression and enabling the synthesis of hSMVT and uptake of biotin 28.
Biotinylation
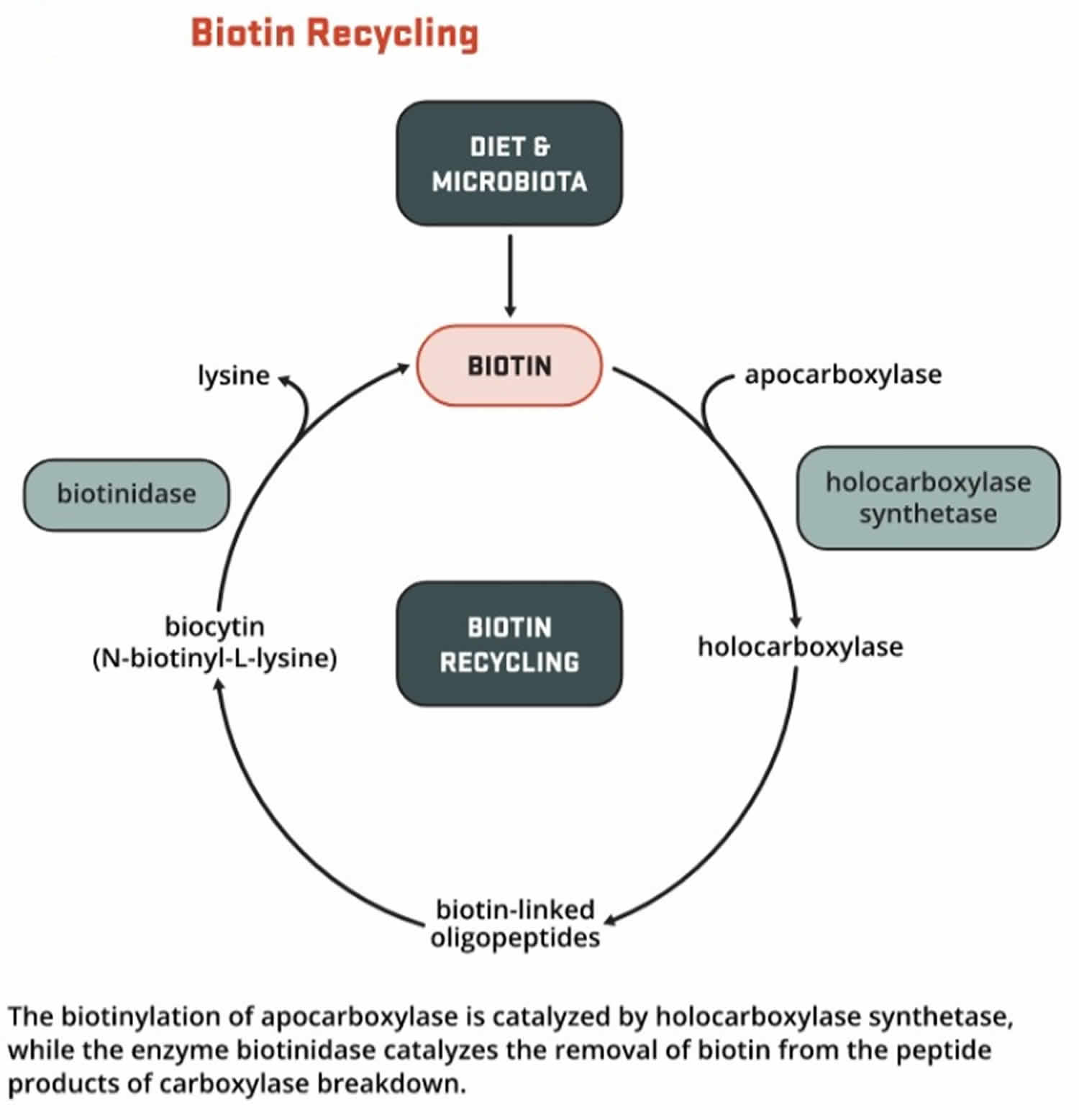
Biotin functions as a covalently bound cofactor required for the biological activity of the five known mammalian biotin-dependent carboxylases (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]). Such non-protein cofactors are termed “prosthetic groups” and are common in water-soluble vitamins. The covalent attachment of biotin to the apocarboxylase (i.e., the carboxylase protein without the biotin prosthetic group and is catalytically inactive) is catalyzed by the enzyme, holocarboxylase synthetase (HCS). The term “biotinylation” refers to the covalent addition of biotin to any molecule, including the apocarboxylases and histones. Holocarboxylase synthetase (HCS) catalyzes the post-translational biotinylation of the epsilon amino group of a lysine residue at the active site of each apocarboxylase, converting the inactive apocarboxylase into a fully active holocarboxylase (Figure 11). Particular lysine residues within the N-terminal tail of specific histone that help package DNA in eukaryotic nuclei can also be biotinylated 26. Biotinidase is the enzyme that catalyzes the release of biotin from biotinylated histones and from the peptide products of holocarboxylase breakdown (Figure 12).
Figure 11. Biotinylation

Figure 12. Biotin recycling
Biotin health benefits
Scientists are studying biotin to understand how it affects health. Biotin is commonly used for hair loss, brittle nails, nerve damage, and many other conditions.
Taking biotin can help treat low blood levels of biotin or biotin deficiency. It can also prevent blood levels of biotin from becoming too low. Low blood levels of biotin can cause thinning of the hair and rash around the eyes, nose, and mouth. Other symptoms include depression, lack of interest, hallucinations, and tingling in the arms and legs. Low biotin levels can occur in people who are pregnant, who have had long-term tube feeding, who are malnourished, who have undergone rapid weight loss, or who have a specific inherited condition. Cigarette smoking might also cause low blood levels of biotin.
Biotin possibly ineffective for:
- Skin rash in infants (seborrheic dermatitis). Taking biotin does not seem to help improve rash in infants.
Insufficient evidence to rate Biotin effectiveness for:
- Hair loss. Taking biotin and zinc by mouth in addition to applying a steroid cream to the skin might help reduce hair loss.
- An inherited disorder called biotin-thiamine-responsive basal ganglia disease. People with this condition experience episodes of altered mental state and muscle problems. Early research shows that taking biotin plus thiamine does not prevent these episodes better than taking thiamine alone. But the combination might shorten how long the episodes last when they do occur.
- Brittle fingernails and toenails. Taking biotin by mouth for up to a year might increase the thickness of fingernails and toenails in people with brittle nails.
- Diabetes. Some early research shows that taking biotin along with chromium might lower blood sugar in people with diabetes. However, taking biotin alone doesn’t seem to improve blood sugar levels in people with diabetes.
- Diabetic nerve pain. Early research shows that taking biotin by mouth or receiving it as a shot might reduce nerve pain in the legs of people with diabetes.
- Muscle cramps related to dialysis. People receiving dialysis tend to have muscle cramps. Early research shows that taking biotin by mouth might reduce muscle cramps in these people.
- Multiple sclerosis. Early research shows that taking high-dose biotin might improve vision and reduce partial paralysis in some people with multiple sclerosis.
- Other conditions.
More evidence is needed to rate biotin for these uses.
Hair, nail, and skin health
Dietary supplements that contain biotin are often promoted to improve the health of your hair, skin, and nails, but there is little scientific evidence to support these claims. In a few small studies, some people with thin and brittle nails who took high doses of biotin had harder nails. Doctors have also reported that in a few cases, high doses of biotin have improved a rare hair disorder in children and skin rash in infants. More research is needed before biotin supplements can be recommended for any of these conditions.
Signs of biotin deficiency include skin rashes, hair loss, and brittle nails 29. Therefore, biotin supplements are often promoted for hair, skin, and nail health 30. However, these claims are supported, at best, by only a few case reports and small studies.
The evidence on biotin supplementation to treat brittle nails includes three small studies that did not include a placebo group, and these reports do not indicate the baseline biotin status of study participants. One of these studies assessed the effects of 2.5 mg/day biotin for 6–15 months in 22 women with brittle, splitting, or soft nails and 10 healthy volunteers 30. In the eight patients with brittle nails whose nail samples were obtained immediately before and after biotin supplementation, nail thickness increased by 25%. In the 14 patients with brittle nails whose nail specimens were obtained 2–4 months after starting treatment and 1–4 months after ending treatment, nail thickness increased by 7%, a difference that was not statistically significant 30. In the second study, 2.5 mg biotin daily for an average of 5.5 months in 45 patients with thin and brittle fingernails resulted in firmer and harder fingernails in 41 of the patients (91%) 31. Finally, the third, retrospective study in 35 patients with brittle nails found that 2.5 mg/day biotin for 6–15 months resulted in clinical improvement in 22 of the 35 patients (63%) 32.
Only case reports are available to support claims that biotin supplements can promote hair health, and these reports were only in children 33, 34. These studies found that 3–5 mg/day biotin in children with uncombable hair syndrome (an inherited condition that is characterized by dry, frizzy hair that cannot be combed flat and is caused by mutations in the PADI3, TGM3, or TCHH gene) significantly improved hair health after 3–4 months 33, 34.
The evidence supporting the use of biotin supplements to support skin health is equally limited to a small number of case reports, all in infants, showing that 100 mcg to 10 mg/day resulted in dramatic improvements in rash or dermatitis as well as alopecia (hair loss) 35, 36.
Future studies are needed to determine whether biotin supplements might improve hair, nail, and skin health, especially among healthy individuals.
Brittle fingernails (onychorrhexis)
The finding that biotin supplements were effective in treating hoof abnormalities in hoofed animals suggested that biotin might also be helpful in strengthening brittle fingernails in humans 37, 38, 39. Three uncontrolled trials examining the effects of biotin supplementation (2.5 mg/day for several months) in women with brittle fingernails have been published 40, 31, 32. In two of the trials, subjective evidence of clinical improvement was reported in 67%-91% of the participants available for follow-up at the end of the treatment period 40, 31. One trial that used scanning electron microscopy to assess fingernail brittleness reported less fingernail splitting and a 25% increase in the thickness of the nail plate in patients supplemented with biotin for 6 to 15 months 32. Biotin supplementation (5 mg/day) was also found to be effective in controlling unruly hair and splitting nails in two toddlers with inherited uncombable hair syndrome 33. Although preliminary evidence suggests that supplemental biotin may help strengthen fragile nails 41, larger placebo-controlled trials are needed to assess the efficacy of high-dose biotin supplementation for the treatment of brittle fingernails.
Hair loss (alopecia)
Biotin administration has been associated with hair loss (alopecia) reversal in children treated with the anticonvulsant valproic acid, as well as with hair regrowth or normal hair growth in some children with inborn errors of biotin metabolism or other genetic disorders (i.e., uncombable hair syndrome) 42. Yet, while hair loss is a symptom of severe biotin deficiency, there are no published scientific studies that support the claim that high-dose biotin supplements are effective in preventing or treating hair loss in men or women 43, 44. Randomized, placebo-controlled trials in healthy individuals would be needed to evaluate this claim.
Diabetes mellitus
Overt biotin deficiency has been shown to impair glucose utilization in mice 45 and cause fatal hypoglycemia in chickens. Overt biotin deficiency likely also causes abnormalities in glucose regulation in humans (see biotin function). One early human study reported lower serum biotin concentrations in 43 patients with type 2 diabetes mellitus compared to 64 control subjects without the disease; an inverse relationship between fasting blood glucose and biotin concentrations was observed as well 46. In a small, randomized, placebo-controlled intervention study in 28 patients with type 2 diabetes, daily supplementation with 9 milligrams (mg) of biotin for one month resulted in a 45% decrease in mean fasting blood glucose concentrations 46. Yet, another small study in 10 patients with type 2 diabetes and 7 controls without diabetes found no effect of biotin supplementation (15 mg/day) for 28 days on fasting blood glucose concentrations in either group 47. A more recent double-blind, placebo-controlled study by the same research group showed that the same biotin regimen lowered plasma triglyceride concentrations in patients with hypertriglyceridemia — independent of whether they had type 2 diabetes 48. In this study, biotin administration did not affect blood glucose concentrations in either patient group. Additionally, a few studies have shown that co-supplementation with biotin and chromium picolinate may be a beneficial adjunct therapy in patients with type 2 diabetes 49, 50, 51, 52.
Potential mechanisms for the glucose and lipid effects have been suggested. As a cofactor of carboxylases required for fatty acid synthesis, biotin may increase the utilization of glucose for fat synthesis. Also, biotin stimulates glucokinase, a liver enzyme that increases synthesis of glycogen, the storage form of glucose. Biotin also triggers the secretion of insulin in the pancreas of rats and improves glucose homeostasis 37. Yet, reduced activity of acetyl-CoA carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2) would be expected to reduce fatty acid synthesis and increase fatty acid oxidation, respectively. Hence, whether pharmacologic doses of biotin benefits the management of hyperglycemia in patients with impaired glucose tolerance remains unclear. Moreover, whether supplemental biotin lowers the risk of cardiovascular complications in patients with diabetes by reducing serum triglycerides and LDL-cholesterol remains to be proven 48, 49, 50.
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease characterized by progressive damage to the myelin sheath surrounding nerve fibers (axons) and neuronal loss in the brain and spinal cord of affected individuals in anatomic locations that vary widely among affected individuals producing variable signs and symptoms. The progression of neurologic disabilities in multiple sclerosis (MS) patients is often assessed by the Expanded Disability Status Scale (EDSS) with scores from 1 to 10, from minimal signs of motor dysfunction (score of 1) to death by multiple sclerosis (score of 10). ATP deficiency due to mitochondrial dysfunction and increased oxidative stress may be partly responsible for the progressive degeneration of neurons in multiple sclerosis 53. Given its role in energy production by intermediary metabolism and fatty acid oxidation and in fatty acid synthesis (required for myelin formation) (see biotin function), high-dose biotin supplementation it has been hypothesized that to exert beneficial effects that would limit or reverse multiple sclerosis-associated functional impairments 53.
The mechanism of action of high-dose biotin has been investigated in a genetic mouse model of chronic axon injury caused by oxidative damage and bioenergetic failure. High-dose biotin restored redox homeostasis, mitochondria biogenesis, and ATP levels, and reversed axonal death and locomotor impairment. Dysregulation of the transcriptional program for lipid synthesis and degradation in the spinal cord was also normalized, possibly as the result of hyperactivation of a nutrient, energy or redox sensor that controls protein synthesis restoring lipid homeostasis.
A nonrandomized, uncontrolled pilot study in 23 patients with progressive multiple sclerosis (MS) found high doses of biotin (100-600 mg/day) to be associated with sustained clinical improvements in five (out of five) patients with progressive visual loss and 16 out of 18 patients with partial paralysis of the limbs after a mean three months following treatment onset 54. Additionally, a multicenter, randomized, placebo-controlled trial in 154 subjects with progressive multiple sclerosis (MS) reported that 13 out of 103 patients supplemented with high-dose, pharmaceutical-grade biotin (300 mg/day) for 12 months achieved MS-related disability reversal — assessed by improved Expanded Disability Status Scale (EDSS) or 25-foot walk time (37). In comparison, none of the 51 patients randomized to the placebo group showed significant clinical improvements 55. However, when this regimen of high-dose biotin supplementation was examined in a larger, international cohort of patients with progressive MS (326 patients receiving biotin and 316 patients receiving placebo), no benefits on EDSS or walk time were seen after 12 months 56. Moreover, a randomized, double-blind, placebo-controlled trial in 93 MS patients with chronic visual loss found that 300 mg/day of pharmaceutical-grade biotin for six months did not improve visual acuity, but an interesting trend favoring the biotin group was observed in the subgroup of patients with progressive optic neuritis 57. Moreover, a meta-analysis of three randomized controlled trials (2 on disability; 3 on adverse effects), involving 889 individuals diagnosed with MS (the preponderance of participants [830] had progressive MS while only 59 had remitting relapsing MS) was conducted 58. Pooling results of two trials found no benefit of high-dose biotin on multiple sclerosis (MS)-related disability, but there was significant heterogeneity between the trials. When the subgroup progressive multiple sclerosis (MS) was analyzed separately, a moderate certainty of evidence suggested a potential benefit in favor of high-dose biotin for the 25-foot minute walk time 58. On balance, studies remain inconclusive but promising.
Biotin-thiamine-responsive basal ganglia disease
Biotin-thiamine-responsive basal ganglia disease also called biotin-responsive basal ganglia disease, thiamine transporter-2 deficiency, and thiamine metabolism dysfunction syndrome-2, is caused by an autosomal recessive mutation in the SLC19A3 gene that codes for thiamin transporter-2 (THTR-2) 59. Biotin-thiamine-responsive basal ganglia disease usually presents around 3 to 10 years of age 59, but an early infantile form of the disease exists with onset as early as one month of age 60. Clinical features include subacute encephalopathy (confusion, drowsiness, altered level of consciousness), ataxia, and seizures.
A retrospective study of 18 affected individuals from the same family or the same tribe in Saudi Arabia showed that biotin monotherapy (5-10 mg/kg/day) efficiently abolished the clinical manifestations of the disease, although one-third of the patients suffered from recurrent acute crises. Often associated with poor outcomes, acute crises were not observed after thiamine (vitamin B1) supplementation started (300-400 mg/day) and during a five-year follow-up period, early diagnosis and immediate treatment with biotin and thiamin led to positive outcomes 61. Although the specific mechanism for therapeutic effects of biotin in biotin-thiamine-responsive basal ganglia disease remains unknown, lifelong high-dose supplementation with a combination of biotin and thiamin is the recommended treatment 59. Early diagnosis and treatment is important to ensure a better prognosis 60, 62.
Birth defects
Current research indicates that at least one-third of women develop marginal biotin deficiency during pregnancy 3. Small observational studies in pregnant women have reported an abnormally high urinary excretion of 3-hydroxyisovaleric acid in both early and late pregnancy, suggesting decreased activity of biotin-dependent methylcrotonyl-CoA carboxylase (MCC) 63, 64. In a randomized, single-blinded intervention study in 26 pregnant women, supplementation with 300 mcg/day of biotin for two weeks limited the excretion of 3-hydroxyisovaleric acid compared to placebo, confirming that increased 3-hydroxyisovaleric acid excretion indeed reflected marginal biotin deficiency in pregnancy 65. A small cross-sectional study in 22 pregnant women reported an incidence of low lymphocyte propionyl-CoA carboxylase (PCC) activity greater than 80% 66. Although these levels of biotin deficiency are not associated with overt signs of deficiency in pregnant women, such observations are sources of concern because subclinical biotin deficiency has been shown to cause cleft palate and limb hypoplasia in several animal species 66. In addition, biotin depletion has been found to suppress the expression of biotin-dependent carboxylases, remove biotin marks from histones, and decrease the proliferation in human embryonic palatal mesenchymal cells in culture 67. Impaired carboxylase activity may result in alterations in lipid metabolism, which have been linked to cleft palate and skeletal abnormalities in animals. Furthermore, biotin deficiency leading to reduced histone biotinylation at specific genomic loci may increase genomic instability and result in chromosome anomalies and fetal malformations 66.
Analogous to pregnant women who are advised to consume supplemental folic acid (vitamin B9) prior to and during pregnancy to prevent neural tube defects, it would also be prudent to ensure adequate biotin intake throughout pregnancy. The current Adequate Intake (intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an Recommended Dietary Allowance [RDA], the average daily level of intake sufficient to meet the nutrient requirements of nearly all (97%–98%) healthy individuals; often used to plan nutritionally adequate diets for individuals) for pregnant women is 30 mcg/day of biotin, and no toxicity has ever been reported at this level of intake.
Lipid metabolism
Consistent with roles of biotin-dependent acetyl-CoA carboxylases 1 (ACC1) and 2 (ACC2), and propionyl-CoA carboxylase (PCC) in lipid metabolism, biotin deficiency causes alterations of the fatty acid profile in liver, skin, and serum of several animal species 68. Biotin deficiency is associated with increased abundance of odd-chain fatty acids, suggesting that odd-chain fatty acid accumulation may be a marker for reduced propionyl-CoA carboxylase (PCC) activity in biotin deficiency. Biotin deficiency does not affect the fatty acid composition in brain tissue to the same extent as in liver 68.
Biotin deficiency also causes abnormalities in fatty acid composition in humans. In patients who developed biotin deficiency during parenteral alimentation, the percentage of odd-chain fatty acids (15:0, 17:0) in serum increased for each of the four major lipid classes, i.e. cholesterol esters, phospholipids, triglycerides, and free fatty acids 68. However, the relative changes in these four classes of lipids have not always been consistent among studies 68.
Immune function and cell stress
Biotin deficiency has adverse effects on cellular and humoral immune functions 69. For example, children with hereditary abnormalities of biotin metabolism developed Candida dermatitis and presented with absent delayed-hypersensitivity skin-tests responses, IgA deficiency, and subnormal percentages of T lymphocytes in peripheral blood 70. Recent studies have shown that biotin also regulates immunological and inflammatory function. Agrawal et al. 71 reported that biotin deficiency facilitates the secretion of tumour necrosis factor (TNF)-α, IL-1, IL-23, and IL-12p40 from dendritic cells, and that biotin-deficient dendritic cells induce significantly higher levels of secretion of IFN-γ, IL-17, and IL-22 from CD4 T cells. These findings suggest that biotin deficiency leads to a shift of Th-cell responses towards Th1/Th17. Among the various skin disorders, the IL-23/Th17 pathway 72 and the IL-1/IL-36 axis 73 have been considered to play a major role in the pathogenesis of psoriasis and pustular psoriasis, respectively.
In rodents, biotin deficiency decreases antibody synthesis 74, decreases the number of spleen cells and the percentage of B lymphocytes in spleen 75, and impairs thymocyte maturation 76. Decreased rates of cell proliferation may cause some of the effects of biotin on immune function 77, 78, 79.
Biotin deficiency is linked also to cell stress, enhancing the nuclear translocation of the transcription factor NF-kappaB in human lymphoid cells 80. NF-kappaB mediates activation of anti-apoptotic genes; this is associated with enhanced survival of biotin-deficient cells in response to cell death signals compared with biotin-sufficient controls 80. Stress-resistant Drosophila can be selected by feeding biotin-deficient diets for multiple generations 81.
How much biotin do I need?
The amount of biotin you need each day depends on your age. Average daily recommended amounts are listed below in micrograms (mcg).
Intake recommendations for biotin and other nutrients are provided in the Dietary Reference Intakes (DRIs) developed by the Food and Nutrition Board at the National Academies of Sciences, Engineering, and Medicine 82. Dietary Reference Intake is the general term for a set of reference values used for planning and assessing nutrient intakes of healthy people. These values, which vary by age and sex, include:
- Recommended Dietary Allowance (RDA): Average daily level of intake sufficient to meet the nutrient requirements of nearly all (97%–98%) healthy individuals; often used to plan nutritionally adequate diets for individuals.
- Adequate Intake (AI): Intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an recommended dietary allowance (RDA).
- Estimated Average Requirement (EAR): Average daily level of intake estimated to meet the requirements of 50% of healthy individuals; usually used to assess the nutrient intakes of groups of people and to plan nutritionally adequate diets for them; can also be used to assess the nutrient intakes of individuals.
- Tolerable Upper Intake Level (UL): Maximum daily intake unlikely to cause adverse health effects.
The Food and Nutrition Board found the available data to be insufficient to derive an Estimated Average Requirement (EAR) and Recommended Dietary Allowance (RDA) for biotin. For this reason, the Food and Nutrition Board established only adequate intakes (AIs) for biotin. The Food and Nutrition Board based its determination of adequate intakes (AIs) for all populations on the amount of biotin in human milk consumed by infants and then used body weight to extrapolate AIs for other groups 83. Table 1 lists the current adequate intakes (AIs) for biotin 82.
Table 1. Adequate Intakes (AIs) for Biotin
| Life Stage | Recommended Amount |
|---|---|
| Birth to 6 months | 5 mcg |
| Infants 7–12 months | 6 mcg |
| Children 1–3 years | 8 mcg |
| Children 4–8 years | 12 mcg |
| Children 9–13 years | 20 mcg |
| Teens 14–18 years | 25 mcg |
| Adults 19+ years | 30 mcg |
| Pregnant teens and women | 30 mcg |
| Breastfeeding teens and women | 35 mcg |
Biotin supplement
Biotin is found in some multivitamin or multimineral supplements, in B-complex supplements, and in supplements containing only biotin. The absorption rate of oral, free biotin is 100%, even when people consume pharmacologic doses of up to 20 mg/day biotin 84.
The following doses have been studied in scientific research:
Adults By mouth
- General: There is no recommended dietary allowance (RDA) established for biotin. The adequate intakes (AI) for biotin are 30 mcg for adults over 18 years and pregnant women, and 35 mcg for breast-feeding women.
- Biotin deficiency: Up to 10 mg daily has been used.
Children By Mouth
- General: There is no recommended dietary allowance (RDA) established for biotin. The adequate intakes (AI) for biotin are 7 mcg for infants 0-12 months, 8 mcg for children 1-3 years, 12 mcg for children 4-8 years, 20 mcg for children 9-13 years, and 25 mcg for adolescents 14-18 years.
- Biotin deficiency: Up to 10 mg daily has been used in infants.
What foods provide Biotin?
Many foods contain some biotin. You can get recommended amounts of biotin by eating a variety of foods, including the following 85:
- Meat, fish, eggs, and organ meats (such as liver)
- Seeds and nuts
- Certain vegetables (such as sweet potatoes, spinach, and broccoli)
Foods that contain the most biotin include organ meats, eggs, fish, meat, seeds, nuts, and certain vegetables (such as sweet potatoes) 5. The biotin content of food can vary; for example, plant variety and season can affect the biotin content of cereal grains, and certain processing techniques (e.g., canning) can reduce the biotin content of foods 86.
Dietary avidin, a glycoprotein in raw egg whites, binds tightly to dietary biotin and prevents biotin’s absorption in the gastrointestinal tract 87. Cooking denatures avidin, making it unable to interfere with biotin absorption 87.
Although there are no nationally representative estimates of biotin intakes in the United States, the average biotin intake from foods in other western populations is about 35–70 mcg/day, indicating that most people in these countries consume adequate amounts of biotin 5.
The U.S. Department of Agriculture’s (USDA’s) FoodData Central (https://fdc.nal.usda.gov) does not list the biotin content of foods or provide lists of foods containing biotin.
Several food sources of biotin are listed in Table 2.
Table 2. Biotin content of selected foods
| Food | Micrograms (mcg) per serving | Percent DV* |
|---|---|---|
| Beef liver, cooked, 3 ounces | 30.8 | 103 |
| Egg, whole, cooked | 10 | 33 |
| Salmon, pink, canned in water, 3 ounces | 5 | 17 |
| Pork chop, cooked, 3 ounces | 3.8 | 13 |
| Hamburger patty, cooked, 3 ounces | 3.8 | 13 |
| Sunflower seeds, roasted, ¼ cup | 2.6 | 9 |
| Sweet potato, cooked, ½ cup | 2.4 | 8 |
| Almonds, roasted, ¼ cup | 1.5 | 5 |
| Tuna, canned in water, 3 ounces | 0.6 | 2 |
| Spinach, boiled, ½ cup | 0.5 | 2 |
| Broccoli, fresh, ½ cup | 0.4 | 1 |
| Cheddar cheese, mild, 1 ounce | 0.4 | 1 |
| Milk, 2%, 1 cup | 0.3 | 1 |
| Plain yogurt, 1 cup | 0.2 | 1 |
| Oatmeal, 1 cup | 0.2 | 1 |
| Banana, ½ cup | 0.2 | 1 |
| Whole wheat bread, 1 slice | 0 | 0 |
| Apple, ½ cup | 0 | 0 |
Footnote: *DV = Daily Value. The U.S. Food and Drug Administration (FDA) developed Daily Values (DVs) to help consumers compare the nutrient contents of foods and dietary supplements within the context of a total diet. The Daily Value (DV) for biotin is 30 mcg for adults and children age 4 years and older 88. FDA does not require food labels to list biotin content unless biotin has been added to the food. Foods providing 20% or more of the DV are considered to be high sources of a nutrient, but foods providing lower percentages of the DV also contribute to a healthful diet.
[Source 89 ]Biotin deficiency causes
There are many causes of biotin deficiency or vitamin B7 deficiency. It can occur in rare inborn errors of metabolism, namely holocarboxylase synthetase deficiency or biotinidase deficiency 6, 90. Biotinidase deficiency is a rare autosomal recessive inherited disorder in which the body is unable to recycle biotin, leading to biotin deficiency despite normal biotin intake from foods such as liver, egg yolks, and milk 9. Biotinidase deficiency can present as severe biotin deficiency with both neurological and dermatological features. Biotinidase deficiency affects endogenous recycling and failure in releasing biotin from dietary protein. This affects the activity of 5 carboxylases that depends on biotin 91, 92.
Gastrointestinal tract bacterial imbalances resulting from broad-spectrum antibiotics use or inflammatory bowel disease can affect biotin synthesis in the intestine and thus lead to biotin deficiency 93.
Biotin deficiency can also occur in patients on parenteral nutrition without added biotin 6, 71, 94. Therefore, recommended daily dose of biotin must be added to total parental nutrition (TPN), particularly if TPN therapy is likely to be given for more than a week. Currently, all hospital pharmacies add biotin to TPN preparations 71, 95.
Low Biotin levels can occur in patients on antiepileptics (anticonvulsants) such as carbamazepine, phenytoin, and phenobarbital. Possible underlying mechanisms include impaired biotin uptake across the intestinal mucosa, exaggerated biotin catabolism, and inhibition of renal reabsorption. Therefore, patients who are likely to be on anticonvulsants for long periods should receive biotin supplementation 96, 97.
Prolonged use of oral antibiotics may also lead to biotin deficiency. The most likely underlying mechanism is the inhibition of intestinal flora, leading to reduced biotin production. Another possible explanation is the antibiotic-driven overgrowth of biotin-consuming bacteria 98.
Likewise, low biotin levels can occur in patients on isotretinoin (a vitamin-A derivative retinoid) for acne treatment, elderly individuals, people with alcohol use disorder, and smokers (particularly women) 99, 100, 101. Some studies have found biotin deficiency in a large percentage of pregnant and lactating women. Some experts argue that there can be teratogenic effects of decreased biotin levels, and a higher intake of biotin should be advised to pregnant women 102, 103, 66, 104, 105.
Reports exist of biotin deficiency in severely malnourished children in developing countries and through the intake of modified milk without biotin supplementation. Biotin deficiency has been observed in infants consuming hypoallergenic formulas 106.
Consuming large amounts of raw egg whites can lead to acquired biotin deficiency. Raw egg contains the glycoprotein avidin. Avidin binds to biotin in the gastrointestinal tract and prevents biotin absorption, also known as “egg white injury” 71, 90, 6, 69.
Groups at risk of biotin deficiency
The following groups are among those most likely to have inadequate biotin status:
Biotinidase deficiency
Biotinidase deficiency is a rare autosomal recessive inherited disorder in which the body is unable to recycle biotin, leading to biotin deficiency despite normal biotin intake from foods such as liver, egg yolks, and milk 9. Biotinidase deficiency is caused by genetic changes (mutations) in the BTD gene. The BTD gene provides instructions for making an enzyme called biotinidase 10. The biotinidase enzyme recycles biotin. Biotinidase removes biotin that is bound to proteins in food, leaving the vitamin in its free (unbound) state. Free biotin is needed by enzymes called biotin-dependent carboxylases to break down fats, proteins, and carbohydrates. Because several of these enzymes are impaired in biotinidase deficiency, the condition is considered a form of multiple carboxylase deficiency 9.
Biotinidase deficiency can be partial (10 to 30% of enzyme activity) or profound (less than 10% of enzyme activity), significantly impacting the treatment approach.
- Profound biotinidase deficiency results when the activity of biotinidase is reduced to less than 10 percent of normal 9. Profound biotinidase deficiency can lead to coma or death if treatment is not initiated rapidly.
- Partial biotinidase deficiency occurs when biotinidase activity is reduced to between 10 percent and 30 percent of normal 9.
- Without enough of Biotinidase enzyme, biotin cannot be recycled. The resulting shortage of free biotin impairs the activity of biotin-dependent carboxylases, leading to a buildup of potentially toxic compounds in the body. If biotinidase deficiency is not treated promptly, this buildup damages various cells and tissues, causing the signs and symptoms described below.
Infants with biotinidase deficiency may be born without signs of the condition 10. Symptoms of biotinidase deficiency usually appear after the first few weeks or months of life 10. Treating biotinidase deficiency with biotin supplements before symptoms show up can prevent them from happening 10. Below is a list of symptoms that infants and children with profound untreated biotinidase deficiency may have. It is important to know that not every person with biotinidase deficiency will show all of these symptoms.
Many of the symptoms of biotinidase deficiency are neurological, which means they affect the brain and nervous system. About 70% of infants with biotinidase deficiency will experience seizures if they are not treated 10. This is often the first symptom of biotinidase deficiency. Seizures in infants may look different than seizures in adults. Some signs of seizures in infants include:
- Staring spells
- Jerking arm or leg movements
- Stiffening of the body
- Flickering of the eyelids
Because the seizures are caused by the body being unable to recycle biotin, they may not stop with seizure medications (anticonvulsants) 10. However, the biotinidase deficiency seizures do respond to biotin therapy and often should stop within minutes to hours of receiving biotin treatment 10.
Some infants with biotinidase deficiency may have weak muscles and low muscle tone called hypotonia. Infants with hypotonia may look abnormally “floppy.” Hypotonia can affect feeding and motor skills such sitting up without assistance. Biotinidase deficient infants and children may experience delays in reaching developmental milestones, including holding one’s head up or pulling up to stand.
Infants with biotinidase deficiency may also have breathing problems, hearing and vision loss, problems with movement and balance (ataxia) 9. These issues can be prevented if biotin therapy is started early. Some other common features of biotinidase deficiency include eye infections, like pink eye (conjunctivitis), hair loss (alopecia), certain type of skin rash called eczema and a fungal infection called candidiasis. Infants with biotinidase deficiency may have specific molecules in their urine, such as lactic acid (lactic aciduria) or low but noticeable amounts of ammonia.
Some infants with biotinidase deficiency may have other symptoms like:
- Trouble controlling their body’s movements (ataxia)
- Breathing problems
- Drowsiness (lethargy)
- Enlarged liver (hepatomegaly)
- Enlarged spleen (splenomegaly)
- Speech problems.
Partial biotinidase deficiency is a milder form of this condition 9. Without treatment, affected children and adults with partial biotinidase who don’t receive biotin supplements may experience hypotonia (weak muscles and low muscle tone), skin rashes, and hair loss (alopecia), but these problems may appear only during illness, infection, or other times of stress 9.
Profound or partial biotinidase deficiency occurs in approximately 1 in 60,000 newborns 9. All newborns in the United States and many other countries are screened for biotinidase deficiency 107. According to the worldwide neonatal screening survey, the incidence of profound biotinidase deficiency is one in 112,271, and the incidence of partial biotinidase deficiency is one in 129,282 93. The combined incidence of profound and partial biotinidase deficiency is one in 60,089 live births 93. In 2006, the incidence of profound biotinidase deficiency was 1 in 80,000, and the incidence of partial biotinidase deficiency cases was from 1 per 31,000 to 1 per 40,000 in the US. Biotinidase deficiency has been diagnosed more commonly in children of the White race. Research has observed a higher incidence of biotin deficiency in Brazil, Turkey, and Saudi Arabia 108, 91.
The estimated carrier frequency is 1 in 123 individuals. The rates vary from country to country 109, 110, 111.
Early diagnosis and treatment of biotinidase deficiency can prevent symptoms from happening. Nearly all infants with either profound or partial biotinidase deficiency can be detected in the US by newborn screening for metabolic disorders. However, not every country has added biotinidase deficiency to its newborn screening program and the late-onset forms of biotinidase deficiency have been recently described 112, 113, 114. Because the newborn screen is a screening test, a positive result does not mean that an infant definitely has biotinidase deficiency. Often, a repeat test must be done to confirm the diagnosis. A clinical diagnosis is possible after birth by testing for biotinidase activity in the blood 10. Usually, this is performed when signs and symptoms of biotinidase deficiency become clearer. In some infants, a genetic test may be ordered to identify the specific gene changes (mutation) that are causing biotinidase deficiency 10. Prenatal testing of sample fluid from the womb for biotinidase activity is available as early as 12 weeks of pregnancy (this includes chorionic villi sampling and amniocentesis) 10.
Biotinidase activity can be measured in serum, plasma, and also in fibroblasts and leukocytes and other tissue extracts by radioassay 115. The measurement of biotinidase activity in plasma or serum by colorimetric assay is the most frequently used method for the diagnosis of biotinidase deficiency 116. Normal serum biotinidase activity in humans ranges from 4.4 to 10 nmol/min/mL with a mean activity and standard deviation of 7.1 ± 1.2 nmol/min/mL 117.
Serum biotinidase activity of carriers may be similar to those with partial biotinidase deficiency, confounding diagnosis based on enzyme analysis 118. Wolf 119 suggested that evaluation of parental biotinidase activity may be helpful. Mutation in the biotinidase deficiency gene results in deficient levels of enzyme activity 120. In some cases the enzyme activity does not differentiate partial deficiency from heterozygosity for profound deficiency, and genetic analysis is necessary 121.
Biochemically, in untreated patients, metabolic ketoacidosis, lactic acidosis, and/or hyperammonemia can occur 109. Elevation of 3-hydroxyisovaleric, 3-hydroxypropionic, lactic acid, and 3-methylcrotonylglycine can be detected in urine organic acid analysis 92, 122. In previous reports, it showed that urinary excretion of 3-hydroxyisovaleric acid was an indicator of biotin status 123.
Without treatment with biotin, infants with profound biotinidase deficiency can lead to coma or death 124. Because treatment with oral biotin with as much as 5 to 20 milligrams (mg) of biotin daily starting at birth (or before symptoms develop) and continuing for the rest of the person’s life can prevent these symptoms and complications from occurring or improve them if they have already developed 109, 125. However, it takes a few hours to days for seizures and movement disorders to improve and some weeks for skin manifestations to improve. Sometimes, this dose will not be adequate, and the clinical signs may persist. Increasing the oral biotin dose to 40 mg/day is recommended in such scenarios 125. Smaller oral biotin doses may be sufficient, especially later in childhood 126, 109.
In addition to oral biotin therapy, children with residual neurologic deficits may need medical interventions for spasticity, developmental delay, and bulbar dysfunction. Dystonia and spasticity may be treated with intrathecal baclofen and neurotoxins 127.
Biotinidase deficiency prognosis is characteristically good when biotin therapy is introduced in infancy or early childhood and reliably continued for life 17, 126. The prognosis for asymptomatic biotinidase deficiency cases is good if they receive treatment before the symptoms appear. For symptomatic biotinidase deficiency patients, pharmacological biotin therapy improves most clinical features but cannot reverse neurologic damage that has already occurred 128.
Chronic alcohol exposure
Chronic exposure to alcohol inhibits the absorption of biotin 100, 101. Plasma biotin concentrations are low in 15% of people with chronic alcoholism 129. People who excessively consume alcohol have a relatively higher incidence of low biotin levels compared to the general population 6.
Pregnant and breastfeeding women
At least a third of pregnant women develop marginal biotin deficiency in spite of normal biotin intakes 63, 65, 130; plasma and breastmilk concentrations of biotin decrease in lactating women, even when their dietary biotin intakes exceed the adequate intake (AI) 131. Additional research is needed to understand the clinical significance of these findings.
Holocarboxylase synthetase deficiency
Holocarboxylase synthetase deficiency is an inherited autosomal recessive disorder in which the body is unable to use the vitamin biotin effectively 132, 133. Holocarboxylase synthetase deficiency is classified as a multiple carboxylase deficiency, which is a group of disorders characterized by impaired activity of certain enzymes that depend on biotin.
Mutations in the HLCS gene cause holocarboxylase synthetase deficiency 132. The HLCS gene provides instructions for making an enzyme called holocarboxylase synthetase. The holocarboxylase synthetase enzyme is important for the effective use of biotin, a B vitamin found in foods such as liver, egg yolks, and milk. Holocarboxylase synthetase attaches biotin to certain enzymes that are essential for the normal production and breakdown of proteins, fats, and carbohydrates in the body. Mutations in the HLCS gene reduce the holocarboxylase synthetase enzyme’s ability to attach biotin to these enzymes, preventing them from processing nutrients properly and disrupting many cellular functions. These defects lead to the serious medical problems associated with holocarboxylase synthetase deficiency.
The signs and symptoms of holocarboxylase synthetase deficiency typically appear within first few days or first 2 months of life, but the age of onset varies. Affected infants often have difficulty feeding, breathing problems (tachypnea), a skin rash (periorificial and intertriginous dermatitis), hair loss (alopecia), and a lack of energy (lethargy) 132. Seborrheic dermatitis, psoriatic dermatitis and ichthyosis are also seen in some patients 13. The pathogenesis of skin manifestations in holocarboxylase synthetase deficiency has not been fully explained. It has been postulated that a defect of fatty acid synthesis due to reduced activity of acetyl CoA carboxylase is implicated in the skin manifestations of multiple carboxylase deficiency and biotin deficiency 134. Nakajima et al. 135 reported that newborn serine palmitoyltransferase (SPT)-knockout mice showed significantly decreased epidermal levels of ceramide. They then developed alopecia and psoriasis-like skin lesions mediated by interleukin (IL)-23-dependent γδ T cells, which produce IL-17 and IL-22. Therefore, ceramide deficiency resulting from a defect of fatty acid synthesis may lead to psoriasis-like lesions in holocarboxylase synthetase deficiency, although the exact mechanism remains to be determined. These medical problems may be life-threatening in some cases.
Immediate treatment and lifelong management with biotin supplements may prevent many of these complications. If left untreated, holocarboxylase synthetase deficiency can lead to delayed development, seizures, and coma.
Holocarboxylase synthetase deficiency results in decreased formation of all holocarboxylases at physiological blood biotin concentrations; thus, high-dose biotin supplementation (10-80 mg of biotin daily) is required 17. Holocarboxylase synthetase deficiency responds to supplementation with pharmacologic doses of biotin in some cases but not others. The prognosis of holocarboxylase synthetase is usually, but not always, good if biotin therapy is introduced early (even antenatally) and continued for life 136, 17.
Biotin transport deficiency
There has been one case report of a child with biotin transport deficiency who responded to high-dose biotin supplementation 137. Of note, the presence of a defective human sodium-dependent multivitamin transporter (hSMVT) was ruled out as a cause of biotin transport deficiency.
Risk factors for biotin deficiency
Aside from prolonged consumption of raw egg white or total intravenous nutritional support lacking biotin, other conditions may increase the risk of biotin depletion. Smoking has been associated with increased biotin catabolism 99. The rapidly dividing cells of the developing fetus require biotin for synthesis of essential carboxylases and for histone biotinylation; hence, the maternal biotin requirement is likely increased during pregnancy. Research suggests that a substantial number of women develop marginal or subclinical biotin deficiency during normal pregnancy 3, 138, 66. Moreover, certain types of liver disease may decrease biotinidase activity and theoretically increase the requirement for biotin. For example, a study of 62 children with chronic liver disease and 27 healthy controls found serum biotinidase activity to be abnormally low in those with severely impaired liver function due to cirrhosis 139. However, this study did not provide evidence of biotin deficiency. Additionally, anticonvulsant medications used to prevent seizures in individuals with epilepsy increase the risk of biotin depletion 140, 141.
Biotin deficiency signs and symptoms
Signs of overt biotin deficiency include hair loss (alopecia) and a scaly red rash around the eyes, nose, mouth, and genital area (seborrheic dermatitis). Neurologic symptoms in adults have included depression, lethargy, hallucinations, numbness and tingling of the extremities (paresthesia), ataxia, and seizures. The characteristic facial rash, together with unusual facial fat distribution, has been termed the “biotin deficient facies” by some investigators 87. Individuals with hereditary disorders of biotin metabolism (e.g., biotinidase deficiency and holocarboxylase synthetase deficiency) that result in functional biotin deficiency often have similar physical findings, impaired immune system function, and increased susceptibility to bacterial and fungal infections 17, 142.
Biotin deficiency complications
Since biotin plays a crucial role in maintaining cell-mediated and humoral immunity, biotin deficiency due to inborn errors of metabolism can cause candidiasis of the skin in infants and children. There may be IgA deficiency and low percentages of T lymphocytes. They may have absent delayed-hypersensitivity skin-test responses 108, 69.
Biotin deficiency can cause encephalopathies. Patients usually respond well to large doses of biotin. Evidence shows that a lack of biotin is teratogenic in animal models. Strains of mice with biotin deficiency developed fetal malformations, most commonly cleft palate, micrognathia, and micromelia 143, 144.
Biotin deficiency diagnosis
The diagnostic tests for biotin deficiency are urinary 3-hydroxyisovaleric acid and biotin and the status of propionyl-CoA carboxylase (PCC) in lymphocytes 69, 108. Biotin-dependent carboxylases in human lymphocytes are reliable markers for determining biotin status. Decreased beta-methylcrotonyl-CoA carboxylase activity shunts the catabolism to alternative pathways, leading to the elevated formation of 3-hydroxyisovaleric acid. The most reliable marker of biotin deficiency is increased excretion of 3-hydroxyisovaleric acid in the urine (over 195 micromol/24 hours) 123. Evidence shows that serum biotin concentration does not decrease in biotin deficiency patients receiving biotin-free total parenteral nutrition. Therefore, serum biotin levels are not reliable indicators of marginal biotin deficiency 6, 69. If biotin deficiency is suspected, it warrants a thorough neurological examination and other investigations, including vision and hearing testing.
Biotinidase deficiency confirmation is done by DNA analysis, either allele-targeted methods or full-gene sequencing. Currently, all newborn screening programs in the U.S. and more than 30 other countries carry out screening for biotinidase deficiency 91, 124, 92.
Various MRI changes have been reported in individuals with biotinidase deficiency 145, 146. Desai et al. 146 noted MRI findings in four patients and observed:
- Encephalopathy
- Low cerebral volume
- Ventriculomegaly
- Widened extracerebral cerebrospinal fluid spaces
A complete reversal of these findings occurred with biotin use in two patients.
Biotin deficiency treatment
Biotin deficiency treatment essentially means treating the underlying cause 93. Lifelong treatment with biotin supplements is required in patients with genetic disorders disrupting biotin metabolism, such as holocarboxylase synthetase deficiency and biotinidase deficiency 93. If biotin deficiency is related to excess consumption of raw eggs, it should be stopped, and biotin replacement should ensue. Change anticonvulsants if the Biotin deficiency is because of the use of a particular anticonvulsant. Similarly, those on prolonged oral antibiotic therapy may benefit from biotin supplementation.
Oral biotin supplements have high bioavailability. Usually, a dose of 5 mg/day is given regardless of the cause of biotin deficiency 6. The Food and Nutrition Board of the National Research Council recommends a range of 5 mcg/day in newborn infants to 35 mcg/day in lactating women.
Healthcare professionals should be aware that biotin requirements may increase during anticonvulsant therapy 69. In biotinidase deficiency, patient therapy typically consists of lifelong doses of biotin. Biotin doses in the range of 5 to 20 mg can treat and prevent clinical signs and biotinidase deficiency symptoms 92, 108.
In the cases of holocarboxylase synthetase deficiency detected antenatally, antenatal biotin treatment has been found to be very useful 136.
Biotin deficiency prognosis
Biotin deficiency is rare and has a relatively good prognosis 93. Children diagnosed with biotinidase deficiency require early intervention and life-long biotin treatment. Children who quit therapy develop symptoms again within weeks to months. When neonates diagnosed by neonatal screening receive biotin, they develop normally without having any symptoms, and those with symptoms respond quickly to biotin treatment. Failure to evaluate and manage biotinidase deficiency at an early stage can cause irreversible neurodevelopmental abnormalities and lead to developmental delay and autistic behavior 91, 92, 108.
References- Perry CA, West AA, Gayle A, Lucas LK, Yan J, Jiang X, et al. Pregnancy and lactation alter biomarkers of biotin metabolism in women consuming a controlled diet. J Nutr 2014;144:1977-84.
- Mock DM. Biotin. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:43-51
- Mock DM. Biotin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:390-398.
- Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Inc.; 2012:359-374.
- Mock DM. Biotin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:390-8.
- Trüeb RM. Serum Biotin Levels in Women Complaining of Hair Loss. Int J Trichology. 2016 Apr-Jun;8(2):73-7. doi: 10.4103/0974-7753.188040
- Seymons K, De Moor A, De Raeve H, Lambert J. Dermatologic signs of biotin deficiency leading to the diagnosis of multiple carboxylase deficiency. Pediatr Dermatol. 2004 May-Jun;21(3):231-5. doi: 10.1111/j.0736-8046.2004.21308.x
- Fujimoto, W., Inaoki, M., Fukui, T., Inoue, Y. and Kuhara, T. (2005), Biotin Deficiency in an Infant Fed with Amino Acid Formula. The Journal of Dermatology, 32: 256-261. https://doi.org/10.1111/j.1346-8138.2005.tb00758.x
- Biotinidase deficiency. https://medlineplus.gov/genetics/condition/biotinidase-deficiency
- Biotinidase Deficiency. https://rarediseases.org/rare-diseases/biotinidase-deficiency
- Rajendiran A, Sampath S. Biotinidase deficiency–clinching the diagnosis rapidly can make all the difference! BMJ Case Rep. 2011 Sep 28;2011:bcr0720114494. doi: 10.1136/bcr.07.2011.4494
- Biotin responsive dermatoses. https://dermnetnz.org/topics/biotin-responsive-dermatoses
- Watabe D, Watanabe A, Akasaka T, Sakamoto O, Amano H. Psoriasis-like Dermatitis in Adulthood: A Skin Manifestation of Holocarboxylase Synthetase Deficiency. Acta Derm Venereol. 2018 Aug 29;98(8):805-806. doi: 10.2340/00015555-2954
- Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:359-74
- Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:359-74.
- Said HM. Biotin: biochemical, physiological and clinical aspects. Subcell Biochem 2012;56:1-19.
- Elrefai S, Wolf B. Disorders of biotin metabolism. In: Rosenberg RN, Pascual JM, eds. Rosenberg’s Molecular and Genetic basis of Neurological and Psychiatric Disease. 5th ed. United States of America: Elsevier; 2015:531-539.
- Regula Baumgartner, E. and Suormala, T. (1999), Inherited defects of biotin metabolism. BioFactors, 10: 287-290. https://doi.org/10.1002/biof.5520100229
- Eng WK, Giraud D, Schlegel VL, Wang D, Lee BH, Zempleni J. Identification and assessment of markers of biotin status in healthy adults. Br J Nutr 2013;110:321-9
- Li D, Radulescu A, Shrestha RT, Root M, Karger AB, Killeen AA, et al. Association of biotin ingestion with performance of hormone and nonhormone assays in healthy adults. JAMA 2017;318:1150-60
- Zempleni J, Mock DM. Biotin biochemistry and human requirements. J Nutr Biochem. 1999 Mar;10(3):128-38. doi: 10.1016/s0955-2863(98)00095-3
- Present Knowledge in Nutrition. Basic Nutrition and Metabolism 11th Edition pp. 289–304, July 17, 2020. ISBN: 9780323661621
- Biotin. https://lpi.oregonstate.edu/mic/vitamins/biotin
- Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253-72. doi: 10.1146/annurev.nutr.28.061807.155434
- Mock D, Matthews N. Biotin and pantothenic acid In: Stipanuk MH, Caudill MA, editors. Biochemical, physiological and molecular aspects of human nutrition. 3rd ed Amsterdam (Netherlands): Elsevier; 2012. p. 610–25.
- Zempleni J, Teixeira DC, Kuroishi T, Cordonier EL, Baier S. Biotin requirements for DNA damage prevention. Mutat Res. 2012 May 1;733(1-2):58-60. doi: 10.1016/j.mrfmmm.2011.08.001
- Zempleni J, Li Y, Xue J, Cordonier EL. The role of holocarboxylase synthetase in genome stability is mediated partly by epigenomic synergies between methylation and biotinylation events. Epigenetics. 2011 Jul;6(7):892-4. doi: 10.4161/epi.6.7.15544
- Zempleni J, Gralla M, Camporeale G, Hassan YI. Sodium-dependent multivitamin transporter gene is regulated at the chromatin level by histone biotinylation in human Jurkat lymphoblastoma cells. J Nutr. 2009 Jan;139(1):163-6. doi: 10.3945/jn.108.091967
- Zempleni J, Wijeratne SSK, Hassan YI. Biotin. Biofactors 2009;35:36-46.
- Colombo VE, Gerber F, Bronhofer M, Floersheim GL. Treatment of brittle fingernails and onychoschizia with biotin: scanning electron microscopy. J Am Acad Dermatol. 1990 Dec;23(6 Pt 1):1127-32. doi: 10.1016/0190-9622(90)70345-i
- Floersheim GL. Behandlung brüchiger Fingernägel mit Biotin [Treatment of brittle fingernails with biotin]. Z Hautkr. 1989 Jan 15;64(1):41-8. German.
- Hochman LG, Scher RK, Meyerson MS. Brittle nails: response to daily biotin supplementation. Cutis. 1993 Apr;51(4):303-5.
- Boccaletti V, Zendri E, Giordano G, Gnetti L, De Panfilis G. Familial Uncombable Hair Syndrome: Ultrastructural Hair Study and Response to Biotin. Pediatr Dermatol. 2007 May-Jun;24(3):E14-6. doi: 10.1111/j.1525-1470.2007.00385.x
- Shelley WB, Shelley ED. Uncombable hair syndrome: observations on response to biotin and occurrence in siblings with ectodermal dysplasia. J Am Acad Dermatol. 1985 Jul;13(1):97-102. doi: 10.1016/s0190-9622(85)70150-8
- Mock DM, Baswell DL, Baker H, Holman RT, Sweetman L. Biotin deficiency complicating parenteral alimentation: diagnosis, metabolic repercussions, and treatment. J Pediatr. 1985 May;106(5):762-9. doi: 10.1016/s0022-3476(85)80350-4
- Fujimoto W, Inaoki M, Fukui T, Inoue Y, Kuhara T. Biotin deficiency in an infant fed with amino acid formula. J Dermatol. 2005 Apr;32(4):256-61. doi: 10.1111/j.1346-8138.2005.tb00758.x
- Randhawa SS, Dua K, Randhawa CS, Randhawa SS, Munshi SK. Effect of biotin supplementation on hoof health and ceramide composition in dairy cattle. Vet Res Commun. 2008 Dec;32(8):599-608. doi: 10.1007/s11259-008-9060-z
- Reilly JD, Cottrell DF, Martin RJ, Cuddeford DJ. Effect of supplementary dietary biotin on hoof growth and hoof growth rate in ponies: a controlled trial. Equine Vet J Suppl. 1998 Sep;(26):51-7. doi: 10.1111/j.2042-3306.1998.tb05122.x
- Zenker W, Josseck H, Geyer H. Histological and physical assessment of poor hoof horn quality in Lipizzaner horses and a therapeutic trial with biotin and a placebo. Equine Vet J. 1995 May;27(3):183-91. doi: 10.1111/j.2042-3306.1995.tb03061.x
- Romero-Navarro G, Cabrera-Valladares G, German MS, Matschinsky FM, Velazquez A, Wang J, Fernandez-Mejia C. Biotin regulation of pancreatic glucokinase and insulin in primary cultured rat islets and in biotin-deficient rats. Endocrinology. 1999 Oct;140(10):4595-600. doi: 10.1210/endo.140.10.7084
- Lipner SR, Scher RK. Biotin for the treatment of nail disease: what is the evidence? J Dermatolog Treat. 2018 Jun;29(4):411-414. doi: 10.1080/09546634.2017.1395799
- Walth CB, Wessman LL, Wipf A, Carina A, Hordinsky MK, Farah RS. Response to: “Rethinking biotin therapy for hair, nail, and skin disorders”. J Am Acad Dermatol. 2018 Dec;79(6):e121-e124. doi: 10.1016/j.jaad.2018.07.055
- Famenini S, Goh C. Evidence for supplemental treatments in androgenetic alopecia. J Drugs Dermatol. 2014 Jul;13(7):809-12. https://jddonline.com/articles/evidence-for-supplemental-treatments-in-androgenetic-alopecia-S1545961614P0809X
- Patel DP, Swink SM, Castelo-Soccio L. A Review of the Use of Biotin for Hair Loss. Skin Appendage Disord. 2017 Aug;3(3):166-169. doi: 10.1159/000462981 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5582478
- Larrieta E, Vega-Monroy ML, Vital P, Aguilera A, German MS, Hafidi ME, Fernandez-Mejia C. Effects of biotin deficiency on pancreatic islet morphology, insulin sensitivity and glucose homeostasis. J Nutr Biochem. 2012 Apr;23(4):392-9. doi: 10.1016/j.jnutbio.2011.01.003
- Maebashi M, Makino Y, Furukawa Y, Ohinata K, Kimura S, Sato T. Therapeutic evaluation of the effect of biotin on hyperglycemia in pateints with non-insulin dependent diabetes mellitus. J Clin Biochem Nutr.1993;14:211-218.
- Báez-Saldaña A, Zendejas-Ruiz I, Revilla-Monsalve C, Islas-Andrade S, Cárdenas A, Rojas-Ochoa A, Vilches A, Fernandez-Mejia C. Effects of biotin on pyruvate carboxylase, acetyl-CoA carboxylase, propionyl-CoA carboxylase, and markers for glucose and lipid homeostasis in type 2 diabetic patients and nondiabetic subjects. Am J Clin Nutr. 2004 Feb;79(2):238-43. doi: 10.1093/ajcn/79.2.238
- Revilla-Monsalve C, Zendejas-Ruiz I, Islas-Andrade S, Báez-Saldaña A, Palomino-Garibay MA, Hernández-Quiróz PM, Fernandez-Mejia C. Biotin supplementation reduces plasma triacylglycerol and VLDL in type 2 diabetic patients and in nondiabetic subjects with hypertriglyceridemia. Biomed Pharmacother. 2006 May;60(4):182-5. doi: 10.1016/j.biopha.2006.03.005
- Geohas J, Daly A, Juturu V, Finch M, Komorowski JR. Chromium picolinate and biotin combination reduces atherogenic index of plasma in patients with type 2 diabetes mellitus: a placebo-controlled, double-blinded, randomized clinical trial. Am J Med Sci. 2007 Mar;333(3):145-53. doi: 10.1097/MAJ.0b013e318031b3c9
- Albarracin C, Fuqua B, Geohas J, Juturu V, Finch MR, Komorowski JR. Combination of chromium and biotin improves coronary risk factors in hypercholesterolemic type 2 diabetes mellitus: a placebo-controlled, double-blind randomized clinical trial. J Cardiometab Syndr. 2007 Spring;2(2):91-7. doi: 10.1111/j.1559-4564.2007.06366.x
- Singer GM, Geohas J. The effect of chromium picolinate and biotin supplementation on glycemic control in poorly controlled patients with type 2 diabetes mellitus: a placebo-controlled, double-blinded, randomized trial. Diabetes Technol Ther. 2006 Dec;8(6):636-43. doi: 10.1089/dia.2006.8.636
- Albarracin CA, Fuqua BC, Evans JL, Goldfine ID. Chromium picolinate and biotin combination improves glucose metabolism in treated, uncontrolled overweight to obese patients with type 2 diabetes. Diabetes Metab Res Rev. 2008 Jan-Feb;24(1):41-51. doi: 10.1002/dmrr.755
- Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2016 Nov;110(Pt B):644-653. doi: 10.1016/j.neuropharm.2015.08.028
- Sedel F, Papeix C, Bellanger A, Touitou V, Lebrun-Frenay C, Galanaud D, Gout O, Lyon-Caen O, Tourbah A. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015 Mar;4(2):159-69. doi: 10.1016/j.msard.2015.01.005
- Tourbah A, Lebrun-Frenay C, Edan G, Clanet M, Papeix C, Vukusic S, De Sèze J, Debouverie M, Gout O, Clavelou P, Defer G, Laplaud DA, Moreau T, Labauge P, Brochet B, Sedel F, Pelletier J; MS-SPI study group. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult Scler. 2016 Nov;22(13):1719-1731. doi: 10.1177/1352458516667568
- Cree BAC, Cutter G, Wolinsky JS, Freedman MS, Comi G, Giovannoni G, Hartung HP, Arnold D, Kuhle J, Block V, Munschauer FE, Sedel F, Lublin FD; SPI2 investigative teams. Safety and efficacy of MD1003 (high-dose biotin) in patients with progressive multiple sclerosis (SPI2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2020 Dec;19(12):988-997. doi: 10.1016/S1474-4422(20)30347-1
- Tourbah A, Gout O, Vighetto A, Deburghgraeve V, Pelletier J, Papeix C, Lebrun-Frenay C, Labauge P, Brassat D, Toosy A, Laplaud DA, Outteryck O, Moreau T, Debouverie M, Clavelou P, Heinzlef O, De Sèze J, Defer G, Sedel F, Arndt C. MD1003 (High-Dose Pharmaceutical-Grade Biotin) for the Treatment of Chronic Visual Loss Related to Optic Neuritis in Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Study. CNS Drugs. 2018 Jul;32(7):661-672. doi: 10.1007/s40263-018-0528-2
- Espiritu AI, Remalante-Rayco PPM. High-dose biotin for multiple sclerosis: A systematic review and meta-analyses of randomized controlled trials. Mult Scler Relat Disord. 2021 Oct;55:103159. doi: 10.1016/j.msard.2021.103159
- Tabarki B, Al-Hashem A, Alfadhel M. Biotin-Thiamine-Responsive Basal Ganglia Disease. 2013 Nov 21 [Updated 2020 Aug 20]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK169615
- Kılıç B, Topçu Y, Dursun Ş, Erol İ, Dolu MH, Taşdemir HA, Aydın K. Single gene, two diseases, and multiple clinical presentations: Biotin-thiamine-responsive basal ganglia disease. Brain Dev. 2020 Sep;42(8):572-580. doi: 10.1016/j.braindev.2020.05.008
- Alfadhel M, Almuntashri M, Jadah RH, Bashiri FA, Al Rifai MT, Al Shalaan H, Al Balwi M, Al Rumayan A, Eyaid W, Al-Twaijri W. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J Rare Dis. 2013 Jun 6;8:83. doi: 10.1186/1750-1172-8-83
- Algahtani H, Ghamdi S, Shirah B, Alharbi B, Algahtani R, Bazaid A. Biotin-thiamine-responsive basal ganglia disease: catastrophic consequences of delay in diagnosis and treatment. Neurol Res. 2017 Feb;39(2):117-125. doi: 10.1080/01616412.2016.1263176
- Mock DM, Stadler DD. Conflicting indicators of biotin status from a cross-sectional study of normal pregnancy. J Am Coll Nutr. 1997 Jun;16(3):252-7. doi: 10.1080/07315724.1997.10718682
- Mock DM, Stadler DD, Stratton SL, Mock NI. Biotin status assessed longitudinally in pregnant women. J Nutr. 1997 May;127(5):710-6. doi: 10.1093/jn/127.5.710
- Mock DM, Quirk JG, Mock NI. Marginal biotin deficiency during normal pregnancy. Am J Clin Nutr. 2002 Feb;75(2):295-9. doi: 10.1093/ajcn/75.2.295
- Mock DM. Marginal biotin deficiency is common in normal human pregnancy and is highly teratogenic in mice. J Nutr. 2009 Jan;139(1):154-7. doi: 10.3945/jn.108.095273
- Takechi R, Taniguchi A, Ebara S, Fukui T, Watanabe T. Biotin deficiency affects the proliferation of human embryonic palatal mesenchymal cells in culture. J Nutr. 2008 Apr;138(4):680-4. doi: 10.1093/jn/138.4.680
- Zempleni J, Mock DM. Marginal biotin deficiency is teratogenic. Proc Soc Exp Biol Med. 2000 Jan;223(1):14-21. doi: 10.1046/j.1525-1373.2000.22303.x
- Zempleni J, Wijeratne SS, Hassan YI. Biotin. Biofactors. 2009 Jan-Feb;35(1):36-46. doi: 10.1002/biof.8
- Cowan MJ, Wara DW, Packman S, Ammann AJ, Yoshino M, Sweetman L, Nyhan W. Multiple biotin-dependent carboxylase deficiencies associated with defects in T-cell and B-cell immunity. Lancet. 1979 Jul 21;2(8134):115-8. doi: 10.1016/s0140-6736(79)90002-3
- Agrawal S, Agrawal A, Said HM. Biotin deficiency enhances the inflammatory response of human dendritic cells. Am J Physiol Cell Physiol. 2016 Sep 1;311(3):C386-91. doi: 10.1152/ajpcell.00141.2016
- Boehncke WH, Schön MP. Psoriasis. Lancet. 2015 Sep 5;386(9997):983-94. doi: 10.1016/S0140-6736(14)61909-7
- Johnston A, Xing X, Wolterink L, Barnes DH, Yin Z, Reingold L, Kahlenberg JM, Harms PW, Gudjonsson JE. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol. 2017 Jul;140(1):109-120. doi: 10.1016/j.jaci.2016.08.056
- Kumar M, Axelrod AE. Cellular antibody synthesis in thiamin, riboflavin, biotin and folic acid-deficient rats. Proc Soc Exp Biol Med. 1978 Mar;157(3):421-3. doi: 10.3181/00379727-157-40068
- Báez-Saldaña A, Díaz G, Espinoza B, Ortega E. Biotin deficiency induces changes in subpopulations of spleen lymphocytes in mice. Am J Clin Nutr. 1998 Mar;67(3):431-7. doi: 10.1093/ajcn/67.3.431
- Báez-Saldaña A, Ortega E. Biotin deficiency blocks thymocyte maturation, accelerates thymus involution, and decreases nose-rump length in mice. J Nutr. 2004 Aug;134(8):1970-7. doi: 10.1093/jn/134.8.1970
- Manthey KC, Griffin JB, Zempleni J. Biotin supply affects expression of biotin transporters, biotinylation of carboxylases and metabolism of interleukin-2 in Jurkat cells. J Nutr. 2002 May;132(5):887-92. doi: 10.1093/jn/132.5.887. Erratum in: J Nutr 2002 Aug;132(8):2326
- Crisp SE, Griffin JB, White BR, Toombs CF, Camporeale G, Said HM, Zempleni J. Biotin supply affects rates of cell proliferation, biotinylation of carboxylases and histones, and expression of the gene encoding the sodium-dependent multivitamin transporter in JAr choriocarcinoma cells. Eur J Nutr. 2004 Feb;43(1):23-31. doi: 10.1007/s00394-004-0435-9
- Dakshinamurti K, Chalifour L, Bhullar RP. Requirement for biotin and the function of biotin in cells in culture. Ann N Y Acad Sci. 1985;447:38-55. doi: 10.1111/j.1749-6632.1985.tb18424.x
- Rodriguez-Melendez R, Schwab LD, Zempleni J. Jurkat cells respond to biotin deficiency with increased nuclear translocation of NF-kappaB, mediating cell survival. Int J Vitam Nutr Res. 2004 May;74(3):209-16. doi: 10.1024/0300-9831.74.3.209
- Smith EM, Hoi JT, Eissenberg JC, Shoemaker JD, Neckameyer WS, Ilvarsonn AM, Harshman LG, Schlegel VL, Zempleni J. Feeding Drosophila a biotin-deficient diet for multiple generations increases stress resistance and lifespan and alters gene expression and histone biotinylation patterns. J Nutr. 2007 Sep;137(9):2006-12. doi: 10.1093/jn/137.9.2006
- Institute of Medicine. Food and Nutrition Board. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academy Press; 1998. https://www.ncbi.nlm.nih.gov/books/NBK114310/pdf/Bookshelf_NBK114310.pdf
- Institute of Medicine. Dietary reference intakes: the essential guide to nutrient requirements. Washington, DC: National Academies Press; 2006. https://www.nap.edu/catalog/11537/dietary-reference-intakes-the-essential-guide-to-nutrient-requirements
- Zempleni J, Mock DM. Bioavailability of biotin given orally to humans in pharmacologic doses. Am J Clin Nutr. 1999 Mar;69(3):504-8. doi: 10.1093/ajcn/69.3.504
- Biotin. https://ods.od.nih.gov/factsheets/Biotin-Consumer
- Combs GF, Jr. Biotin. In: Combs GF, Jr., ed. The vitamins: fundamental aspects in nutrition and health. Third ed. Burlington, MA: Elsevier Academic Press; 2008:331-44.
- Mock DM. Biotin. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:43-51.
- Food Labeling: Revision of the Nutrition and Supplement Facts Labels. https://www.federalregister.gov/documents/2016/05/27/2016-11867/food-labeling-revision-of-the-nutrition-and-supplement-facts-labels
- Staggs CG, Sealey WM, McCabe BJ, Teague AM, Mock DM. Determination of the biotin content of select foods using accurate and sensitive HPLC/avidin binding. J Food Compost Anal. 2004 Dec;17(6):767-776. doi: 10.1016/j.jfca.2003.09.015
- Ogawa Y, Kinoshita M, Sato T, Shimada S, Kawamura T. Biotin Is Required for the Zinc Homeostasis in the Skin. Nutrients. 2019 Apr 24;11(4):919. doi: 10.3390/nu11040919
- Hsu RH, Chien YH, Hwu WL, Chang IF, Ho HC, Chou SP, Huang TM, Lee NC. Genotypic and phenotypic correlations of biotinidase deficiency in the Chinese population. Orphanet J Rare Dis. 2019 Jan 7;14(1):6. doi: 10.1186/s13023-018-0992-2
- Strovel ET, Cowan TM, Scott AI, Wolf B. ERRATUM: Laboratory diagnosis of biotinidase deficiency, 2017 update: a technical standard and guideline of the American College of Medical Genetics and Genomics. Genet Med. 2018 Feb;20(2):282. https://doi.org/10.1038/gim.2017.201 Erratum Genet Med online publication 6 July 2017; doi:https://doi.org/10.1038/gim.2017.84
- Saleem F, Soos MP. Biotin Deficiency. [Updated 2023 Feb 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK547751
- Innis SM, Allardyce DB. Possible biotin deficiency in adults receiving long-term total parenteral nutrition. Am J Clin Nutr. 1983 Feb;37(2):185-7. doi: 10.1093/ajcn/37.2.185
- Khalidi N, Wesley JR, Thoene JG, Whitehouse WM Jr, Baker WL. Biotin deficiency in a patient with short bowel syndrome during home parenteral nutrition. JPEN J Parenter Enteral Nutr. 1984 May-Jun;8(3):311-4. doi: 10.1177/0148607184008003311
- Krause KH, Bonjour JP, Berlit P, Kochen W. Biotin status of epileptics. Ann N Y Acad Sci. 1985;447:297-313. doi: 10.1111/j.1749-6632.1985.tb18447.x
- Said HM. Biotin: biochemical, physiological and clinical aspects. Subcell Biochem. 2012;56:1-19. doi: 10.1007/978-94-007-2199-9_1
- Hayashi A, Mikami Y, Miyamoto K, Kamada N, Sato T, Mizuno S, Naganuma M, Teratani T, Aoki R, Fukuda S, Suda W, Hattori M, Amagai M, Ohyama M, Kanai T. Intestinal Dysbiosis and Biotin Deprivation Induce Alopecia through Overgrowth of Lactobacillus murinus in Mice. Cell Rep. 2017 Aug 15;20(7):1513-1524. doi: 10.1016/j.celrep.2017.07.057
- Sealey WM, Teague AM, Stratton SL, Mock DM. Smoking accelerates biotin catabolism in women. Am J Clin Nutr. 2004 Oct;80(4):932-5. doi: 10.1093/ajcn/80.4.932
- Srinivasan P, Kapadia R, Biswas A, Said HM. Chronic alcohol exposure inhibits biotin uptake by pancreatic acinar cells: possible involvement of epigenetic mechanisms. Am J Physiol Gastrointest Liver Physiol. 2014 Nov 1;307(9):G941-9. doi: 10.1152/ajpgi.00278.2014
- Subramanya SB, Subramanian VS, Kumar JS, Hoiness R, Said HM. Inhibition of intestinal biotin absorption by chronic alcohol feeding: cellular and molecular mechanisms. Am J Physiol Gastrointest Liver Physiol. 2011 Mar;300(3):G494-501. doi: 10.1152/ajpgi.00465.2010
- Mock DM. Adequate intake of biotin in pregnancy: why bother? J Nutr. 2014 Dec;144(12):1885-6. doi: 10.3945/jn.114.203356
- Mock DM. Biotin: From Nutrition to Therapeutics. J Nutr. 2017 Aug;147(8):1487-1492. doi: 10.3945/jn.116.238956
- Czeizel AE, Dudás I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992 Dec 24;327(26):1832-5. doi: 10.1056/NEJM199212243272602
- Mock DM, Mock NI, Stewart CW, LaBorde JB, Hansen DK. Marginal biotin deficiency is teratogenic in ICR mice. J Nutr. 2003 Aug;133(8):2519-25. doi: 10.1093/jn/133.8.2519
- Hayashi H, Tokuriki S, Okuno T, Shigematsu Y, Yasushi A, Matsuyama G, Sawada K, Ohshima Y. Biotin and carnitine deficiency due to hypoallergenic formula nutrition in infants with milk allergy. Pediatr Int. 2014 Apr;56(2):286-8. doi: 10.1111/ped.12319
- Küry S, Ramaekers V, Bézieau S, Wolf B. Clinical utility gene card for: Biotinidase deficiency-update 2015. Eur J Hum Genet. 2016 Jul;24(7). doi: 10.1038/ejhg.2015.246
- Zempleni J, Hassan YI, Wijeratne SS. Biotin and biotinidase deficiency. Expert Rev Endocrinol Metab. 2008 Nov 1;3(6):715-724. doi: 10.1586/17446651.3.6.715
- Canda E, Kalkan Uçar S, Çoker M. Biotinidase Deficiency: Prevalence, Impact And Management Strategies. Pediatric Health Med Ther. 2020 May 4;11:127-133. doi: 10.2147/PHMT.S198656
- Pindolia K, Chen J, Cardwell C, Cui X, Chopp M, Wolf B. Neurological deficits in mice with profound biotinidase deficiency are associated with demylination and axonal degeneration. Neurobiol Dis. 2012 Sep;47(3):428-35. doi: 10.1016/j.nbd.2012.04.016
- Wolf B. Biotinidase Deficiency. 2000 Mar 24 [Updated 2023 May 25]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1322
- Yang Y, Yang JY, Chen XJ. Biotinidase deficiency characterized by skin and hair findings. Clin Dermatol. 2020 Jul-Aug;38(4):477-483. doi: 10.1016/j.clindermatol.2020.03.004
- Mohite K, Nair KV, Sapare A, Bhat V, Shukla A, Kekatpure M, Patil SJ. Late Onset Subacute Profound Biotinidase Deficiency Caused by a Novel Homozygous Variant c.466-3T>G in the BTD Gene. Indian J Pediatr. 2022 Jun;89(6):594-596. doi: 10.1007/s12098-021-04000-3
- Radelfahr F, Riedhammer KM, Keidel LF, Gramer G, Meitinger T, Klopstock T, Wagner M. Biotinidase deficiency: A treatable cause of hereditary spastic paraparesis. Neurol Genet. 2020 Oct 13;6(6):e525. doi: 10.1212/NXG.0000000000000525
- Wolf B, Secor McVoy J. A sensitive radioassay for biotinidase activity: deficient activity in tissues of serum biotinidase-deficient individuals. Clin Chim Acta. 1983;135(3):275–281. doi: 10.1016/0009-8981(83)90286-3
- Wolf B, Grier RE, Allen RJ, Goodman SI, Kien CL. Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta. 1983;131(3):273–281. doi: 10.1016/0009-8981(83)90096-7
- Wolf B, Grier RE, Secor McVoy JR, Heard GS. Biotinidase deficiency: a novel vitamin recycling defect. J Inherit Metab Dis. 1985;8(Suppl 1):53–58. doi: 10.1007/BF01800660
- Gannavarapu S, Prasad C, DiRaimo J, et al. Biotinidase deficiency: spectrum of molecular, enzymatic and clinical information from newborn screening Ontario, Canada (2007–2014). Mol Genet Metab. 2015;116(3):146–151. doi: 10.1016/j.ymgme.2015.08.010
- Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med. 2012;14(6):565–575. doi: 10.1038/gim.2011.6
- Hymes J, Stanley CM, Wolf B. Mutations in BTD causing biotinidase deficiency. Hum Mutat. 2001;18(5):375–381. doi: 10.1002/(ISSN)1098-1004
- Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100(1):6–13. doi: 10.1016/j.ymgme.2010.01.003
- Schubiger G, Caflisch U, Baumgartner R, Suormala T, Bachmann C. Biotinidase deficiency: clinical course and biochemical findings. J Inherit Metab Dis. 1984;7(3):129-30. doi: 10.1007/BF01801771
- Horvath TD, Matthews NI, Stratton SL, Mock DM, Boysen G. Measurement of 3-hydroxyisovaleric acid in urine from marginally biotin-deficient humans by UPLC-MS/MS. Anal Bioanal Chem. 2011;401(9):2805–2810. doi: 10.1007/s00216-011-5356-x
- Wolf B. Biotinidase deficiency and our champagne legacy. Gene. 2016 Sep 10;589(2):142-50. doi: 10.1016/j.gene.2015.10.010
- Saleem H, Simpson B. Biotinidase Deficiency. [Updated 2023 Feb 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560607
- Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med. 2012 Jun;14(6):565-75. doi: 10.1038/gim.2011.6
- Armstrong RW, Steinbok P, Cochrane DD, Kube SD, Fife SE, Farrell K. Intrathecally administered baclofen for treatment of children with spasticity of cerebral origin. J Neurosurg. 1997 Sep;87(3):409-14. doi: 10.3171/jns.1997.87.3.0409
- Szymańska E, Średzińska M, Ługowska A, Pajdowska M, Rokicki D, Tylki-Szymańska A. Outcomes of oral biotin treatment in patients with biotinidase deficiency – Twenty years follow-up. Mol Genet Metab Rep. 2015 Oct 6;5:33-35. doi: 10.1016/j.ymgmr.2015.09.004
- Combs GF, Jr. Biotin. In: Combs GF, Jr., ed. The vitamins: fundamental aspects in nutrition and health. Third ed. Burlington, MA: Elsevier Academic Press; 2008:331-44
- Stratton SL, Bogusiewicz A, Mock MM, Mock NI, Wells AM, Mock DM. Lymphocyte propionyl-CoA carboxylase and its activation by biotin are sensitive indicators of marginal biotin deficiency in humans. Am J Clin Nutr. 2006 Aug;84(2):384-8. doi: 10.1093/ajcn/84.1.384
- Perry CA, West AA, Gayle A, Lucas LK, Yan J, Jiang X, et al. Pregnancy and lactation alter biomarkers of biotin metabolism in women consuming a controlled diet. J Nutr 2014;144:1977-84
- Holocarboxylase synthetase deficiency. https://medlineplus.gov/genetics/condition/holocarboxylase-synthetase-deficiency
- Kalter, H. (1972), The metabolic basis of inherited disease. 3rd Ed. J. B. Stanbury, J. B. Wyngaarden, and D. S. Fredrickson, eds. McGraw-Hill, New York. 1778 pp. 1972. Teratology, 6: 362-362. https://doi.org/10.1002/tera.1420060320
- Mock, Donald M.. Evidence for a Pathogenic Role of ω6 Polyunsaturated Fatty Acid in the Cutaneous Manifestations of Biotin Deficiency. Journal of Pediatric Gastroenterology and Nutrition 10(2):p 222-229, February 1990. doi: 10.1097/00005176-199002000-00013
- Nakajima K, Terao M, Takaishi M, Kataoka S, Goto-Inoue N, Setou M, Horie K, Sakamoto F, Ito M, Azukizawa H, Kitaba S, Murota H, Itami S, Katayama I, Takeda J, Sano S. Barrier abnormality due to ceramide deficiency leads to psoriasiform inflammation in a mouse model. J Invest Dermatol. 2013 Nov;133(11):2555-2565. doi: 10.1038/jid.2013.199
- Bandaralage SP, Farnaghi S, Dulhunty JM, Kothari A. Antenatal and postnatal radiologic diagnosis of holocarboxylase synthetase deficiency: a systematic review. Pediatr Radiol. 2016 Mar;46(3):357-64. doi: 10.1007/s00247-015-3492-8
- Mardach R, Zempleni J, Wolf B, Cannon MJ, Jennings ML, Cress S, Boylan J, Roth S, Cederbaum S, Mock DM. Biotin dependency due to a defect in biotin transport. J Clin Invest. 2002 Jun;109(12):1617-23. doi: 10.1172/JCI13138
- Perry CA, West AA, Gayle A, Lucas LK, Yan J, Jiang X, Malysheva O, Caudill MA. Pregnancy and lactation alter biomarkers of biotin metabolism in women consuming a controlled diet. J Nutr. 2014 Dec;144(12):1977-84. doi: 10.3945/jn.114.194472
- Pabuçcuoğlu A, Aydoğdu S, Baş M. Serum biotinidase activity in children with chronic liver disease and its clinical significance. J Pediatr Gastroenterol Nutr. 2002 Jan;34(1):59-62. doi: 10.1097/00005176-200201000-00014
- Mock DM. Biotin status: which are valid indicators and how do we know? J Nutr. 1999 Feb;129(2S Suppl):498S-503S
- Schulpis KH, Karikas GA, Tjamouranis J, Regoutas S, Tsakiris S. Low serum biotinidase activity in children with valproic acid monotherapy. Epilepsia. 2001 Oct;42(10):1359-62. doi: 10.1046/j.1528-1157.2001.47000.x
- Baumgartner ER, Suormala T. Inherited defects of biotin metabolism. Biofactors. 1999;10(2-3):287-90. doi: 10.1002/biof.5520100229
- Misir R, Blair R, Doige CE. Development of a system for clinical evaluation of the biotin status of sows. Can Vet J. 1986 Jan;27(1):6-12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1680218/pdf/canvetj00589-0012.pdf
- BROWN A. The effect of egg white and crystalline biotin methyl ester on a skin lesion in three infants. Glasgow Med J. 1948 Sep;29(9):309-16. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5966187/pdf/glasgowmedj76301-0017.pdf
- Bunch M, Singh A. Peculiar neuroimaging and electrophysiological findings in a patient with biotinidase deficiency. Seizure. 2011 Jan;20(1):83-6. doi: 10.1016/j.seizure.2010.10.001
- Desai S, Ganesan K, Hegde A. Biotinidase deficiency: a reversible metabolic encephalopathy. Neuroimaging and MR spectroscopic findings in a series of four patients. Pediatr Radiol. 2008 Aug;38(8):848-56. doi: 10.1007/s00247-008-0904-z





{kind=link}