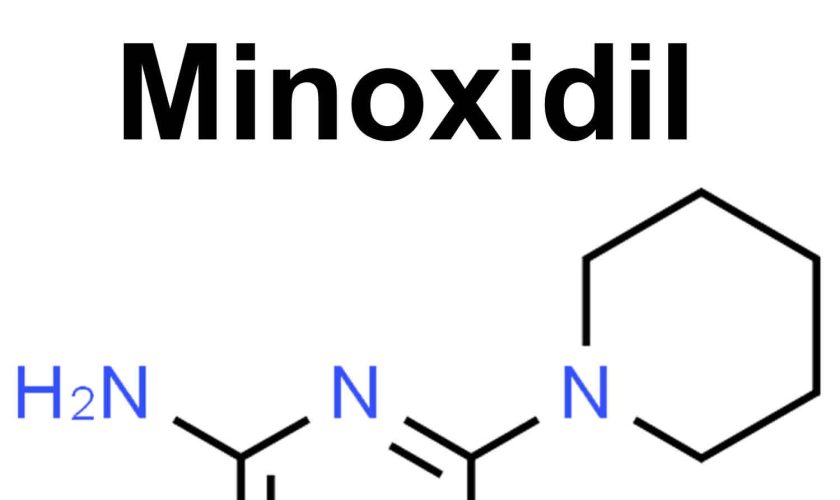
Minoxidil
Minoxidil liquid to be applied to your scalp is used to stimulate hair growth and to slow hair loss and Minoxidil tablet to be taken by mouth is used with other medications to treat high blood pressure not responding to maximum doses of 3 different antihypertensive medications also called resistant hypertension. In some countries, Minoxidil is available in other formulations such as foam or gel. Minoxidil liquid to be applied to your scalp is most effective for people under 40 years of age whose hair loss is recent. Minoxidil liquid to be applied to your scalp has no effect on receding hairlines. Minoxidil liquid to be applied to your scalp does not cure baldness; most new hair is lost within a few months after Minoxidil is stopped. In United States, Minoxidil Foam or Minoxidil Solution is available without a prescription. Furthermore, Minoxidil is marketed in an oral tablet formulation and as a topical agent in foam and solution formulations for both male and female patients.
Minoxidil is in a class of medications called vasodilators (medicines that open blood vessels). Minoxidil works by relaxing the blood vessels so that blood can flow more easily through your body. Minoxidil was originally marketed in a tablet form for the treatment of high blood pressure (hypertension). When Minoxidil tablet was used to treat high blood pressure (hypertension) one of the side effects of Minoxidil was unwanted hair growth. This side effect promoted researchers to produce Minoxidil topical solution for treating hair loss.
Topical Minoxidil is used in the treatment of male pattern hair loss in men (MPHL), and female pattern hair loss in women (FPHL) commonly known as androgenetic alopecia (androgenic alopecia). Topical Minoxidil is occasionally useful for other forms of hair loss (alopecia), including alopecia areata, and after hair replacement surgery or chemotherapy. Minoxidil effectiveness may be enhanced by concurrent use of a topical retinoid (medications derived from vitamin A formulated as a cream, lotion, foam, emulsion, or gel).
Minoxidil is marketed as Rogaine Topical Solution (5%) and Headway Topical Solution (2% and 5%). Minoxidil 5% solution is probably more effective but it may result in greater irritation than Minoxidil 2% solution. Minoxidil Topical Solution is applied to the affected area of the scalp once or twice daily for a minimum of 6 months. If it is found to be effective, it may be continued long term.
Follow the directions on your package or prescription label carefully, and ask your doctor or pharmacist to explain any part you do not understand. Use minoxidil exactly as directed. Do not use more or less of it or use it more often than directed by your doctor.
Exceeding the recommended dosage does not produce greater or faster hair growth and may cause increased side effects. You must use Minoxidil Topical Solution for at least 4 months, and possibly for up to 1 year, before you see any effect.
Three special applicators are provided: a metered-spray applicator for large scalp areas, an extender spray applicator (used with the metered-spray applicator) for small areas or under the hair, and a rub-on applicator.
Remove the outer and inner caps from the bottle, choose an applicator, and screw it tightly onto the bottle.
To use the extender spray applicator, first assemble the metered-spray applicator and then follow the instructions provided to attach the extender spray applicator. Pump the metered-spray or extender spray applicator six times for each dose. Try not to inhale the mist. Place the large cap on the metered-spray bottle or the small cap on the extender spray nozzle when not in use.
To use the rub-on applicator, hold the bottle upright and squeeze it until the upper chamber of the applicator is filled to the black line. Then turn the bottle upside down and rub on the medication. Place the large cap on the bottle when not in use. If you use your fingertips to apply the medication, wash them thoroughly afterwards.
Apply Minoxidil Topical Solution to dry hair and scalp only. Do not apply it to other body areas, and keep it away from your eyes and sensitive skin. If it accidentally comes in contact with these areas, wash them with lots of cool water; call your doctor if they become irritated.
Do not apply minoxidil to a sunburned or irritated scalp.
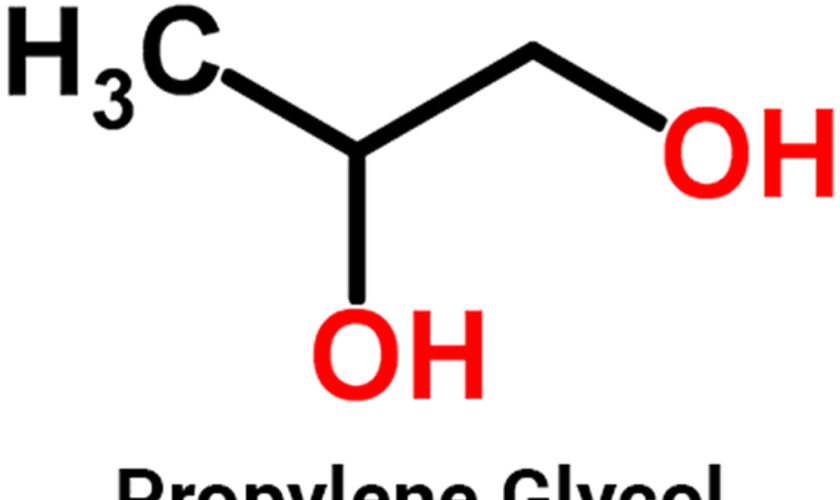
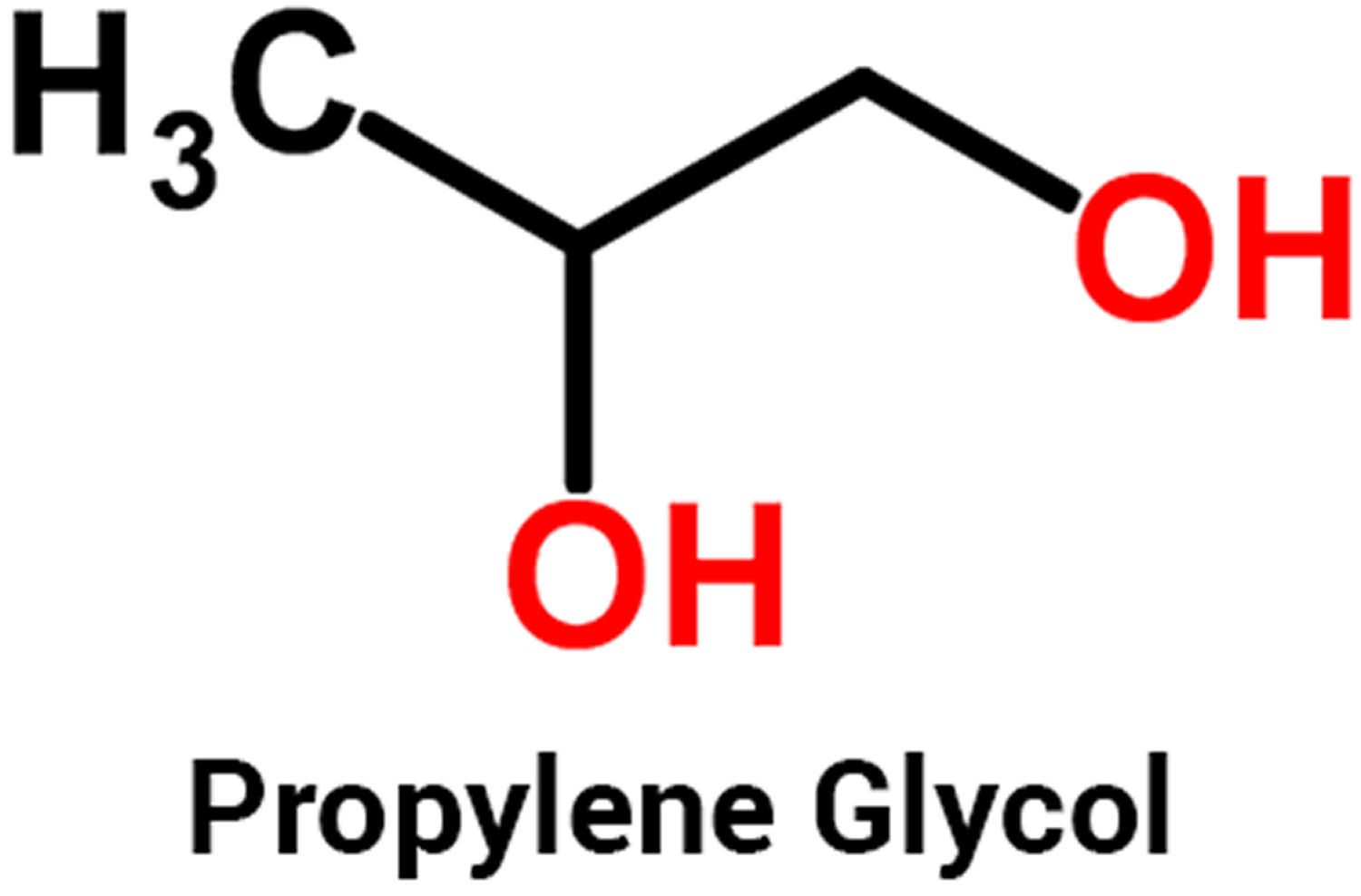
Minoxidil side effects include local irritation most often resulting in itching or stinging. Contact allergy to minoxidil or propylene glycol (the main component of Minoxidil solution) occurs rarely, and results in dermatitis.
Unwanted hair growth might occur (hypertrichosis), for example if Minoxidil solution is dripped onto the forehead.
Internal effects are not likely as there is minimal absorption into your body. However, hair growth in distant sites such as your arms, chest and lower back has occasionally been reported when Minoxidil 5% solution has been used.
Hypotension (low blood pressure) is a rare occurrence in patients using topical minoxidil. Nonetheless, some clinicians advise consistent monitoring of blood pressure, heart rate, and electrocardiographic changes for patients undergoing topical minoxidil therapy. Clinicians should conduct thorough assessments to eliminate secondary causes of hypertension, such as renal artery stenosis, primary aldosteronism, pheochromocytoma, and Cushing syndrome, warranting minoxidil treatment. Regular monitoring of fundoscopic examination, renal function, echocardiogram, and ankle-brachial index is recommended to assess potential target organ damage for patients taking minoxidil 1.
Minoxidil is best avoided during pregnancy and breastfeeding (Pregnancy Category C, i.e., Drugs that, owing to their pharmacological effects, have caused or maybe suspected of causing harmful effects on the human fetus or neonate without causing malformations) 2.
Minoxidil may increase chest pain (angina) or cause other heart problems including pericardial effusion, that can advance to cardiac tamponade 3. If chest pain occurs or worsens while you are taking this medication, call your doctor immediately. Your doctor may prescribe other medications as part of your minoxidil therapy. Do not stop taking these medications unless your doctor tells you to do so.
You should not use minoxidil if you have pheochromocytoma (adrenal gland tumor).
Patients receiving guanethidine should be admitted to a hospital for monitoring when minoxidil is administered due to the substantial risk of severe orthostatic hypotension 4. Coadministration of beta-blockers with minoxidil is advised to help manage the elevated myocardial workload and rapid heart rate 5. Oral minoxidil should be used with a loop diuretic to prevent severe fluid accumulation in a patient’s body 6.
Keep all appointments with your doctor and the laboratory. Your blood pressure should be checked regularly to determine your response to minoxidil. Your doctor may order other tests such as EKG (electrocardiogram) to monitor your heart function.
What causes hair loss in men and women?
Male pattern hair loss also called androgenetic alopecia (androgenic alopecia) is an inherited condition caused by a genetically determined excessive sensitivity to the effects of dihydrotestosterone (DHT) in some areas of the scalp that affects up to 50% of males and females 7, 8, 9. Male pattern hair loss (androgenetic alopecia) is characterized by progressive loss of terminal hair of the scalp any time after puberty, in a characteristic distribution in both males and females. In males, hair loss is most prominent in the crown (the vertex, the highest point on your scalp) and frontotemporal regions, while in women the frontal hairline is typically spared with diffuse hair loss at the crown and top of head, with loss often marked by a wider center part 10, 11, 12.
Dihydrotestosterone (DHT) is believed to shorten the growth or anagen, phase of the hair cycle, from a usual duration of 3 to 6 years to just weeks or months 13, 14. This occurs together with miniaturization of the hair follicles and progressively produces fewer and finer hairs. The production of dihydrotestosterone (DHT) is regulated by an enzyme called 5-alpha reductase (5α-reductase). 5-alpha reductase (5α-reductase) enzyme is involved in processing androgens, which are sex hormones that direct male sexual development that help start puberty and play a role in reproductive health and body development. Specifically, the steroid 5-alpha reductase 2 enzyme is responsible for a chemical reaction that converts the hormone Testosterone to Dihydrotestosterone (DHT). DHT has a critical role in male sexual development, and a shortage of this hormone disrupts the formation of the external sex organs before birth.
Several genes are involved, accounting for differing age of onset, progression, pattern and severity of hair loss in family members. The hair loss genes are inherited from both mother and father. At this time, genetic testing for prediction of balding is unreliable.
A few women present with male pattern hair loss because they have excessive levels of androgens as well as genetic predisposition. These women also tend to suffer from acne, irregular menses and excessive facial and body hair. These symptoms are characteristic of polycystic ovarian syndrome (PCOS) although the majority of women with PCOS (polycystic ovarian syndrome) do not experience hair loss. Less often, congenital adrenal hyperplasia (CAH or 21-hydroxylase deficiency) may be responsible. Females that are losing their hair with age are more likely to present with female pattern hair loss, in which hormone tests are normal.
Figure 1. Hair loss in women (androgenic alopecia in women)
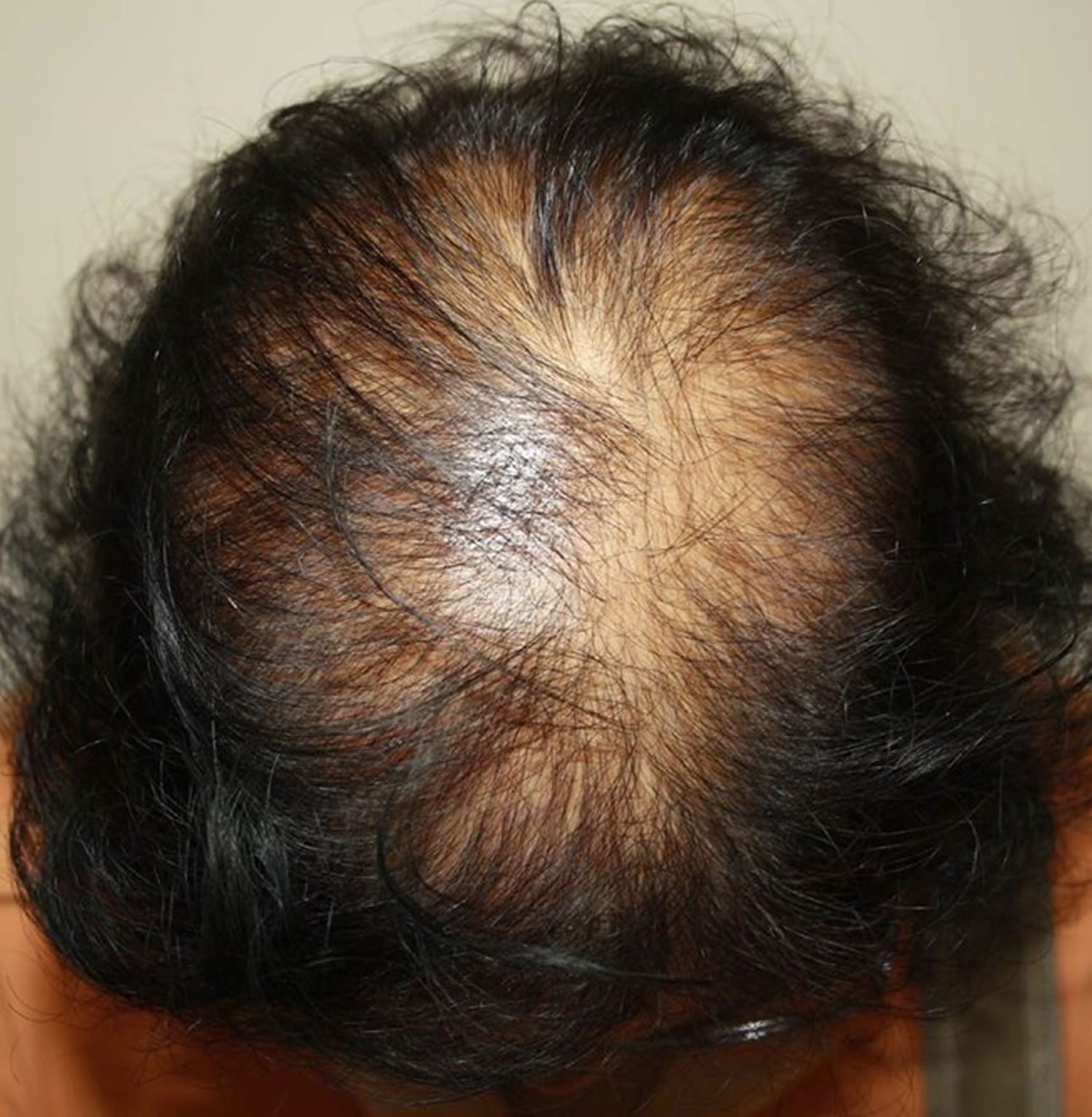
Footnote: Female pattern hair loss (androgenic alopecia in women) showing hair thinning mostly confined to the crown (top of scalp) with retention of frontal hairline.
[Source 15 ]Figure 2. Androgenetic alopecia female (female pattern baldness)
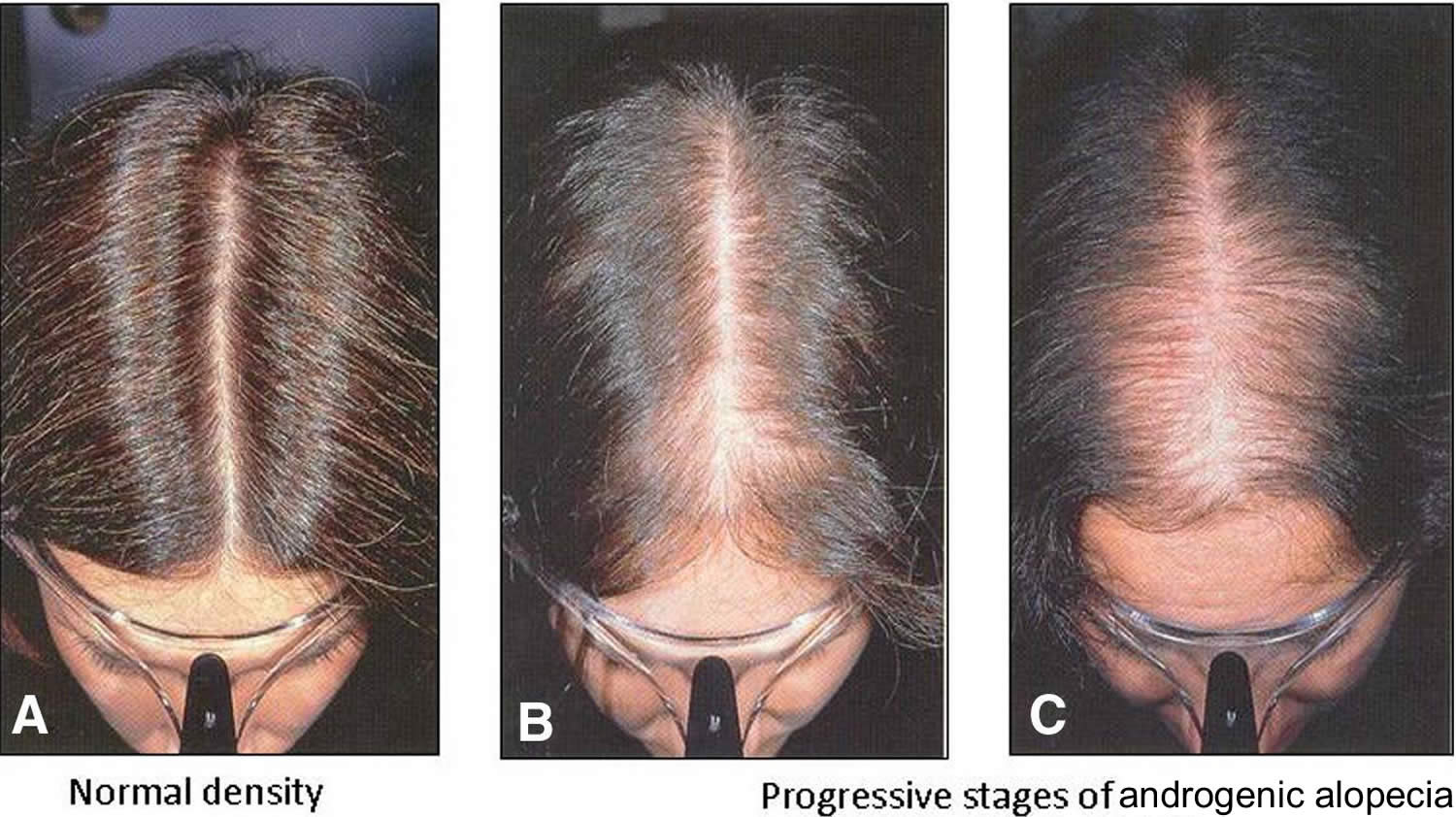
Footnote: Androgenetic alopecia in women with ‘Christmas Tree Pattern’
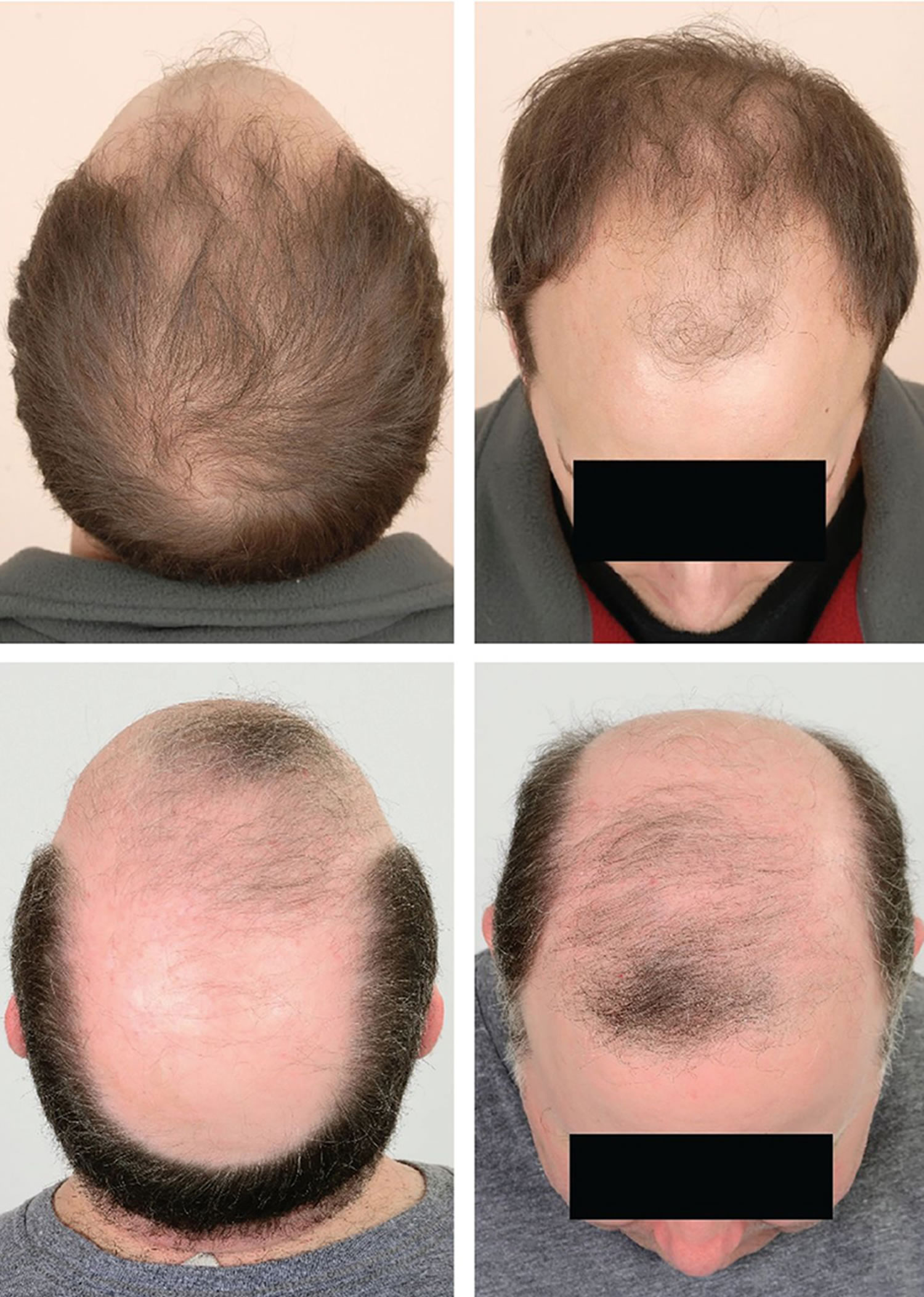
[Source 16 ]Figure 3. Male pattern hair loss
What is hair growth cycle?
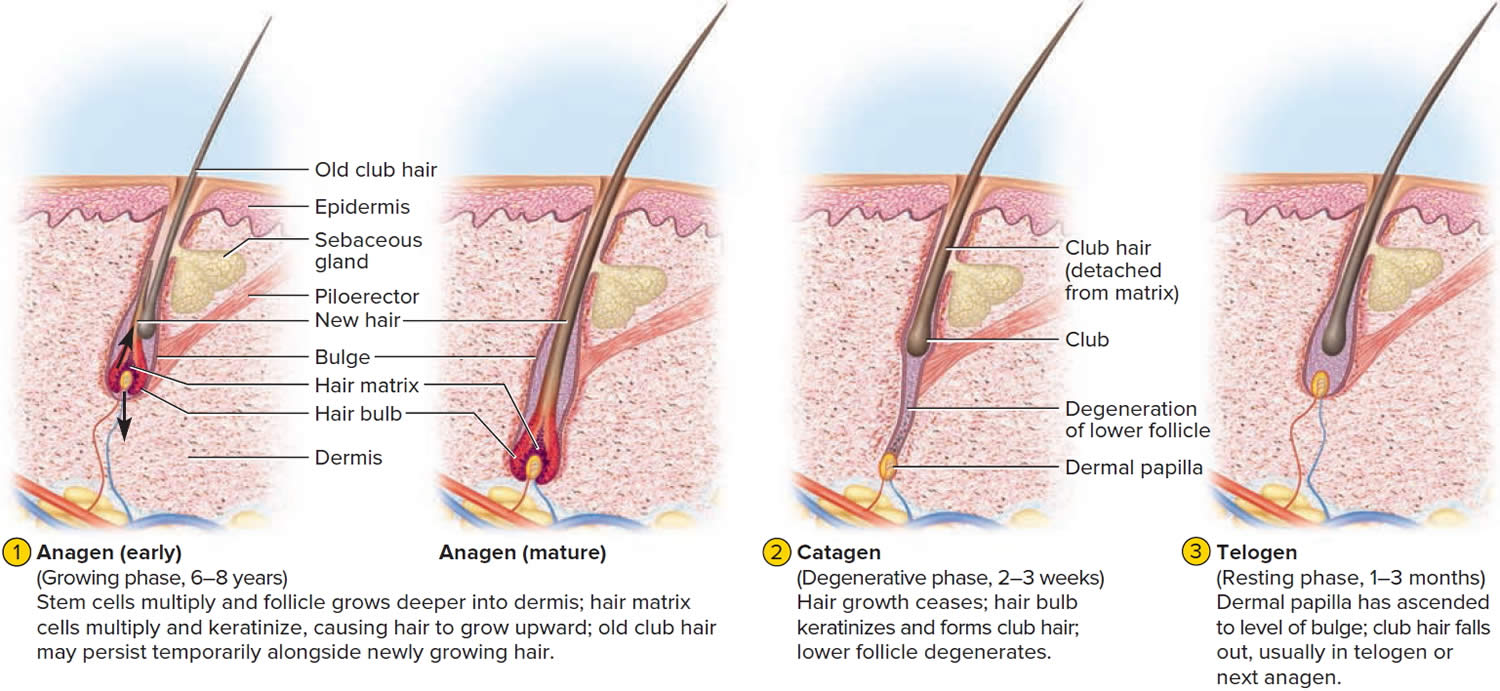
The human scalp contains about 100,000 hair follicles. These anchor the hair to the skin and contain the cells that produce new hairs. A hair in the scalp grows for two to five years, at a rate of around 0.33 mm/day (about 1/64 inch). Variations in hair growth rate and the duration of the hair growth cycle account for individual differences in uncut hair length. Hair grows in 3 developmental stages:
- Anagen. The anagen phase or actively hair growing phase starts the growing of new hair. The anagen phase is genetically determined and can vary from 2 to 6 years (the average is just under 3 years). Most hair follicles on the scalp are in the anagen phase.
- Catagen. The catagen phase is a transition stage (in-between phase) between the growing and resting phases and lasts 2-3 weeks. The catagen phase is when hair growth stops and the hair follicle shrinks. About 1–3% of hairs are in the catagen phase (in-between phase).
- Telogen. The telogen phase or resting phase is a mature hair with a root, which is held very loosely in the follicle. The telogen phase (resting phase) generally lasts about 4-5 months. Up to 10% of hairs in a normal scalp are in the telogen phase (resting phase). About 100 telogen hairs are lost from the human scalp each day.
Everyone is born with a fixed number of hair follicles on the scalp that produce hairs throughout life. Hair grows from the base of the follicle at a rate of about one centimetre a month for about three years. This growth phase is called anagen. After anagen, the hair dies (catagen hair) and no longer grows. It sits dormant in the follicle for a three-month phase called telogen. After telogen, the hair follicle undergoes another anagen phase to produce new hair that grows out of the same follicle. As it grows, the old telogen hair is dislodged or pushed out. The hair cycle continues throughout life.
At any given time, about 90% of the scalp follicles are in the Anagen stage. In this stage, stem cells from the bulge in the hair follicle multiply and travel downward, pushing the dermal papilla deeper into the skin and forming the epithelial root sheath. Root sheath cells directly above the papilla form the hair matrix. Here, sheath cells transform into hair cells, which synthesize keratin and then die as they are pushed upward away from the papilla. The new hair grows up the follicle, often alongside an old club hair left from the previous cycle.
Hair length depends on the duration of anagen stage. Short hairs (eyelashes, eyebrows, hair on arms and legs) have a short anagen phase of around one month. Anagen lasts up to 6 years or longer in scalp hair.
In the Catagen stage, mitosis in the hair matrix ceases and sheath cells below the bulge die. The follicle shrinks and the dermal papilla draws up toward the bulge. The base of the hair keratinizes into a hard club and the hair, now known as a club hair, loses its anchorage. Club hairs are easily pulled out by brushing the hair, and the hard club can be felt at the hair’s end. When the papilla reaches the bulge, the hair goes into a resting period called the Telogen stage. Eventually, anagen begins anew and the cycle repeats itself. A club hair may fall out during catagen or telogen, or as it is pushed out by the new hair in the next anagen phase.
You lose about 50 to 100 scalp hairs daily. In a young adult, scalp follicles typically spend 6 to 8 years in anagen, 2 to 3 weeks in catagen, and 1 to 3 months in telogen. Scalp hairs grow at a rate of about 1 mm per 3 days (10–18 cm/yr) in the anagen phase.
Hair grows fastest from adolescence until the 40s. After that, an increasing percentage of follicles are in the catagen and telogen phases rather than the growing anagen phase. Hair follicles also shrink and begin producing wispy vellus hairs instead of thicker terminal hairs. Thinning of the hair or baldness, is called alopecia. It occurs to some degree in both sexes and may be worsened by disease, poor nutrition, fever, emotional stress, radiation, or chemotherapy. In the great majority of cases, however, it is simply a matter of aging.
Pattern baldness is the condition in which hair is lost unevenly across the scalp rather than thinning uniformly. It results from a combination of genetic and hormonal influences. The relevant gene has two alleles: one for uniform hair growth and a baldness allele for patchy hair growth. The baldness allele is dominant in males and is expressed only in the presence of the high level of testosterone characteristic of men. In men who are either heterozygous or homozygous for the baldness allele, testosterone causes terminal hair to be replaced by vellus hair, beginning on top of the head and later the sides. In women, the baldness allele is recessive. Homozygous dominant and heterozygous women show normal hair distribution; only homozygous recessive women are at risk of pattern baldness. Even then, they exhibit the trait only if their testosterone levels are abnormally high for a woman (for example, because of a tumor of the adrenal gland, a woman’s principal source of testosterone). Such characteristics in which an allele is dominant in one sex and recessive in the other are called sex-influenced traits.
Excessive or undesirable hairiness in areas that are not usually hairy, especially in women and children, is called hirsutism. It tends to run in families and usually results from either masculinizing ovarian tumors or hypersecretion of testosterone by the adrenal cortex. It is often associated with menopause.
Contrary to popular misconceptions, hair and nails do not continue to grow after a person dies, cutting hair does not make it grow faster or thicker, and emotional stress cannot make the hair turn white overnight.
Different causes of hair loss affect the hair follicles in different phases of growth.
Figure 4. Hair growth cycle
How does minoxidil work?
Topical minoxidil functions as a stimulant for hair growth. The precise mechanism of action for minoxidil remains inadequately understood. The sulfotransferase enzyme in the human scalp converts minoxidil into minoxidil sulfate, which is the active form of minoxidil 17. Differences in sulfotransferase activity among individuals can affect minoxidil’s effectiveness, leading to inconsistencies in minoxidil therapy 18.
Minoxidil shortens the telogen phase to prompt the dormant hair follicles for premature transition into the anagen phase. The shortening of the telogen phase might lead to telogen effluvium following minoxidil therapy. Furthermore, minoxidil extends the anagen phase, resulting in increased hair length and thickness, thereby representing the observable outcomes of minoxidil therapy 19.
The initial outcomes of minoxidil become apparent after approximately 8 weeks of initiating the treatment, with the maximum effects manifesting around 4 months. Minoxidil affects the potassium channels present in vascular smooth muscles and hair follicles 19. This potassium channel activity may induce the following effects:
- Stimulation of the microcirculation around the hair follicles induces arteriolar vasodilation, thereby encouraging conditions conducive to hair growth.
- Induction of the vascular endothelial growth factor expression leads to heightened vascularization around the hair follicles, thereby promoting hair growth.
- Activation of the prostaglandin-endoperoxide synthase-1 enzyme leads to the enhancement of hair growth.
- Inhibition of androgen-related effects on androgen-sensitive hair follicles.
- Direct stimulation of the hair follicles as the drug acts as an epidermal growth factor on the matrix cells, slowing their aging process and extending their anagen phase. This process is achieved through the activation of the beta-catenin pathway.
- Display of antifibrotic characteristics due to its impact on collagen synthesis.
Hypertension
Minoxidil exerts its antihypertensive effect by opening adenosine triphosphate (ATP) sensitive potassium channels. As a result, when the smooth muscle vasculature is relaxed, it reduces peripheral resistance, which ultimately helps to lower blood pressure levels. Consequently, this process increases plasma renin activity, which, in turn, triggers salt and water retention in a patient’s body. Additionally, minoxidil triggers the activation of the sympathetic nervous system, resulting in tachycardia and heightened cardiac output. Therefore, clinicians prescribe beta-blockers and diuretics alongside minoxidil to mitigate potential adverse effects 20.
What happens if minoxidil treatment is stopped?
If Minoxidil treatment is stopped, the pretreatment appearance will normally return within 3 or 4 months. If Minoxidil is stopped after several years of use, the hair that was genetically programmed to be lost during that time will fall out.
Minoxidil special precautions
Before using minoxidil:
- tell your doctor and pharmacist if you are allergic to minoxidil or any other drugs.
- tell your doctor and pharmacist what prescription and nonprescription medications you are taking, especially guanethidine (Ismelin), other medications for high blood pressure, and vitamins.
- tell your doctor if you have or have ever had heart, kidney, liver, or scalp disease.
- you should not use minoxidil if you have pheochromocytoma (adrenal gland tumor).
- tell your doctor if you are pregnant, plan to become pregnant, or are breast-feeding. If you become pregnant while using minoxidil, call your doctor.
- plan to avoid unnecessary or prolonged exposure to sunlight and to wear protective clothing, sunglasses, and sunscreen. Minoxidil may make your skin sensitive to sunlight.
It is important that your doctor check your progress at regular visits to make sure that minoxidil is working properly and to check for unwanted effects.
Your hair loss may continue for 2 weeks after you start using minoxidil. Tell your doctor if your hair loss continues after 2 weeks. Also, tell your doctor if your hair growth does not increase after using minoxidil for 4 months.
Allergies
Tell your doctor if you have ever had any unusual or allergic reaction to this medicine or any other medicines. Also tell your health care professional if you have any other types of allergies, such as to foods, dyes, preservatives, or animals. For non-prescription products, read the label or package ingredients carefully.
Children
Appropriate studies have not been performed on the relationship of age to the effects of topical minoxidil in children. Safety and efficacy have not been established. The recommended initial dosage of oral minoxidil in patients younger than 12 is 0.2 mg/kg daily. Typically, the maintenance dosage of minoxidil ranges from 0.25 to 1 mg/kg per day, and the maximum recommended dosage is 50 mg per day 21. Clinicians may suggest topical minoxidil in children; however, it is considered an off-label drug indication 22.
Elderly patients
Appropriate studies performed to date have not demonstrated geriatrics-specific problems that would limit the usefulness of topical minoxidil in the elderly. However, studies have shown that minoxidil works best in younger patients who have a short history of hair loss. Minoxidil has not been studied in patients older than 65 years of age.
Oral minoxidil should be initiated at a lower dose in older patients as clinicians recommend, considering comorbidities, polypharmacy, and increased risk of falls in this population.
Pregnant women
As animal reproductive studies have indicated specific adverse effects of minoxidil in pregnant women, it is not advisable to use this drug. Minoxidil holds an U.S. Food and Drug Administration (FDA) pregnancy category C classification 2.
Breastfeeding women
Studies in women suggest that minoxidil poses minimal risk to the infant when used during breastfeeding. As minoxidil is excreted in breast milk, its use is not recommended in breastfeeding women 19.
Drug Interactions
Although certain medicines should not be used together at all, in other cases two different medicines may be used together even if an interaction might occur. In these cases, your doctor may want to change the dose, or other precautions may be necessary. Tell your healthcare professional if you are taking any other prescription or nonprescription (over-the-counter [OTC]) medicine.
- Systemic cyclosporine can exacerbate adverse effects such as hypertrichosis when combined with topical minoxidil. Notably, hypertrichosis symptoms significantly improved after discontinuing topical minoxidil for 2 months 23.
- Coadministration of low-dose aspirin and minoxidil may diminish the effectiveness of topical minoxidil. This decrease in effectiveness is attributed to the inhibitory effect of low-dose aspirin on sulfotransferase enzymes in human hair 24.
- Concurrent use of guanethidine with minoxidil can cause severe hypotension 4
Medical Problems
The presence of other medical problems may affect the use of minoxidil. Make sure you tell your doctor if you have any other medical problems, especially:
- Any other skin problems, an irritation, or a sunburn on the scalp: These conditions may cause too much topical minoxidil to be absorbed into the body and may increase the chance of side effects.
- Heart disease or
- Hypertension (high blood pressure): Topical minoxidil has not been studied in patients who have these conditions, but more serious problems may develop for these patients if they use more medicine than is recommended over a large area and too much minoxidil is absorbed into the body.
Minoxidil uses
Minoxidil was initially developed as a potent peripheral vasodilator agent for treating severe difficult to control high blood pressure (Resistant hypertension). However, owing to the significant side effects of oral minoxidil use, Minoxidil tablet is currently used only for patients with resistant hypertension who do not adequately respond to the maximum doses of 3 different antihypertensive medications 25.
Androgenic alopecia and female pattern hair loss is the only indication approved by the U.S. Food and Drug Administration (FDA) for topical minoxidil 19.
Off-label uses of topical minoxidil are as follows 25:
- Alopecia areata: Minoxidil has demonstrated the ability to elicit a favorable clinical response when used as a standalone treatment for alopecia areata or in conjunction with other medications, such as corticosteroids.
- Chemotherapy-induced alopecia: In this case, minoxidil has exhibited the capacity to reduce hair loss and expedite the process of hair regrowth.
- Hair transplant: Hair loss, also known as telogen effluvium, is a common observation after a hair transplant. Minoxidil can be used before and after hair transplantation to reduce hair loss in patients. However, the treatment should be temporarily suspended for 3 days before the hair transplant to prevent excessive bleeding.
- Scarring alopecia: Minoxidil has displayed evidence of having an antifibrotic effect on this condition. Consequently, topical minoxidil treatment could be a viable therapeutic option during the initial stages of dermatoses, leading to scarring alopecia, such as those stemming from scalp burns.
- Monilethrix: Minoxidil induces the synchronization of hair follicles entering the anagen phase in patients experiencing this condition.
- Loose anagen hair syndrome: Loose anagen hair syndrome is due to defective keratinization of the inner root sheath, rather than the hair shaft itself. The loose hair shaft can be easily pulled out of the follicle, leaving localized or diffuse bald areas of the scalp. Loose anagen hair syndrome results in short, brittle hair. Although both sexes may be affected, it most often presents in young girls over the age of 2 years. They usually have blond or red colored hair. It may be diagnosed by the painless extraction of at least 10 hairs on a hair pull test of which over 80% of the hairs are in the anagen phase.
- Telogen effluvium: Telogen effluvium is the name for a common cause of temporary hair loss due to the excessive shedding of resting or telogen hair after some shock to your system. New hair continues to grow. Telogen hair is also known as a club hair due to the shape of the root. Regrowth usually occurs after removal of the trigger causing telogen effluvium. However, repeated episodes of acute telogen effluvium can sometimes evolve into female pattern hair loss.
- Hereditary alopecia or hypotrichosis: Minoxidil has demonstrated its beneficial effects by promoting hair shaft thickening in hypotrichosis.
Topical Minoxidil
Minoxidil applied to the scalp is used to stimulate hair growth in adult men and women with androgenic alopecia. If hair growth is going to occur with the use of Minoxidil, it usually occurs after the medicine has been used for 4 to 6 months and lasts only as long as the medicine continues to be used. Hair loss will begin again within a few months after minoxidil treatment is stopped.
It is very important that you use minoxidil only as directed. Do not use more of it and do not use it more often than your doctor ordered. To do so may increase the chance of it being absorbed through your skin. For the same reason, do not apply minoxidil to other parts of your body. Absorption into the body may affect your heart and blood vessels and cause unwanted side effects.
Do not use any other skin products on the same skin area on which you use minoxidil. Hair coloring, hair permanents, and hair relaxers may be used during minoxidil therapy as long as the scalp is washed just before applying the hair coloring, permanent, or relaxer. Minoxidil should not be used 24 hours before and after the hair treatment procedure. Be sure to not double your doses of minoxidil to make up for any missed doses.
To apply minoxidil topical solution:
- Make sure your hair and scalp are completely dry before applying minoxidil.
- Apply the amount prescribed to the area of the scalp being treated, beginning in the center of the area. Follow your doctor’s instructions on how to apply the solution, using the applicator provided.
- Do not shampoo your hair for 4 hours after applying minoxidil.
- Immediately after using minoxidil, wash your hands to remove any medicine that may be on them.
- Do not use a hairdryer to dry the scalp after you apply minoxidil solution. Blowing with a hairdryer on the scalp may make the treatment less effective.
- Allow the minoxidil to completely dry for 2 to 4 hours after applying it, including before going to bed. Minoxidil can stain clothing, hats, or bed linen if your hair or scalp is not fully dry after using the medicine.
- Avoid transferring the medicine while wet to other parts of the body. This can occur if the medicine gets on your pillowcase or bed linens or if your hands are not washed after applying minoxidil.
To apply minoxidil topical foam:
- Open the container by matching the arrow on can ring with the arrow on cap. Pull off the cap.
- Part the hair into one or more rows to expose the hair thinning area on the scalp.
- Hold the can upside down and press the nozzle to put foam on your fingers.
- Use your fingers to spread the foam over the hair loss area and gently massage into your scalp.
- Immediately after using minoxidil, wash your hands to remove any medicine that may be on them .
If your scalp becomes abraded, irritated, or sunburned, check with your doctor before applying minoxidil.
Minoxidil topical foam or solution is for use on the scalp only. Keep minoxidil away from the eyes, nose, and mouth. If you should accidentally get some in your eyes, nose, or mouth, flush the area thoroughly with cool tap water. If you are using the pump spray, be careful not to breathe in the spray .
Do not use the foam near heat or open flame, or while smoking. Do not puncture, break, or burn the aerosol can.
Oral Minoxidil
According to the American College of Cardiology and the American Heart Association, resistant hypertension is characterized by elevated blood pressure levels exceeding the target range, despite the simultaneous utilization of 3 different classes of antihypertensive medications 26, 27, 28. The classes of antihypertensive medications typically include a combination of a calcium channel blocker, a renin-angiotensin-aldosterone system blocker (angiotensin-converting enzyme inhibitor or angiotensin receptor blocker), and a diuretic. These drugs are administered at the highest daily doses a patient can tolerate. The FDA approves minoxidil for maintaining blood pressure levels in patients with resistant hypertension 20.
Many forms of alopecia (hair loss) are off-label indications for oral or sublingual minoxidil. Recently, multiple studies have explored the use of low-dose oral minoxidil (less than 5mg daily) for treating many forms of alopecia, including male-patterned and female-patterned hair loss, with the goal to treat hair loss without adverse reactions 29. The low adverse-effect profile of low-dose oral minoxidil aids in long-term adherence to treatment and positive clinical response 30, 31. More studies are needed to test the effectiveness of oral and sublingual minoxidil when treating various types of alopecia (hair loss) to improve patient outcomes and to provide an alternative for those having difficulty with topical formulations 29.
Minoxidil dosage
The dose of minoxidil will be different for different patients. Follow your doctor’s orders or the directions on the label. The following information includes only the average doses of minoxidil. If your dose is different, do not change it unless your doctor tells you to do so.
The amount of minoxidil that you take depends on the strength of the minoxidil. Also, the number of doses you take each day, the time allowed between doses, and the length of time you take the medicine depend on the medical problem for which you are using the medicine.
Hair loss
Minoxidil topical solution dosage:
- Adults: Apply 1 milliliter (mL) to the scalp two times a day.
- Women should apply 1 mL of the 2% topical minoxidil solution to their scalp twice daily.
- Men should apply 1 mL of 2% or 5% topical minoxidil solution to their scalp twice daily.
- Children: Use and dose must be determined by the doctor.
Minoxidil topical foam dosage form:
- Adults: Apply half a capful to the scalp two times a day.
- Patients can apply a 5% aerosol formulation of topical minoxidil directly onto their scalp at a one-half capful dose. The medication should be used twice daily by male patients and once daily by female patients.
- Children: Use and dose must be determined by your doctor.
Scalp massage is not required following the application of minoxidil. Minoxidil uptake reaches approximately 50% within an hour and increases to 75% after 4 hours. Although certain practitioners use microneedling with topical minoxidil to potentially enhance minoxidil’s efficacy, further studies are necessary to evaluate the potential significance of this practice 32, 33, 34.
A 2% topical minoxidil solution is effective for male patients experiencing androgenic alopecia in their frontotemporal and vertex regions. However, the 5% topical minoxidil solution provides a more significant clinical benefit than the 2% solution. The clinical response to minoxidil is notably more pronounced when the onset of alopecia occurs within 5 years, primarily among young adults, and when the hair follicles are not extensively miniaturized 35.
Oral minoxidil
Oral minoxidil is supplied in tablet formulations, with options of 2.5 mg and 10 mg doses. Although the U.S. Food and Drug Administration (FDA) does not approve the oral minoxidil for treating hair loss in individuals, clinical trials have demonstrated its effectiveness at dosages ranging from 0.25 to 2.5 mg daily for maintaining blood pressure levels in patients with resistant hypertension 31.
The recommended initial dosage of minoxidil for hypertensive therapy is 5 mg, administered once daily to patients younger than 12, which can be gradually escalated up to a maximum of 40 mg per day 25. Higher doses should be equally divided and administered to individuals 2 or 3 times daily to prevent excessive hypotension. The maximum recommended dosage of oral minoxidil is 100 mg per day 5. As per the American Heart Association’s guidelines for managing resistant hypertension, minoxidil should be prescribed alongside a loop diuretic and a beta-blocker to reduce potential adverse effects in patients 1.
The recommended off-label dosing of oral minoxidil for treating alopecia (hair loss) is 0.25 to 2.5 mg, administered once or twice daily in 1 or 2 equally divided doses.
What should I do if I forget a dose?
Skip the missed dose and continue your regular dosing schedule. Do not apply a double dose to make up for a missed one.
Minoxidil side effects
Minoxidil may cause side effects. Tell your doctor if any of these symptoms are severe or do not go away:
- scalp itching, dryness, scaling, flaking, irritation, or burning
Tell your doctor if you notice continued itching, redness, or burning of your scalp after you apply minoxidil. If the itching, redness, or burning is severe, wash the minoxidil off and check with your doctor before using it again.
Rare side effects:
- Minoxidil-induced telogen effluvium: The shortening of the telogen phase caused by minoxidil can result in excessive hair shedding in individuals.
- Skin irritation: This condition can lead to erythema, discomfort, and a burning sensation on the scalp.
- Scaly changes of the scalp: This condition can entail irritation or a potential worsening of seborrheic dermatitis.
- Isolated itch: appears in the area of application.
- Allergic contact dermatitis: This can lead to symptoms such as erythema, eczematous skin reactions, and pruritus. Minoxidil and propylene glycol are the primary allergens implicated in cases of allergic contact dermatitis. Patch testing can aid in identifying the underlying causative agent. If allergic contact dermatitis arises due to propylene glycol, topical minoxidil as a foam formulation can be used as an alternative option, as it does not contain propylene glycol.
- Acne at site of application
- Facial hair growth
- Inflammation or soreness at root of hair
- Reddened skin
- Swelling of face
Signs and symptoms of too much minoxidil topical being absorbed into your body:
- blurred vision or other changes in vision
- chest pain
- dizziness
- fainting
- fast or irregular heartbeat
- flushing
- headache
- lightheadedness
- numbness or tingling of hands, feet, or face
- swelling of face, hands, feet, or lower legs
- weight gain (rapid)
If you experience any of the following symptoms, see your doctor immediately:
- new or worsening chest pain
- chest pain spreading to your jaw or shoulder
- fast or pounding heartbeats
- swelling in your legs, ankles, or feet
- rapid weight gain, especially in your face and midsection
- shortness of breath
- a light-headed feeling, like you might pass out
- fluid build-up in the lungs–pain when you breathe, feeling short of breath while lying down, wheezing, gasping for breath, cough with foamy mucus
- severe skin reaction–fever, sore throat, swelling in your face or tongue, burning in your eyes, skin pain, followed by a red or purple skin rash that spreads (especially in the face or upper body) and causes blistering and peeling.
Oral minoxidil is also associated with significant adverse effects:
- Rare but severe reactions include pericarditis, pericardial effusion, cardiac tamponade, exacerbating congestive heart failure, and worsening angina.
- Oral minoxidil administration can lead to significant hypotension and potential complications such as thrombocytopenia and leukopenia in individuals taking the medication 36.
- Breast tenderness and gynecomastia have also been reported as adverse effects of the medication 37.
- Hypertrichosis, edema, tachycardia, and weight gain are also caused by oral minoxidil.
Patients using topical minoxidil should undergo regular monitoring for any alterations in the scalp and the occurrence of localized or generalized hypertrichosis (excessive hair growth over and above the normal for the age, sex and race of an individual) 38. Both oral and topical minoxidil usage can lead to hypertrichosis. However, hypertrichosis is more frequently observed with the oral minoxidil formulation and when the 5% topical minoxidil solution is used compared to the 2% minoxidil solution. Research indicates that hypertrichosis is linked to minoxidil extending the anagen phase. In addition, cases of hypertrichosis have been documented in infants due to unintentional direct skin contact 39.
Other side effects not listed may also occur in some patients. If you notice any other effects, check with your healthcare professional.
Minoxidil Overdose
Minoxidil overdose is a rare occurrence following standard topical minoxidil application. In cases of significant oral minoxidil ingestion, there is currently no recognized antidote for minoxidil overdose. Unintentional oral consumption of minoxidil may lead to vomiting and, in rare instances, necessitate hospitalization. Instances of hypotension, tachycardia (fast heart beat) and changes in the electrocardiogram have been reported following accidental ingestion. Intravenous fluids and pharmaceutical vasopressors can address malignant hypotension. In substantial inadvertent minoxidil ingestion, gastric lavage technique and activated charcoal administration might be essential to avert systemic toxicity in patients 40.
A case was reported where a young girl unintentionally ingested a topical solution of minoxidil prescribed to her father. After ingesting the medication, the girl encountered adverse effects such as hypotension and prolonged tachycardia 41. Intravenous fluids are essential in cases of oral minoxidil overdose, causing hypotension in individuals. Vasopressors should be administered to patients for hypotension that does not respond to standard treatment 42, 43.
References- Carey RM, Calhoun DA, Bakris GL, Brook RD, Daugherty SL, Dennison-Himmelfarb CR, Egan BM, Flack JM, Gidding SS, Judd E, Lackland DT, Laffer CL, Newton-Cheh C, Smith SM, Taler SJ, Textor SC, Turan TN, White WB; American Heart Association Professional/Public Education and Publications Committee of the Council on Hypertension; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Genomic and Precision Medicine; Council on Peripheral Vascular Disease; Council on Quality of Care and Outcomes Research; and Stroke Council. Resistant Hypertension: Detection, Evaluation, and Management: A Scientific Statement From the American Heart Association. Hypertension. 2018 Nov;72(5):e53-e90. doi: 10.1161/HYP.0000000000000084
- Smorlesi C, Caldarella A, Caramelli L, Di Lollo S, Moroni F. Topically applied minoxidil may cause fetal malformation: a case report. Birth Defects Res A Clin Mol Teratol. 2003 Dec;67(12):997-1001. doi: 10.1002/bdra.10095
- Oye M, Oye M, Ali A. Signs of early cardiac tamponade induced by Minoxidil. Am J Emerg Med. 2021 Feb;40:226.e1-226.e2. doi: 10.1016/j.ajem.2020.07.050
- Mutterperl RE, Diamond FB, Lowenthal DT. Long-term effects of minoxidil in the treatment of malignant hypertension in chronic renal failure. J Clin Pharmacol. 1976 Oct;16(10 Pt 1):498-509.
- Sica DA. Minoxidil: an underused vasodilator for resistant or severe hypertension. J Clin Hypertens (Greenwich). 2004 May;6(5):283-7. doi: 10.1111/j.1524-6175.2004.03585.x
- Gbadamosi WA, Melvin J, Lopez M. Atypical Case of Minoxidil-Induced Generalized Anasarca and Pleuropericardial Effusion. Cureus. 2021 Jun 3;13(6):e15424. doi: 10.7759/cureus.15424
- Ho CH, Sood T, Zito PM. Androgenetic Alopecia. [Updated 2022 Oct 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430924
- Chan L, Cook DK. Female pattern hair loss. Aust J Gen Pract. 2018 Jul;47(7):459-464. doi: 10.31128/AJGP-02-18-4498
- Tanaka Y, Aso T, Ono J, Hosoi R, Kaneko T. Androgenetic Alopecia Treatment in Asian Men. J Clin Aesthet Dermatol. 2018 Jul;11(7):32-35. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6057731
- Sasaki GH. Review of Human Hair Follicle Biology: Dynamics of Niches and Stem Cell Regulation for Possible Therapeutic Hair Stimulation for Plastic Surgeons. Aesthetic Plast Surg. 2019 Feb;43(1):253-266. doi: 10.1007/s00266-018-1248-1
- Neuhaus K, Schiestl C, Adelsberger R, Weibel L, Meuli M, Böttcher-Haberzeth S. Bold to do – bald to be? Outcomes decades after harvesting the scalp in burned children. Burns. 2019 May;45(3):543-553. doi: 10.1016/j.burns.2018.09.023
- Almohanna HM, Perper M, Tosti A. Safety concerns when using novel medications to treat alopecia. Expert Opin Drug Saf. 2018 Nov;17(11):1115-1128. doi: 10.1080/14740338.2018.1533549
- Bienenfeld A, Azarchi S, Lo Sicco K, Marchbein S, Shapiro J, Nagler AR. Androgens in women: Androgen-mediated skin disease and patient evaluation. J Am Acad Dermatol. 2019 Jun;80(6):1497-1506. doi: 10.1016/j.jaad.2018.08.062
- Sadick NS, Callender VD, Kircik LH, Kogan S. New Insight Into the Pathophysiology of Hair Loss Trigger a Paradigm Shift in the Treatment Approach. J Drugs Dermatol. 2017 Nov 1;16(11):s135-s140. https://jddonline.com/articles/new-insight-into-the-pathophysiology-of-hair-loss-trigger-a-paradigm-shift-in-the-treatment-approach-S1545961617S0135X
- Iamsumang W, Leerunyakul K, Suchonwanit P. Finasteride and Its Potential for the Treatment of Female Pattern Hair Loss: Evidence to Date. Drug Des Devel Ther. 2020 Mar 2;14:951-959. doi: 10.2147/DDDT.S240615
- Monselise A, Cohen DE, Wanser R, Shapiro J. What Ages Hair? Int J Womens Dermatol. 2017 Feb 16;3(1 Suppl):S52-S57. doi: 10.1016/j.ijwd.2017.02.010
- Ramos PM, Goren A, Sinclair R, Miot HA. Oral minoxidil bio-activation by hair follicle outer root sheath cell sulfotransferase enzymes predicts clinical efficacy in female pattern hair loss. J Eur Acad Dermatol Venereol. 2020 Jan;34(1):e40-e41. doi: 10.1111/jdv.15891
- Freire PCB, Riera R, Martimbianco ALC, Petri V, Atallah AN. Minoxidil for patchy alopecia areata: systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2019 Sep;33(9):1792-1799. doi: 10.1111/jdv.15545
- Suchonwanit P, Thammarucha S, Leerunyakul K. Minoxidil and its use in hair disorders: a review. Drug Des Devel Ther. 2019 Aug 9;13:2777-2786. doi: 10.2147/DDDT.S214907. Erratum in: Drug Des Devel Ther. 2020 Feb 10;14:575.
- Mundt HM, Matenaer M, Lammert A, Göttmann U, Krämer BK, Birck R, Benck U. Minoxidil for Treatment of Resistant Hypertension in Chronic Kidney Disease–A Retrospective Cohort Analysis. J Clin Hypertens (Greenwich). 2016 Nov;18(11):1162-1167. doi: 10.1111/jch.12847
- Meyers RS, Siu A. Pharmacotherapy review of chronic pediatric hypertension. Clin Ther. 2011 Oct;33(10):1331-56. doi: 10.1016/j.clinthera.2011.09.003
- Chu JJ, Chen XJ, Shen SS, Zhang XF, Chen LY, Zhang JM, He J, Zhao JF. A poor performance in comprehensive geriatric assessment is associated with increased fall risk in elders with hypertension: a cross-sectional study. J Geriatr Cardiol. 2015 Mar;12(2):113-8. doi: 10.11909/j.issn.1671-5411.2015.02.006
- Sever MS, Sonmez YE, Kocak N. Limited use of minoxidil in renal transplant recipients because of the additive side-effects of cyclosporine on hypertrichosis. Transplantation. 1990 Sep;50(3):536. doi: 10.1097/00007890-199009000-00042
- Goren A, Sharma A, Dhurat R, Shapiro J, Sinclair R, Situm M, Kovacevic M, Lukinovic Skudar V, Goldust M, Lotti T, McCoy J. Low-dose daily aspirin reduces topical minoxidil efficacy in androgenetic alopecia patients. Dermatol Ther. 2018 Nov;31(6):e12741. doi: 10.1111/dth.12741
- Patel P, Nessel TA, Kumar D D. Minoxidil. [Updated 2023 Dec 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482378
- Champaneria MK, Patel RS, Oroszi TL. When blood pressure refuses to budge: exploring the complexity of resistant hypertension. Front Cardiovasc Med. 2023 Jun 21;10:1211199. doi: 10.3389/fcvm.2023.1211199
- Myat A, Redwood SR, Qureshi AC, Spertus JA, Williams B. Resistant hypertension. Br Med J. (2012) 345:e7473. 10.1136/bmj.e7473
- Carey RM, Calhoun DA, Bakris GL, Brook RD, Daugherty SL, Dennison-Himmelfarb CR, et al. Resistant hypertension: detection, evaluation, and management: a scientific statement from the American heart association. Hypertension. (2018) 72(5):e53–e90. 10.1161/HYP.0000000000000084
- Ramírez-Marín HA, Tosti A. Role of Oral Minoxidil in Patterned Hair Loss. Indian Dermatol Online J. 2022 Oct 12;13(6):729-733. doi: 10.4103/idoj.idoj_246_22
- Gupta AK, Talukder M, Venkataraman M, Bamimore MA. Minoxidil: a comprehensive review. J Dermatolog Treat. 2022 Jun;33(4):1896-1906. doi: 10.1080/09546634.2021.1945527
- Randolph M, Tosti A. Oral minoxidil treatment for hair loss: A review of efficacy and safety. J Am Acad Dermatol. 2021 Mar;84(3):737-746. doi: 10.1016/j.jaad.2020.06.1009
- Sharma A, Goren A, Dhurat R, Agrawal S, Sinclair R, Trüeb RM, Vañó-Galván S, Chen G, Tan Y, Kovacevic M, Situm M, McCoy J. Tretinoin enhances minoxidil response in androgenetic alopecia patients by upregulating follicular sulfotransferase enzymes. Dermatol Ther. 2019 May;32(3):e12915. doi: 10.1111/dth.12915
- Sung CT, Juhasz ML, Choi FD, Mesinkovska NA. The Efficacy of Topical Minoxidil for Non-Scarring Alopecia: A Systematic Review. J Drugs Dermatol. 2019 Feb 1;18(2):155-160. https://jddonline.com/articles/the-efficacy-of-topical-minoxidil-for-non-scarring-alopecia-a-systematic-review-S1545961619P0155X
- Fabbrocini G, Cantelli M, Masarà A, Annunziata MC, Marasca C, Cacciapuoti S. Female pattern hair loss: A clinical, pathophysiologic, and therapeutic review. Int J Womens Dermatol. 2018 Jun 19;4(4):203-211. doi: 10.1016/j.ijwd.2018.05.001
- Kanti V, Messenger A, Dobos G, Reygagne P, Finner A, Blumeyer A, Trakatelli M, Tosti A, Del Marmol V, Piraccini BM, Nast A, Blume-Peytavi U. Evidence-based (S3) guideline for the treatment of androgenetic alopecia in women and in men – short version. J Eur Acad Dermatol Venereol. 2018 Jan;32(1):11-22. doi: 10.1111/jdv.14624
- Sánchez-Díaz M, López-Delgado D, Montero-Vílchez T, Salvador-Rodríguez L, Molina-Leyva A, Tercedor-Sánchez J, Arias-Santiago S. Systemic Minoxidil Accidental Exposure in a Paediatric Population: A Case Series Study of Cutaneous and Systemic Side Effects. J Clin Med. 2021 Sep 20;10(18):4257. doi: 10.3390/jcm10184257
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012-. Minoxidil. [Updated 2020 Jan 28]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK548394
- Gargallo V, Gutierrez C, Vanaclocha F, Guerra-Tapia A. Generalized Hypertrichosis Due to Topical Minoxidil. Actas Dermosifiliogr. 2015 Sep;106(7):599-600. English, Spanish. doi: 10.1016/j.ad.2014.12.016
- Rica Echevarría I, García Del Monte J, Delgado Rubio A, Arcangeli F, Lotti T. Severe hypertrichosis in infants due to transdermic exposure to 5% and 7% topical minoxidil. Dermatol Ther. 2020 Nov;33(6):e14230. doi: 10.1111/dth.14230
- MacMillan AR, Warshawski FJ, Steinberg RA. Minoxidil overdose. Chest. 1993 Apr;103(4):1290-1. doi: 10.1378/chest.103.4.1290
- Topical minoxidil: accidental poisoning in children. Prescrire Int. 2015 Apr;24(159):97.
- Garrard A, Wood A, Sollee D, Aaronson P. Refractory hypotension due to Rogaine® (minoxidil) ingestion managed with midodrine. Clin Toxicol (Phila). 2011 Dec;49(10):907-9. doi: 10.3109/15563650.2011.624988
- Chakar B, Salter M, Roberts DM. Minoxidil overdose with hypotension effectively managed with norepinephrine, rather than dopamine. Clin Toxicol (Phila). 2023 Feb;61(2):133-134. doi: 10.1080/15563650.2022.2159831





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}