What is benzoyl peroxide
Benzoyl peroxide is a commonly used topical (for the skin) treatment for mild acne. It is safe for adults and children, and can be used in pregnancy. Benzoyl peroxide may also be used for other skin conditions as determined by your doctor. Benzoyl peroxide is an oxidizing agent that possesses antibacterial properties and is classified as a keratolytic. Benzoyl peroxide is meant to help remove the layer of dead cells on the outer surface of your skin. This makes it easier for oil (sebum) to leave the skin pore, preventing the sebaceous glands from clogging. Benzoyl peroxide also has antibacterial properties and, unlike with antibiotics, there is no risk that bacteria will become resistant if you use it a lot.
Benzoyl peroxide has the following properties:
- Antiseptic i.e. it reduces the number of skin surface bacteria (but it does not cause bacterial resistance and in fact can reduce bacterial resistance if this has arisen from antibiotic therapy). It also reduces the number of yeasts on the skin surface.
- Oxidizing agent – this makes it keratolytic and comedolytic i.e. it reduces the number of comedones.
- Anti-inflammatory action.
In mild to moderate acne, benzoyl peroxide can lead to an improvement within a few weeks. Results has been noted in as soon as 5 days 1. But it can also irritate your skin, causing problems like redness and itching. If benzoyl peroxide comes into contact with clothes and hair it may bleach them, so it is advisable to be cautious when applying it. The effectiveness of benzoyl peroxide does not depend on which form it is used in. Benzoyl peroxide is available in the form of gels, lotions and creams. Benzoyl peroxide comes in various concentrations: 2.5%, 5% and 10%. Products with higher concentrations do not work better than those with lower concentrations, but side effects are more common when 10% benzoyl peroxide is used.
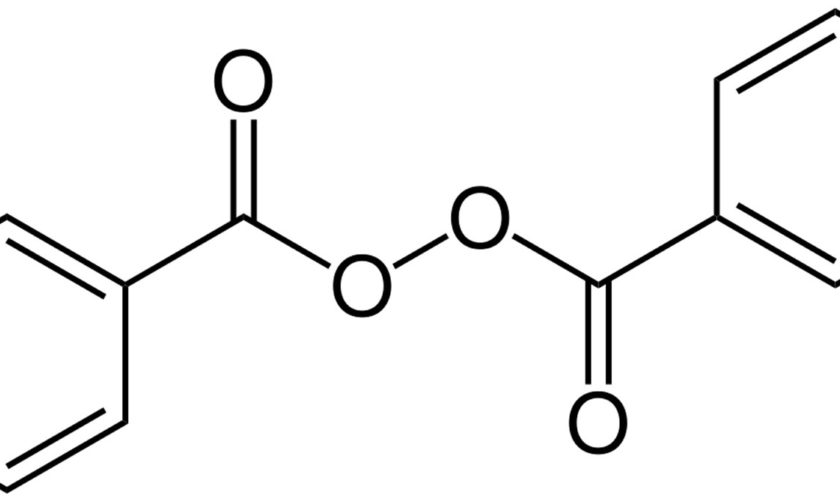
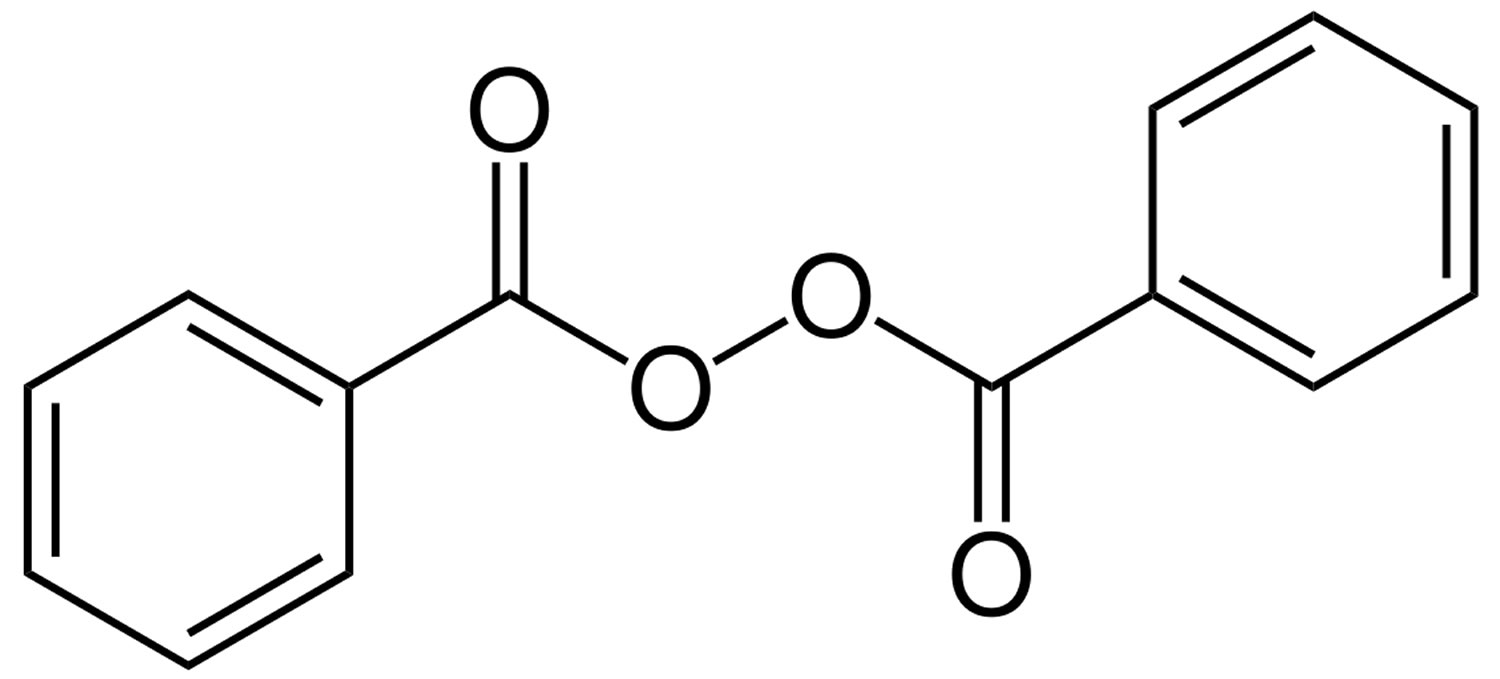
Benzoyl peroxide (C14H10O4) is represented by the structure in Figure 1. Benzoyl peroxide is available without prescription over the counter and benzoyl peroxide is also available on prescription in combination with other active agents.
A meta-analysis (a statistical procedure for combining data from multiple studies in an effort to increase power over individual studies to improve estimates of the size of the effect) comparing the effectiveness of multiple treatments containing 5% benzoyl peroxide, 1% to 1.2% clindamycin, 5% benzoyl peroxide with salicylic acid preparation and combination benzoyl peroxide plus clindamycin in acne lesion reduction 2. Benzoyl peroxide and clindamycin combination is used to treat acne. It works by killing the bacteria that cause acne and by keeping the skin pores clean (tiny openings on the skin). The study authors found at 2 to 4 weeks, 5% benzoyl peroxide + salicylic acid had statistically greater percent acne lesion reductions over other groups (weighted mean inflammatory lesion reduction: benzoyl peroxide = 33.4%, clindamycin = 21.5%, benzoyl peroxide + salicylic acid = 55.2%, benzoyl peroxide + clindamycin = 40.7%, placebo = 7.3%; weighted mean noninflammatory lesion reduction: benzoyl peroxide = 19.1%, clindamycin = 10.0%, benzoyl peroxide + salicylic acid = 42.7%, benzoyl peroxide/clindamycin = 26.2%, placebo = 6.7%). At 10- to 12-week end points, 5% benzoyl peroxide + salicylic acid and benzoyl peroxide/clindamycin were similar, with overlapping confidence intervals (weighted mean inflammatory lesion reduction: benzoyl peroxide = 43.7%, clindamycin = 45.9%, benzoyl peroxide + salicylic acid = 51.8%, benzoyl peroxide/clindamycin = 55.6%, placebo = 26.8%; weighted mean noninflammatory lesion reduction: benzoyl peroxide = 30.9%, clindamycin = 32.6%, benzoyl peroxide + salicylic acid = 47.8%, benzoyl peroxide + clindamycin = 40.3%, placebo = 17.0%). In another word, the authors concluded that at two to four weeks, combination 5 percent benzoyl peroxide plus salicylic acid had the best profile for treating acne vulgaris; at 10 to 12 weeks, this combination treatment was similar to 5 percent benzoyl peroxide + clindamycin treatment. 5 percent benzoyl peroxide + clindamycin was only incrementally better than 5 percent benzoyl peroxide alone but was superior to 1% to 1.2% clindamycin alone. Potential limitations with the review process and the uncertain quality of included trials suggest that the authors’ conclusions should be treated with caution.
Topical antibiotics for acne accumulate in the hair follicle and have been postulated to work through antiinflammatory mechanisms and via antibacterial effects 3. These agents are best used in combination with benzoyl peroxide (wash-off or leave-on), which increases efficacy and decreases the development of resistant bacterial strains. Monotherapy with topical antibiotics in the management of acne is not recommended because of the development of antibiotic resistance. Clindamycin 1% solution or gel is currently the preferred topical antibiotic for acne therapy 4. Topical erythromycin in 2% concentration is available as a cream, gel, lotion, or pledget, but has reduced efficacy in comparison with clindamycin because of resistance of cutaneous Staphylococci and Propionibacterium acnes 3. Stable, fixed-combination agents are available with erythromycin 3%/benzoyl peroxide 5%, clindamycin 1%/benzoyl peroxide 5%, and clindamycin 1%/benzoyl peroxide 3.75% 5. Combination agents may enhance compliance with treatment regimens. Rare reports of diarrhea or Clostridium difficile–related colitis with clindamycin topically have appeared in the literature, but the risk appears low 6. Tolerance of these agents is excellent; clindamycin alone is pregnancy category B (Pregnancy category B means animal reproduction studies have failed to demonstrate a risk to the fetus and there are no adequate and well-controlled studies in pregnant women).
How to use benzoyl peroxide products:
- Make sure your skin is clean and dry before applying
- Apply a thin smear to areas of skin affected by acne, initially every second night, then build up to once or twice daily as tolerated
- It can be used on the face as well as the trunk
- Be patient: acne responds very slowly to treatment. Results has been noted in as soon as 5 days 1, but it may take several months to notice improvement
Problems with benzoyl peroxide products:
- Dryness of the treated area can be expected and is usually mild. If the skin is visibly scaly, apply a light non-oily moisturizer.
- Skin irritation is rarely severe. Occasionally, irritation means that product must be discontinued. Consider applying it less frequently.
- Contact dermatitis (red, dry, itchy skin) can be due to irritation or allergy. It can be treated with a topical steroid such as hydrocortisone cream.
- Rarely, serious allergic reactions to benzoyl peroxide, including anaphylaxis, have been reported.
- Bleaching of clothing. Make sure the benzoyl peroxide has completely dried before the treated skin touches clothes or bedding. It is more likely to stain cotton and linen fabrics than polyester and fleece fabrics 7.
Figure 1. Benzoyl peroxide
Other treatment options for acne
There are many treatment options for acne, including topical and oral medication, and over-the-counter or prescription-only products. But what is the right product for you?
The treatment options for acne will depend on a number of different things. For instance:
- How severe is your acne?
- What is your skin type (dry, oily or combination)?
- How upsetting do you find your acne?
- Are you susceptible to acne scarring?
- Do you have other health problems?
- Are you male or female?
- Which treatments have you already tried out and how well did they work?
- What kinds of side effects do the different products have, and how unpleasant do you think those side effects are?
Nearly all acne treatments require a lot of patience. Many of them only start working after several weeks or months. But it can be worth the wait, and is better than constantly switching treatments, which can sometimes make you feel like nothing will help.
People with acne sometimes also use complementary or alternative medicines to try to improve their skin. These include things like herbal and homeopathic products, tea tree oil and purified bee venom. Acupuncture, cupping and special massages are offered for the treatment of acne too. But none of these products or approaches have been clearly proven to work yet.
Other Topical medications
There are a variety of creams, lotions and gels that can be applied directly to the skin (topically) with different drugs in them. All of these treatments need to be used for several weeks or months before they start working. To prevent new pimples from forming, they have to be applied to the skin surrounding existing pimples too.
Some medications can irritate the skin, causing things like redness and itching. You can reduce this risk by starting with a low dose and then gradually increasing it. If your skin becomes irritated, lowering the dose can help. If your skin stays irritated or if the medication has not worked after some time, you could try a different medication.
- Summary of acne and acne vulgaris topical treatment options for adolescents to adults 8:Benzoyl peroxide and/ or combinations with erythromycin or clindamycin are effective acne treatments and are recommended as monotherapy for mild acne, or in conjunction with a topical retinoid, or systemic antibiotic therapy for moderate to severe acne.
- Benzoyl peroxide is effective in the prevention of bacterial resistance and is recommended for patients on topical or systemic antibiotic therapy.
- Topical antibiotics (e.g., erythromycin and clindamycin) are effective acne treatments but are not recommended as monotherapy due risk of bacterial resistance.
- Topical retinoids are important in addressing the development and maintenance of acne and are recommended as monotherapy in primarily comedonal acne, or in combination with topical or oral antimicrobials in patients with mixed or primarily inflammatory acne lesions.
- Employing multiple topical agents that affect different aspects of acne pathogenesis can be useful. Combination therapy should be used in the majority of patients with acne.
- Topical adapalene, tretinoin and benzoyl peroxide can be safely used in the management of preadolescent acne in children.
- Azelaic acid is a useful adjunctive acne treatment and is recommended in the treatment of postinflammatory dyspigmentation.
- Topical dapsone 5% gel is recommended for inflammatory acne, particularly in adult females with acne.
- There is limited evidence to support recommendations for sulfur, nicotinamide, resorcinol, sodium sulfacetamide, aluminum chloride, and zinc in the treatment of acne.
Other products for topical use
Azelaic acid helps prevent oil glands in the skin from becoming clogged and can improve acne. It has an antibacterial and anti-inflammatory effect. The possible side effects include skin irritations such as itching and burning. Azelaic acid 20% is mildly effective as a comedolytic, antibacterial, and antiinflammatory agent. Azelaic acid has use in patients with sensitive skin or of Fitzpatrick skin types IV or greater because of the lightening effect of the product on dyspigmentation 9. Azelaic acid is category B in pregnancy.
Pregnancy Category B: Animal reproduction studies have failed to demonstrate a risk to the fetus and there are no adequate and well-controlled studies in pregnant women.
Many acne products, such as cleansing toners and creams, contain salicylic acid. This ingredient is believed to work by removing dead skin cells from blocked pores (comedolytic agent). It is not clear whether products containing salicylic acid help reduce acne. Salicylic acid can also have side effects such as redness, dryness and peeling. Salicylic acid is a comedolytic agent that is available over the counter in 0.5% to 2% strengths for the therapy of acne vulgaris. Both wash-off and leave-on preparations are well tolerated. Clinical trials demonstrating the efficacy of salicylic acid in acne are limited 10.
The sulfone agent, dapsone 5% gel, is available as a twice-daily agent for the therapy of acne vulgaris. In clinical trials, topical dapsone showed modest to moderate efficacy, primarily in the reduction of inflammatory lesions 11. Combination with topical retinoids may be indicated if comedonal components are present. The mechanism of action is poorly understood, and its ability to kill Propionibacterium acnes (is a gram-positive human skin commensal bacteria and is involved in the pathogenesis of acne) has been poorly studied. It is generally thought to work as an antiinflammatory agent. The benefit in women seems to exceed the benefit in male and adolescent patients 12. Topical dapsone may be oxidized by the coapplication of benzoyl peroxide, causing orange-brown coloration of the skin which can be brushed or washed off. Topical dapsone 5% gel is pregnancy category C and has efficacy and safety data down to patients 12 years of age. Glucose-6-phosphate dehydrogenase testing is not required before starting topical dapsone.
Pregnancy Category C: Animal reproduction studies have shown an adverse effect on the fetus and there are no adequate and well-controlled studies in humans, but potential benefits may warrant use of the drug in pregnant women despite potential risks.
Topical Antibiotics
In inflammatory forms of acne, the skin is infected with bacteria. Antibiotics that are applied to the skin have an anti-inflammatory effect and can reduce inflammatory forms of acne. They are not effective in the treatment of non-inflammatory acne.
Topical antibiotics need to be used for quite some time before they can have an effect. In US and other countries, they are only available on prescription. The treatment takes at least three weeks, and many people only see an improvement after three to six months. One problem with antibiotics is that there is always a risk of bacteria becoming resistant. In other words, the bacteria may get used to the drug if it is used too often. As a result, the antibiotics do not work as well the next time you use them, or they may not work at all. So antibiotics are not suitable for repeated long-term use.
Topical Retinoids
Topical retinoids are vitamin A derivatives that are prescription agents with randomized, double-blind, placebo-controlled trials supporting their use for acne treatment 13. The retinoids used in the topical treatment of acne include adapalene, isotretinoin and tretinoin. Three active agents are available: tretinoin (0.025-0.1% in cream, gel, or microsphere gel vehicles), adapalene (0.1%, 0.3% cream, or 0.1% lotion73,74), and tazarotene (0.05%, 0.1% cream, gel or foam). Each retinoid binds to a different set of retinoic acid receptors: tretinoin to alpha, beta, and gamma, and tazarotene and adapalene, selectively, to beta and gamma—thereby conferring slight differences in activity, tolerability, and efficacy. Retinoids are the core of topical therapy for acne because they are comedolytic, resolve the precursor microcomedone lesion, and are antiinflammatory. In US and other countries, topical retinoids are prescription-only and are available as creams, gels or solutions. They can help in both inflammatory and non-inflammatory acne. Treatment with retinoids can lead to a visible improvement within a few weeks.
Side effects such as redness, burning and itching may occur. Retinoids have NOT been approved for use in women who are pregnant or breastfeeding.
Topical retinoids enhance any topical acne regimen and allow for maintenance of clearance after discontinuation of oral therapy. Retinoids are ideal for comedonal acne and, when used in combination with other agents, for all acne variants. Three topical agents are available that contain retinoids in combination with other products: adapalene 0.1%/benzoyl peroxide 2.5%, approved for use in patients ≥9 years of age, and 2 agents with fixed combination clindamycin phosphate 1.2%/tretinoin 0.025% gel, approved for patients ≥12 years of age 14.
Retinoid use may be limited by side effects, including dryness, peeling, erythema, and irritation, which can be mitigated by reduced frequency of application 15. Given any single agent, higher concentrations may be more efficacious, but with greater side effects 15. Some formulations of tretinoin (primarily generic products) are not photostable and should be applied in the evening. Tretinoin also may be oxidized and inactivated by the coadministration of benzoyl peroxide. It is recommended that the 2 agents be applied at different times. Tretinoin microsphere formulation, adapalene, and tazarotene do not have similar restrictions. Topical retinoids have been associated with an increased risk of photosensitivity; concurrent daily sunscreen can be used to reduce the risk of sunburn.
There are several head-to-head studies with retinoid products. Some support greater efficacy of tazarotene over adapalene and tretinoin, and adapalene over tretinoin, but the concentrations and formulations used were varied 16. Data suggest that adapalene is better tolerated than multiple concentrations of tretinoin, but this is based on older formulations 16. Overall, the limitations of the existing studies prohibit direct efficacy comparisons of topical retinoids.
Tretinoin and adapalene are pregnancy category C, while tazarotene is category X; therefore, patients should be counseled on these pregnancy risks when starting a retinoid or if a woman patient desires pregnancy.
Pregnancy Category C: Animal reproduction studies have shown an adverse effect on the fetus and there are no adequate and well-controlled studies in humans, but potential benefits may warrant use of the drug in pregnant women despite potential risks.
Pregnancy Category X: Studies in animals or humans have demonstrated fetal abnormalities and/or there is positive evidence of human fetal risk based on adverse reaction data from investigational or marketing experience, and the risks involved in use of the drug in pregnant women clearly outweigh potential benefits.
Oral medications
Oral medications are usually considered in people with moderate to severe acne, or if topical treatment has not led to a big enough improvement.
Oral Antibiotics
Antibiotic tablets can help improve inflammatory acne when taken for several weeks or months.
Oral antibiotics can have side effects, including dizziness, digestive problems and allergic reactions such as rashes. The antibiotics called tetracycline and minocycline are not suitable for anyone who is pregnant or breastfeeding. People who take minocycline for longer than three weeks should have a blood test before starting the treatment, as well as regular blood tests during the treatment. This is done in order to detect any problems with the liver, kidney or the formation of blood as soon as possible.
As with topical antibiotics, there is a risk that the bacteria may become resistant and that oral antibiotics will then stop working if they are used too often.
Oral Hormones
One of the main causes of acne is higher levels of, or an increased sensitivity to, the hormone androgen. Certain hormone products can reduce the production and effect of androgen, leading to better skin.
Some hormone products can be prescribed especially for the treatment of acne. These medicines also have a contraceptive effect. Three combinations have been approved for the treatment of acne in girls and women:
- Ethinyl estradiol / cyproterone acetate
- Ethinyl estradiol / chlormadinone acetate
- Ethinyl estradiol / dienoges
In women with moderate to severe acne, these hormone products are often used together with a topical treatment in order to improve the overall effect.
Hormone products such as birth control pills, on the other hand, are intended for contraceptive use and are usually not approved for the treatment of acne. But if girls and women who have acne use the contraceptive pill as a form of contraception, it may also have a positive effect on their acne. This is only true, though, if they take a pill that has the hormones estrogen and progestin in it.
Hormone products can also cause side effects, such as headaches and nausea. They increase the risk of deep vein thrombosis too – some more so than others.
Oral Retinoids
Retinoid tablets are the most effective medications for the treatment of acne, but they also have the most side effects. Because of this, they are generally only used if other medications have not worked. Retinoid tablets can lead to noticeable improvement, or might even make acne clear up completely. Acne sometimes comes back again after a while, though.
Because retinoids lower the production of oil in the skin, people who take them might have dry lips, skin and eyes. Other side effects include headaches, achy joints and backache. The higher the dose of retinoids you take, the more likely you are to experience side effects. Pregnant and breastfeeding women should not take retinoids.
Retinoids will only be considered as a treatment option for sexually active women if they use at least one contraceptive method, or preferably two at the same time. For example, if they take the pill and use condoms as well, to be on the safe side. Women must keep using contraception for at least four weeks after they stop taking retinoids. This is because retinoids can be harmful to unborn babies.
A few years ago there were a number of reports that taking isotretinoin led to a higher risk of suicide. This has not been confirmed by scientific studies. Still, it is important to look out for any unusual changes in mood if you are taking retinoids, and inform your doctor if you have any. In any case, it always makes sense to seek medical and/or psychological help if acne is a major problem for someone or if it causes mental health problems.
Light-based therapy
Besides topical and oral medications, different types of light-based therapy can be used to treat acne. Generally speaking, there has not been enough good-quality research on these approaches.
Laser treatment and intense pulsed light therapy
There is some evidence that laser treatment and intense pulsed light therapy can lead to a short-term improvement in inflammatory acne. But there is a lack of good research on the long-term benefits of these treatment approaches. So it is not clear whether they represent an alternative to other therapies that have been proven to be effective.
Phototherapy
Somewhat more research has been done on phototherapy. This treatment approach involves shining UV light on the affected areas of skin under medical supervision. This is meant to help kill bacteria in the acne. Research suggests that this treatment can improve acne, at least in the short term. Phototherapy is not the same as using a tanning bed.
Is benzoyl peroxide safe?
Yes, benzoyl peroxide is safe for skin (topical) use for acne. However, like any medicine benzoyl peroxide is not right for everyone. Do not use benzoyl peroxide if you had an allergic reaction to benzoyl peroxide.
Topical benzoyl peroxide has not been studied during breastfeeding. Because only about 5% is absorbed following topical application, it is considered a low risk to the nursing infant 17. Ensure that your baby’s skin does not come into direct contact with the areas of skin that have been treated. Only water-miscible cream or gel products should be applied to the breast because ointments may expose the infant to high levels of mineral paraffins via licking 18.
There is inadequate evidence in humans for the carcinogenicity of benzoyl peroxide. There is limited evidence in experimental animals for the carcinogenicity of benzoyl peroxide. Overall evaluation: Benzoyl peroxide is NOT classifiable as a human carcinogen 19. The International Agency for Research on Cancer 20.
What does benzoyl peroxide do
Benzoyl peroxide is an oxidizing agent that possesses antibacterial properties and is classified as a keratolytic. Benzoyl peroxide is meant to help remove the layer of dead cells on the outer surface of your skin (comedolytic) 21. This makes it easier for oil (sebum) to leave the skin pore, preventing the sebaceous glands from clogging. Benzoyl peroxide also has antibacterial properties that kills Propionibacterium acnes bacteria and, unlike with antibiotics, there is no risk that bacteria will become resistant if you use it a lot. Propionibacterium acnes is a gram-positive human skin commensal that prefers anaerobic growth conditions and is involved in the development of acne 22. Benzoyl peroxide is an antibacterial agent that kills Propionibacterium acnes through the release of free oxygen radicals. The addition of benzoyl peroxide to regimens of antibiotic therapy enhances results and may reduce antibiotic resistance development. Benzoyl peroxide is available as topical washes, foams, creams, or gels, and can used as leave-on or wash-off agents. Strengths available for acne therapy range from 2.5% to 10%. Benzoyl peroxide therapy is limited by concentration-dependent irritation, staining and bleaching of fabric, and uncommon contact allergy. Total skin contact time and formulation can also affect efficacy. Lower concentrations (e.g, 2.5-5%), water-based, and wash-off benzoyl peroxide agents may be better tolerated in patients with more sensitive skin 23. Results can be noted in as soon as 5 days 1.
Benzoyl peroxide products
Acne-Clear, Benzac AC, BenzePrO, Benziq, BPO, Brevoxyl Acne Wash Kit, Clearplex, Clearskin, Desquam-X Wash, Fostex Wash 10%, NeoBenz Micro, Neutrogena Acne Mask, Oscion, Oxy Balance, Oxy Daily Wash, Oxy-10, Pacnex, PanOxyl, Persa-Gel, Riax, SoluCLENZ Rx, Triaz, Benzac, Desquam-X 10, Benzashave 5, Benzashave 10, Panoxyl AQ 2.5, Desquam-E, Benzac W, Brevoxyl, Panoxyl AQ 5, Panoxyl 5, Desquam-X 5, Persa-Gel W, Benzagel-5, Panoxyl 10, Panoxyl AQ 10, Benzagel-10, Peroxin A 10, Triaz Cleanser, Benoxyl 5, Acne Treatment, Ben-Aqua, Del-Aqua, Peroxin A, Zeroxin, Acne-10, Benoxyl 10, Fostex Bar 10%, Fostex Gel 10%, Clear By Design, Loroxide, Vanoxide, Oxy Vanishing Gel, Neutrogena On Spot Acne Treatment, Seba-Gel, Brevoxyl Creamy Wash, Clinac BPO, Benzagel Wash, Ethexderm, Oxy 10 Balance, Zaclir, Benziq LS, Benziq Wash, ZoDerm Redi-Pads (obsolete), Panoxyl Aqua Gel, Oscion Cleanser, Inova, NeoBenz Micro SD, Lavoclen-4 Creamy Wash, Lavoclen-8 Creamy Wash, Oxy Daily Wash Chill Factor, Oxy Spot Treatment, PanOxyl Maximum Strength Foaming Acne Wash, Lavoclen-4, Lavoclen-8, Breze, Brevoxyl-4 Creamy Wash Complete Pack, Brevoxyl-8 Creamy Wash Complete Pack, NeoBenz Micro Wash, NeoBenz Micro Wash Plus Pack, Triaz Foaming Cloths, Triaz Pads, BenzEFoam, Pacnex MX Wash, NeoBenz Micro Cream Plus Pack, Benzac AC Wash, BPO Foaming Cloths, Pacnex HP, Pacnex LP, BenzEFoam Ultra, BP Cleansing Lotion, BP Wash, OC8, Delos, PR Benzoyl Peroxide Wash and Differin Daily Deep Cleanser
Benzoyl peroxide uses
Benzoyl peroxide topical (for the skin) is used to treat acne.
The therapy of acne in children <12 years of age with products approved by the FDA has expanded. Fixed combination benzoyl peroxide 2.5%/adapalene 1% gel is approved for patients ≥9 years of age, and tretinoin 0.05% micronized tretinoin gel for patients ≥10 years of age. All other retinoids are approved by the FDA for patients ≥12 years of age. Current data show that retinoids in younger patients are effective and are not associated with increased irritation or risk.
How to use benzoyl peroxide
Ask your doctor or pharmacist before using any other medicine, including over-the-counter medicines, vitamins, and herbal products.
Benzoyl peroxide is not right for everyone. Do not use benzoyl peroxide if you had an allergic reaction to benzoyl peroxide.
Bar, Cream, Foam, Gel/Jelly, Liquid, Lotion, Pad, Soap
When you first begin to use benzoyl peroxide, apply it to 1 or 2 small affected areas of the skin for 3 days. If no discomfort occurs, follow the directions on the product label or use benzoyl peroxide as directed by your doctor.
Use benzoyl peroxide only on your skin. Rinse it off right away if benzoyl peroxide gets on a cut or scrape. Do not get benzoyl peroxide in your eyes, nose, or mouth.
Missed dose: Apply a dose as soon as you can. If it is almost time for your next dose, wait until then and apply a regular dose. Do not apply benzoyl peroxide to make up for a missed dose.
Store benzoyl peroxide in a closed container at room temperature, away from heat, moisture, and direct light. Do not freeze.
Warnings
Tell your doctor if you are pregnant or breastfeeding, or if you ever had an allergic reaction to an acne product.
Benzoyl peroxide may bleach your hair or clothes.
Benzoyl peroxide may make your skin more sensitive to sunlight. Wear sunscreen. Do not use sunlamps or tanning beds.
Call your doctor if your symptoms do not improve or if they get worse.
Keep all medicine out of the reach of children. Never share your medicine with anyone.
Benzoyl peroxide side effects
Benzoyl peroxide topical can cause a rare but serious allergic reaction or severe skin irritation. These reactions may occur just a few minutes after you apply the medicine, or within a day or longer afterward.
Stop using benzoyl peroxide topical and get emergency medical help if you have signs of an allergic reaction: hives, itching; difficult breathing, feeling light-headed; swelling of your face, lips, tongue, or throat.
Some side effects of benzoyl peroxide topical may occur that usually do not need medical attention. These side effects may go away during treatment as your body adjusts to the medicine. Also, your health care professional may be able to tell you about ways to prevent or reduce some of these side effects. Check with your health care professional if any of the following side effects continue or are bothersome or if you have any questions about them:
Less common
- dryness or peeling of the skin (may occur after a few days)
- feeling of warmth, mild stinging, and redness of the skin
Common side effects may include:
- mild stinging or burning;
- itching or tingly feeling;
- skin dryness, peeling, or flaking; or
- redness or other irritation.
Stinging or burning sensation for a brief time after benzoyl peroxide topical application, with continuous use these effects mostly disappear. After 1 or 2 weeks of benzoyl peroxide topical use there may be a sudden excess of dryness of your skin and peeling 24.
This is not a complete list of side effects and others may occur. Call your doctor for medical advice about side effects.
Call your doctor right away if you notice any of these side effects:
- Allergic reaction: Itching or hives, swelling in your face or hands, swelling or tingling in your mouth or throat, chest tightness, trouble breathing
- Burning, blistering, swollen, or peeling skin
- Fainting
- Swelling of the eyes, face, lips, or tongue
Stop using benzoyl peroxide and call your doctor at once if you have any of these side effects on the treated skin:
- severe itching or burning;
- severe stinging or redness;
- swelling; or
- peeling.
Less common or rare side effects of benzoyl peroxide:
- painful irritation of skin, including burning, blistering, crusting, itching, severe redness, or swelling
- skin rash
Incidence not known
- difficult breathing
- fainting
- hives
- itching
- swelling of the eyes, face, lips, or tongue
- tightness in the throat
Get emergency help immediately if any of the following symptoms of overdose occur while taking benzoyl peroxide topical:
Symptoms of Overdose
- Burning, itching, scaling, redness, or swelling of skin (severe).
- Schutte H, Cunliffe WJ, Forster RA. The short-term effects of benzoyl peroxide lotion on the resolution of inflamed acne lesions. Br J Dermatol. 1982;106:91-94.
- Meta-analysis comparing efficacy of benzoyl peroxide, clindamycin, benzoyl peroxide with salicylic acid, and combination benzoyl peroxide/clindamycin in acne. JAAD July 2010, Volume 63, Issue 1, Pages 52–62 https://www.jaad.org/article/S0190-9622(09)00987-6/fulltext
- Mills O Jr, Thornsberry C, Cardin CW, Smiles KA, Leyden JJ. Bacterial resistance and therapeutic outcome following three months of topical acne therapy with 2% erythromycin gel versus its vehicle. Acta Derm Venereol. 2002;82:260-265.
- Padilla RS, McCabe JM, Becker LE. Topical tetracycline hydrochloride vs. topical clindamycin phosphate in the treatment of acne: a comparative study. Int J Dermatol. 1981;20:445-448.
- Pariser DM, Rich P, Cook-Bolden FE, Korotzer A. An aqueous gel fixed combination of clindamycin phosphate 1.2% and benzoyl peroxide 3.75% for the once-daily treatment of moderate to severe acne vulgaris. J Drugs Dermatol. 2014;13:1083-1089.
- Mills OH, Jr., Kligman AM, Pochi P , Comite H. Comparing 2.5%, 5%, and 10% benzoyl peroxide on inflammatory acne vulgaris. International journal of dermatology 1986;25:664-7.
- Edwards T, Cardwell L, Patel N, Feldman SR, Title: Benzoyl Peroxide Gel Stains Synthetic Fabrics less than Cotton, Journal of the American Academy of Dermatology (2018), doi: 10.1016/j.jaad.2018.05.008.
- Topical therapies: Recommendations. https://www.aad.org/practicecenter/quality/clinical-guidelines/acne/topical-therapies
- Kircik LH. Efficacy and safety of azelaic acid (AzA) gel 15% in the treatment of post-inflammatory hyperpigmentation and acne: a 16-week, baseline-controlled study. J Drugs Dermatol. 2011;10:586-590.
- Zouboulis CC, Derumeaux L, Decroix J, Maciejewska-Udziela B, Cambazard F , Stuhlert A. A multicentre, single-blind, randomized comparison of a fixed clindamycin phosphate/tretinoin gel formulation (Velac) applied once daily and a clindamycin lotion formulation (Dalacin T) applied twice daily in the topical treatment of acne vulgaris. Br J Dermatol 2000;143:498-505.
- Cunliffe WJ, Caputo R, Dreno B, Forstrom L, Heenen M, Orfanos CE et al. Clinical efficacy and safety comparison of adapalene gel and tretinoin gel in the treatment of acne vulgaris: Europe and U.S. multicenter trials. Journal of the American Academy of Dermatology 1997;36:S126-34.
- Del Rosso JQ, Kircik L, Gallagher CJ. Comparative efficacy and tolerability of dapsone 5% gel in adult versus adolescent females with acne vulgaris. J Clin Aesthet Dermatol. 2015;8:31-37.
- Lucky AW, Cullen SI, Funicella T, et al. Double-blind, vehicle-controlled, multicenter comparison of two 0.025% tretinoin creams in patients with acne vulgaris. J Am Acad Dermatol. 1998;38:S24-S30.
- Dreno B, Bettoli V, Ochsendorf F, et al. Efficacy and safety of clindamycin phosphate 1.2%/tretinoin 0.025% formulation for the treatment of acne vulgaris: pooled analysis of data from three randomised, double-blind, parallel-group, phase III studies. Eur J Dermatol. 2014;24:201-209.
- Pedace FJ, Stoughton R. Topical retinoic acid in acne vulgaris. Br J Dermatol. 1971;84:465-469.
- Lucky AW, Cullen SI, Funicella T, Jarratt MT, Jones T , Reddick ME. Double-blind, vehicle-controlled, multicenter comparison of two 0.025% tretinoin creams in patients with acne vulgaris. Journal of the American Academy of Dermatology 1998;38:S24-30.
- Leachman SA, Reed BR. The use of dermatologic drugs in pregnancy and lactation. Dermatol Clin. 2006;24:167-97. https://www.ncbi.nlm.nih.gov/pubmed/16677965
- Noti A, Grob K, Biedermann M et al. Exposure of babies to C(15)-C(45) mineral paraffins from human milk and breast salves. Regul Toxicol Pharmacol. 2003;38:317-25. https://www.ncbi.nlm.nih.gov/pubmed/14623482
- American Conference of Governmental Industrial Hygienists TLVs and BEIs. Threshold Limit Values for Chemical Substances and Physical Agents and Biological Exposure Indices. Cincinnati, OH, 2008, p. 14
- BENZOYL PEROXIDE. IARC. Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. Geneva: World Health Organization, International Agency for Research on Cancer, 1972-PRESENT. https://monographs.iarc.fr/ENG/Monographs/vol71/mono71-13.pdf
- Cunliffe WJ, Dodman B, Ead R. Benzoyl peroxide in acne. Practitioner. 1978;220:479-482.
- Kirschbaum JO, Kligman AM. The pathogenic role of Corynebacterium acnes in acne vulgaris. Archives of Dermatology. 1963;88:832–833
- Fyrand O, Jakobsen HB. Water-based versus alcohol-based benzoyl peroxide preparations in the treatment of acne vulgaris. Dermatologica. 1986;172:263-267.
- Osol, A. (ed.). Remington’s Pharmaceutical Sciences. 16th ed. Easton, Pennsylvania: Mack Publishing Co., 1980., p. 728







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}