Cohen syndrome
Cohen syndrome also called Pepper syndrome, is a rare inherited disorder that affects many parts of the body and is characterized by developmental delay, intellectual disability, small head size (microcephaly), and weak muscle tone (hypotonia) 1. Other features common in this condition include worsening nearsightedness (myopia), breakdown (degeneration) of the light-sensitive tissue at the back of the eye (retinal dystrophy), an unusually large range of joint movement (hypermobility), and distinctive facial features. These facial features typically include thick hair and eyebrows, long eyelashes, unusually-shaped eyes (down-slanting and wave-shaped), a bulbous nasal tip, a smooth or shortened area between the nose and the upper lip (philtrum), and prominent upper central teeth. The combination of the last two facial features results in an open mouth.
The features of Cohen syndrome vary widely among affected individuals. Additional signs and symptoms in some individuals with this disorder include low levels of white blood cells (neutropenia), overly friendly behavior, and obesity that develops in late childhood or adolescence. When obesity is present, it typically occurs around the torso, with the arms and legs remaining slender called truncal obesity. Individuals with Cohen syndrome may also have narrow hands and feet, and slender fingers.
Cohen syndrome affects males and females in about equal numbers. The exact incidence of Cohen syndrome is unknown. More than 150 cases have been reported in the medical literature and an estimated 500-1,000 individuals have been diagnosed with the disorder worldwide 2. More cases are likely undiagnosed or misdiagnosed, making it difficult to determine the true frequency of the disorder in the general population. Cohen syndrome appears to occur more frequently in people of Finnish, Amish, Greek/Mediterranean and Irish ancestry.
There is no cure for Cohen syndrome. Treatment is focused on improving or alleviating the signs and symptoms in the patient. Typically, when a person is first diagnosed with Cohen syndrome, he or she will undergo an eye and blood examination. If vision problems are detected, early correction of the problems, usually with glasses, often leads to general improvement of cognitive skills. If neutropenia (a condition in which an abnormally low number of white blood cells called neutrophils are present, which may result in an increased risk for infections) is discovered when the blood is examined, treatment should be given. Follow-up should include annual eye exams and repeat testing of white blood cell count. Early intervention and physical, occupational, and speech therapy can address developmental delay, hypotonia, joint hyperextensibility, and motor clumsiness 3.
Figure 1. Cohen syndrome
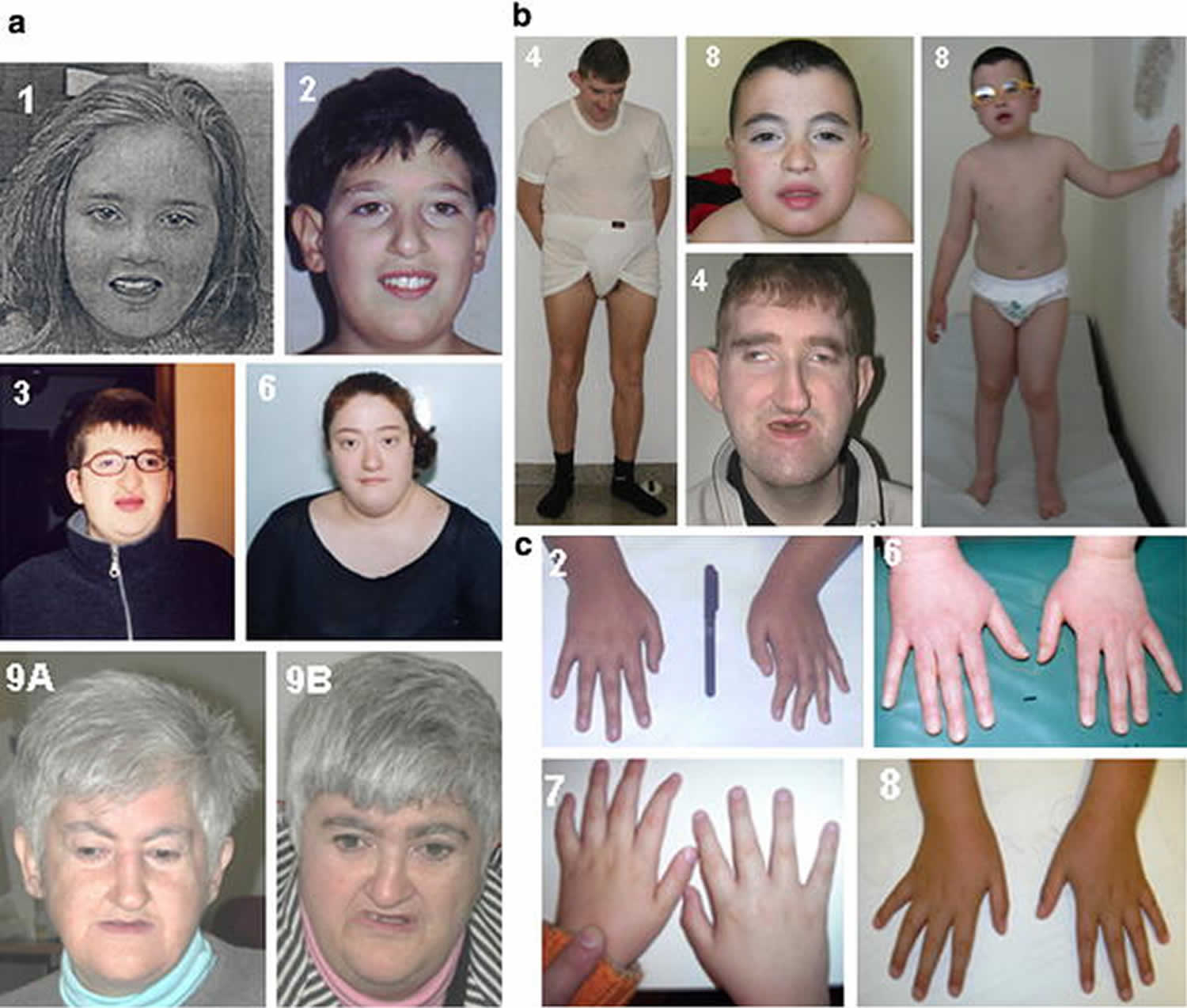
Footnote: Features of Italian patients with Cohen syndrome. (a) Patients 1, 2, 3, 6, 9A and 9B are shown. Note the characteristic facial gestalt of these patients. (b) Frontal view of patients 4 and 8. Note the truncal obesity of both cases. (c) Hands of cases 2, 6, 7 and 8. The fingers are slender and tapering from the proximal interfalangeal joint.
[Source 4 ]Figure 2. Cohen syndrome eye
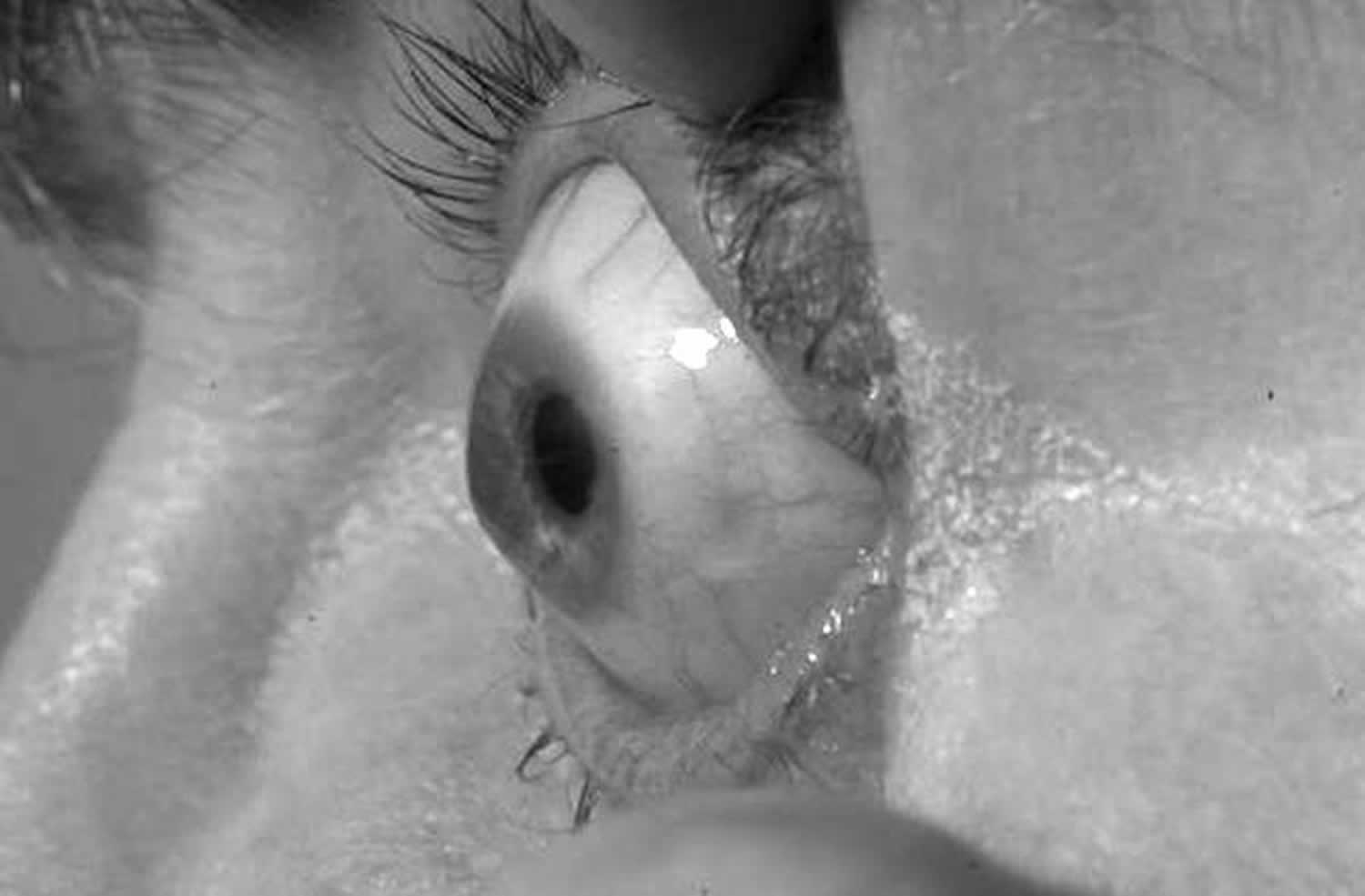
Footnote: Corneal ectasia in patient with Cohen syndrome with COH1 mutations confirmed on molecular analysis.
[Source 5 ]Cohen syndrome causes
Mutations in the VPS13B gene (COH1 gene) cause Cohen syndrome. The protein produced from COH1 gene (VPS13B gene) is a part of the Golgi apparatus, which is a cell structure in which newly produced proteins are modified so they can carry out their functions. In particular, the VPS13B protein is involved in a modification called glycosylation, which is the attachment of sugar molecules to proteins. The VPS13B protein also appears to be involved in the sorting and transporting of proteins inside the cell. This protein is thought to be involved in normal growth and development of nerve cells (neurons) and fat cells (adipocytes), and may play a role in the storage and distribution of fats in the body.
Most mutations in the VPS13B gene are believed to prevent the production of functional VPS13B protein. Studies suggest that a loss of this protein disrupts the organization of the Golgi apparatus and impairs normal glycosylation. However, it is not known how a lack of functional VPS13B protein or these cellular changes lead to the signs and symptoms of Cohen syndrome. Researchers speculate that problems with neuron development underlie microcephaly, intellectual disability, and retinal dystrophy and that abnormal fat storage may cause truncal obesity in people with Cohen syndrome.
Cohen syndrome inheritance pattern
Cohen syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 3 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 3. Cohen syndrome autosomal recessive inheritance pattern

People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Cohen syndrome symptoms
The signs and symptoms of Cohen syndrome may vary from one individual to another. Although researchers have been able to establish a clear syndrome with characteristic or “core” features, much about the disorder is not fully understood. Several factors including the small number of identified cases and the lack of large clinical studies, prevent physicians from developing a complete picture of associated symptoms and prognosis. Therefore, it is important to note that affected individuals may not have all of the symptoms discussed below. Parents should talk to their children’s physician and medical team about their specific case, associated symptoms and overall prognosis.
Newborns with Cohen syndrome usually have diminished muscle tone (hypotonia). Feeding and breathing difficulties due to hypotonia may be present in the first few days of life. Some newborns may have a weak or high-pitched cry. Some infants may exhibit a failure to gain weight and grow as would otherwise be expected based upon gender and age (failure to thrive). An infant’s joints may be ‘loose’, meaning that they have an abnormally large range of motion (joint hypermobility). Mild to moderate microcephaly often develops within the first year of life and continues into adulthood.
As infants grow older, they may exhibit delays in reaching normal developmental milestones such as sitting up or rolling over (developmental delays). The degree of such delays is highly variable, even among members of the same family. Walking is often delayed until 2-5 years of age. Speech delays are also common; an infant’s or child’s first words or ability to speak in sentences are often delayed.
Mild to moderate intellectual disability is non-progressive and affected individuals show an ability to learn new concepts. Most children are described as sociable with a cheerful disposition. In some instances, children may exhibit behavioral issues that fall within the autistic spectrum. Although rare, seizures have been reported in a minority of individuals.
During childhood, often around the age of 5, distinctive facial features may become apparent. Such features include large ears; a prominent root of the nose (the part of nose between the eyes); a low hairline; highly-arched or wave-shaped eyelids; long, thick eyelashes; thick eyebrows; a high, narrow roof of the mouth (palate); an abnormally short groove in the middle of the upper lip (philtrum); and prominent upper central incisors. Some individuals may develop recurrent, small rounded ulcers in the mouth (aphthous ulcers) and inflammation or infection of the gums (gingivitis) may occur. In the medical literature, the range of distinctive facial features is highly variable and specific features appear to be more likely to occur in individuals of specific ethnic backgrounds.
Affected individuals often develop a variety of abnormalities affecting the eyes and may experience vision problems early in childhood. Such abnormalities include decreased clarity of vision (visual acuity), nearsightedness (myopia), and crossed eyes (strabismus). Myopia usually becomes progressively worse throughout childhood.
Affected individuals may also have chorioretinal dystrophy, a condition characterized by abnormalities affecting the choroid and retina including degeneration of the retina. The choroid is the middle layer of the eye that consists of blood vessels that supply blood to the retina. The retina is a membranous layer of light-sensing cells in the back of the eye that converts light to specific nerve signals, which are then transmitted to the brain to form images. Chorioretinal dystrophy is progressive and can cause poor vision in dim light and eventually night blindness (nyctalopia) and a decreased field of vision with a decreased ability to see to the left or right when looking straight ahead (constriction of the peripheral field of vision; sometimes referred to as tunnel vision). Loss of peripheral vision may cause individuals to trip or fall easily.
Less often, additional abnormalities of the eyes are associated with Cohen syndrome including abnormal curvature of the cornea (astigmatism), reduced size of the cornea (microcornea), abnormally small eyeballs (microphthalmia), clouding (opacity) of the lenses, degeneration of the iris (iris atrophy), degeneration of the optic nerve, which carries impulses from the eyes to the brain (optic atrophy), and a cleft of missing tissue (colobomas) in the retina or eyelids.
Some individuals develop obesity of the trunk or torso of the body that occurs during mid-childhood. The arms and legs can remain slender or thin. Individuals may be below average height for their age and gender (short stature). Some individuals may also have small, narrow hands and feet. Delayed puberty has also been reported and some males exhibit undescended testicles (cryptorchidism).
Abnormal curvature of the spine is common. Affected individuals may develop abnormal front-to-back curvature of the spine (kyphosis), or a combination of kyphosis with abnormal sideways curvature of the spine (scoliosis).
Individuals with Cohen syndrome may have a condition called neutropenia, in which there are abnormally low levels of certain white blood cells called neutrophils. Neutrophils are essential in helping the body to fight off infection by surrounding and destroying bacteria that enter the body. Episodes of neutropenia are usually mild or moderate. Some individuals may experience repeated infections such as respiratory infections or minor skin infections. Children with Cohen syndrome may be prone to developing middle ear infections (otitis media). Chronic development of aphthous ulcers and gingivitis may be partly due to neutropenia.
Individuals with Cohen syndrome appear to be at an increased risk of developing autoimmune disorders, especially diabetes mellitus, but also thyroid disorders and celiac disease. Autoimmune disorders occur when the body’s immune system mistakenly attacks healthy tissue.
Cohen syndrome diagnosis
The diagnosis of Cohen syndrome is based on the symptoms present in the patient, but because the symptoms vary greatly from person to person, no consensus diagnostic criteria exist 6. Genetic testing is available for COH1 gene (VPS13B gene), the only gene known to be associated with Cohen syndrome. However, the rate at which mutations are detected via genetic testing varies by ethnicity. For example, the mutation detection rate in COH1 is higher among the Finnish and Old Amish compared to individuals of from other populations 6.
Cohen syndrome should be suspected in individuals with the following findings 7:
- Retinal dystrophy appearing by mid-childhood
- Progressive high myopia
- Acquired microcephaly
- Non-progressive intellectual disability and global developmental delay
- Hypotonia
- Joint hypermobility
- Typical Cohen syndrome facial gestalt: thick hair and eyebrows, long eyelashes, wave-shaped palpebral fissures, broad nasal tip, smooth or short philtrum, and hypotonic appearance
- Short stature
- Small or narrow hands and feet
- Truncal obesity appearing in or after mid-childhood
- Friendly disposition
- Neutropenia
Cohen syndrome cardinal features 6:
- Retinal dystrophy and high myopia
- Microcephaly
- Developmental delay
- Joint hypermobility
- Typical Cohen syndrome facial gestalt
- Truncal obesity with slender extremities
- Overly sociable behavior
- Neutropenia
Cohen syndrome treatment
The treatment of Cohen syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, pediatric neurologists, orthopedists, ophthalmologists, psychiatrists, speech pathologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling is recommended for affected individuals and their families.
Treatment options that may be used to treat individuals with Cohen syndrome are complex and varied. The specific treatment plan will need to be highly individualized. Decisions concerning the use of specific treatments should be made by physicians and other members of the health care team in careful consultation with an affected child’s parents or with an adult patient based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
Early developmental intervention is important to ensure that affected children reach their potential. Most affected children will benefit from occupational, physical and speech therapy. Various methods of rehabilitative and behavioral therapy may be beneficial. Additional medical, social and/or vocational services including special remedial education may be necessary. Psychosocial support for the entire family is essential as well.
Specific treatments for Cohen syndrome include spectacles and eyeglasses to help with vision. In later years, low vision training as needed in individuals with visual impairment. Recurrent infections can be treated with standard therapies including antibiotics.
In some instances, neutropenia may be treated with the administration of granulocyte-colony stimulating factors (G-CSF). G-CSF is a manufactured version of the natural hormones that stimulate the bone marrow to produce neutrophils. G-CSF increases the number of neutrophils generated by the bone marrow and improves the efficacy of their bacteria-killing ability.
Cohen syndrome prognosis
Obesity progresses over time, along with the orthopedic alterations and oral problems, though the patient life expectancy is not altered in any significant way 8.
References- Cohen syndrome. https://ghr.nlm.nih.gov/condition/cohen-syndrome
- Cohen syndrome. https://rarediseases.org/rare-diseases/cohen-syndrome/
- García Ballesta C, Pérez Lajarin L, Cortés Lillo O. Cohen syndrome. Orphanet. 2004. https://www.orpha.net/data/patho/GB/uk-cohen.pdf
- Katzaki, E., Pescucci, C., Uliana, V. et al. Clinical and molecular characterization of Italian patients affected by Cohen syndrome. J Hum Genet 52, 1011–1017 (2007). https://doi.org/10.1007/s10038-007-0208-4
- Khan A, Chandler K, Pimenides D, Black GC, Manson FD. Corneal ectasia associated with Cohen syndrome: a role for COH1 in corneal development and maintenance?. Br J Ophthalmol. 2006;90(3):390-391. doi:10.1136/bjo.2005.080085 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1856973
- Wang H, Falk MJ, Wensel C, et al. Cohen Syndrome. 2006 Aug 29 [Updated 2016 Jul 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1482
- El Chehadeh-Djebbar S, Blair E, Holder-Espinasse M, Moncla A, Frances AM, Rio M, Debray FG, Rump P, Masurel-Paulet A, Gigot N, Callier P, Duplomb L, Aral B, Huet F, Thauvin-Robinet C, Faivre L. Changing facial phenotype in Cohen syndrome: towards clues for an earlier diagnosis. Eur J Hum Genet. 2013;21:736–42.
- Cruz M, Bosch J. Atlas de síndromes pediátricos . Barcelona: Spaxs publicaciones médicas.1998; 118-119.





{kind=link}