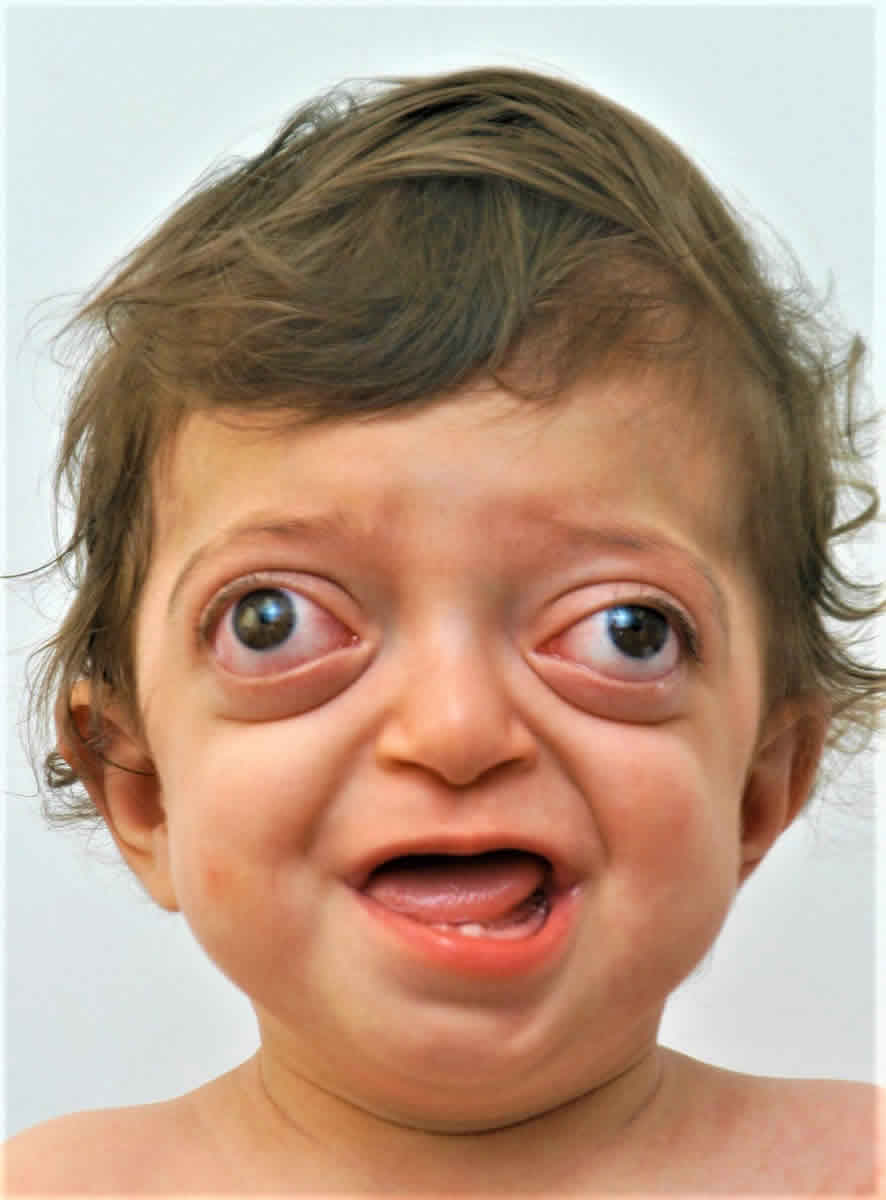
What is Crouzon syndrome
Crouzon syndrome also known as craniofacial dysostosis, is a genetic disorder characterized by the premature fusion of certain skull bones (craniosynostosis). This early fusion of certain skull bones prevents the skull from growing normally and affects the shape of the head and face. Crouzon syndrome is the most common type of craniosynostosis syndrome. Crouzon syndrome is seen in about 1.6 per 100,000 newborns.
Crouzon syndrome shares many of the same features as Apert syndrome due to the result of the premature fusion of the skull bones, including abnormal development of the eye sockets, prominent and widely spaced eyes, bulging eyes and vision problems caused by shallow eye sockets, eyes that do not point in the same direction (strabismus); a beaked nose; and an underdeveloped upper jaw and slow development of the midface. In addition, people with Crouzon syndrome may have dental problems and hearing loss, which is sometimes accompanied by narrow ear canals. A few people with Crouzon syndrome have an opening in the lip and the roof of the mouth (cleft lip and palate). The severity of these signs and symptoms varies among affected people.
In addition to the physical characteristics common to Crouzon syndrome, your child may have the following problems:
- dental problems due to crowded teeth and a narrow palate
- poor vision
- ear disease and hearing loss in about 50% of children
- difficulty breathing due to small airway
- possible fluid on the brain (hydrocephalus)
The severity of signs and symptoms can vary among affected people, even within a family. Intelligence is usually normal, but intellectual disability may be present 1. Crouzon syndrome is caused by changes (mutations) in the FGFR2 gene and is inherited in an autosomal dominant manner.
The child with Crouzon syndrome usually enters a programme of care involving many different clinical specialities, which often continues from birth to the later teenage years. Treatment may involve surgeries to prevent complications, improve function, and aid in healthy psychosocial development 2.
Depending on the severity of Crouzon syndrome, your child may have some or all of the following surgeries:
- frontal orbital advancement to allow the skull to grow properly and to increase the size of the eye sockets
- jaw surgery
- orthodontics work
- surgical advancement of the mid-face
New advances in procedures to correct Crouzon syndrome are constantly being developed. It is important for patients with Crouzon syndrome to be treated by a multidisciplinary care team that specializes in caring for children with these complex disorders.
Figure 1. Crouzon syndrome
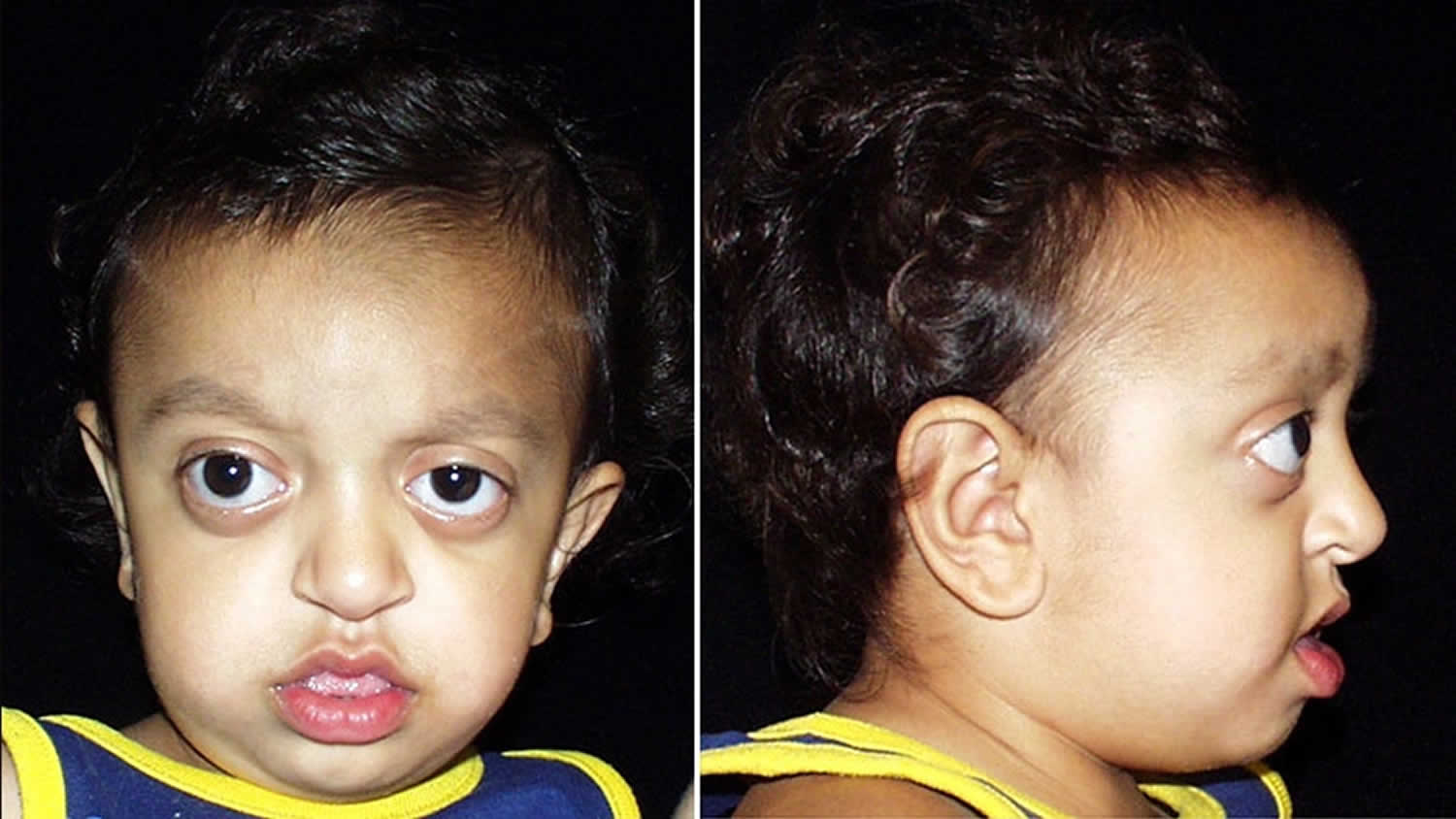
Footnote: 18-month-old with Crouzon syndrome and characteristic rounded forehead, sunken midface and prominent eyes.
[Source 3 ]What is Crouzon syndrome with acanthosis nigricans?
Crouzon syndrome with acanthosis nigricans is found in an estimated 5-10% of all Crouzon cases, it is very rare. Crouzon syndrome with acanthosis nigricans has an estimated prevalence of 1 per 1,000,000 newborns. Fewer than 70 cases have been described in the medical literature. A female-to-male sex ratio of 2.4:1 has been reported. In addition to the facial characteristics, Crouzon syndrome with acanthosis nigricans includes some of the following:
- Darkened, rough patches of skin found in the folds of the body (armpits, neck, groin, elbows, knees, chin/mouth area, eye area, or stomach).
- Signs of this begin between the ages of 2-4.
- The acanthosis nigricans generally does not advance after the age of 12. So, the texture and color stay the same from then on.
Crouzon syndrome with acanthosis nigricans is caused by a specific p.Ala391Glu mutation in the fibroblast growth-factor receptor 3 FGFR3 gene (4p16.3), involved in regulation of cell proliferation, differentiation and apoptosis. Acanthosis nigricans is associated with inadequate stimulation of various fibroblast growth-factor receptors.
Normal newborn skull anatomy and physiology
It is important to have an understanding of skull anatomy and growth in order to understand craniosynostosis. An infant’s skull has 7 bone plates that relate to each other through specialized joints called “cranial sutures”. Sutures are made of tough, elastic fibrous tissue and separate the bones from one another. Sutures meet up (intersect) at two spots on the skull called fontanelles, which are better known as an infant’s “soft spots”. Although there are several major and minor sutures, the sutures that potentially have the most clinical significance are the singular metopic and sagittal sutures, as well as the paired (right and left) coronal and lambdoid sutures (see Figure 2). The seven bones of an infant’s skull normally do not fuse together until around age two or later. The sutures normally remain flexible until this point. In infants with primary craniosynostosis, the sutures abnormally stiffen or harden causing one or more of the bones of the skull to prematurely fuse together. This in turn, may lead to asymmetric skull growth.
Cranial sutures are very unique and specialized joints (syndesmosis joints). Their primary purpose is to grow bone in response to the rapidly developing brain within the protective skull compartment (cranial cavity or calvarium). The skull not only needs to be firm to protect the brain from accidental blows, but should also be expansile to accommodate its rapid growth. The brain doubles in volume in the first year of life and almost triples in volume by the age of three. The sutures of the skull allow for this important but almost contradictory balance of protection and growth.
Each cranial suture is designed to generate growth in the skull in a very specific area and configuration, ultimately reflecting the size and shape of the underlying brain structure. The overall bone development from cranial sutures occurs in a direction perpendicular to the long axis of the suture. Understanding these two facts, it makes sense that a fusion of each suture independently would cause a unique head shape.
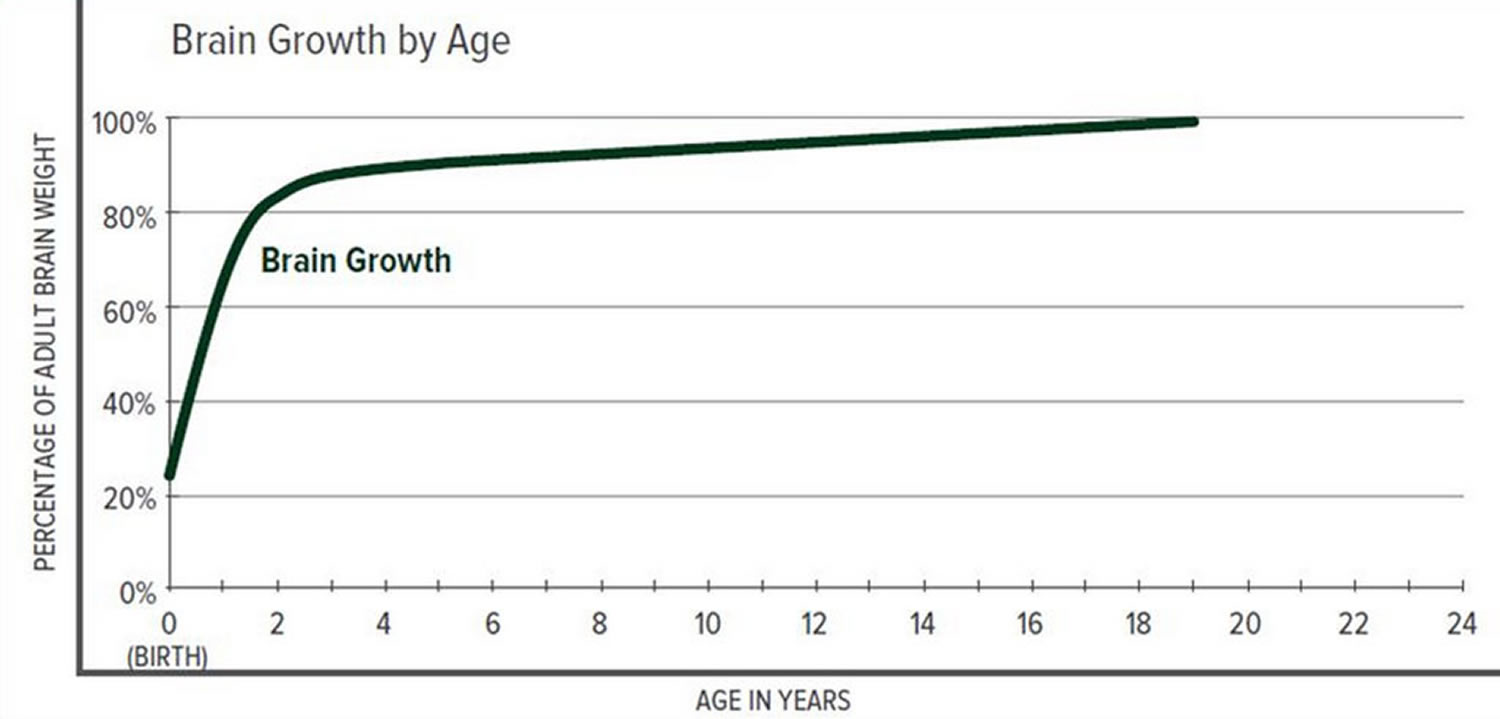
The greatest increase in brain volume (brain growth) occurs from 0 to 14 months of age. The size of a child’s brain typically reaches 80% of adult size by the age of 2. The growth in head circumference after that age is more related to growth in the thickness of the skull and scalp but not actual brain growth. “Hat size” increases but not necessarily “brain size”.
The various cranial sutures close at different ages. The metopic suture closes earliest, around 6 months to 2 years. The rest of the sutures stay open into the 20’s and 30’s. The brain and fluid cavities of the brain do continue to grow in volume as you go into early adulthood, albeit not nearly as rapidly as the first couple years of life. Since the skull is much firmer (calcified or “rock-like”) and thicker, the skull needs the sutures to grow bone for any increase in volume.
Interestingly, there is a lot of variability here. Doctors have operated on adults in their 30’s for reasons unrelated to their skull sutures and have coincidentally found open metopic sutures. They have also seen young adults with closed coronal, lambdoid, and sagittal sutures, but with normal head shapes and often, no indication or symptoms of high pressure.
Scientists have learned that a cranial suture’s purpose is to grow bone to accommodate a growing brain, and that most brain growth occurs in the first two years of life. The brain reaches 85% of adult size by age 3 years (see Figure 3. Brain size vs. age diagram).
So it makes sense that the sutures are vitally important in the first two years of life. The earlier the fusion, the more severe the restriction in growth and, consequently, volume provided to the brain.
Figure 2. Normal skull of a newborn
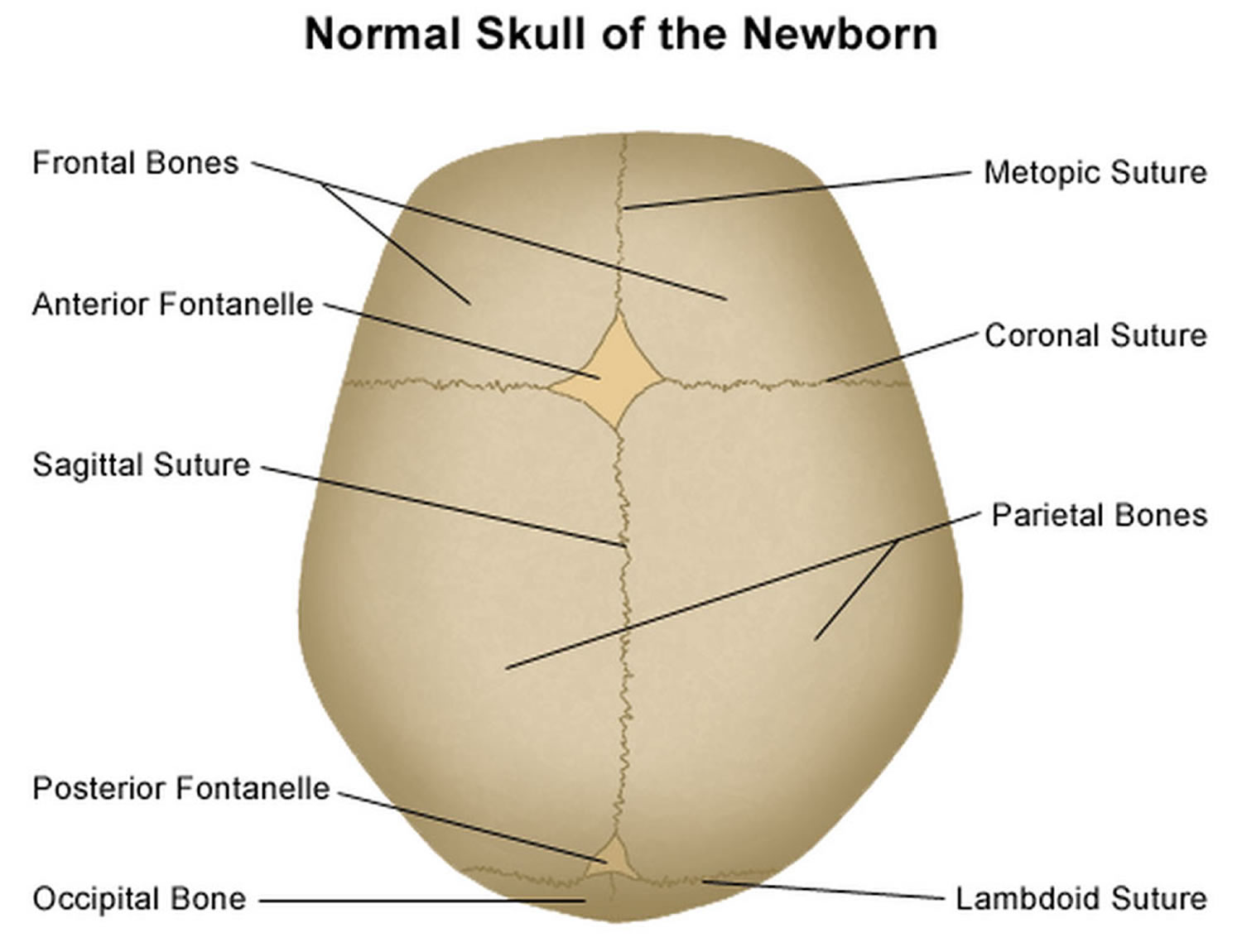
Figure 3. Brain size versus age diagram
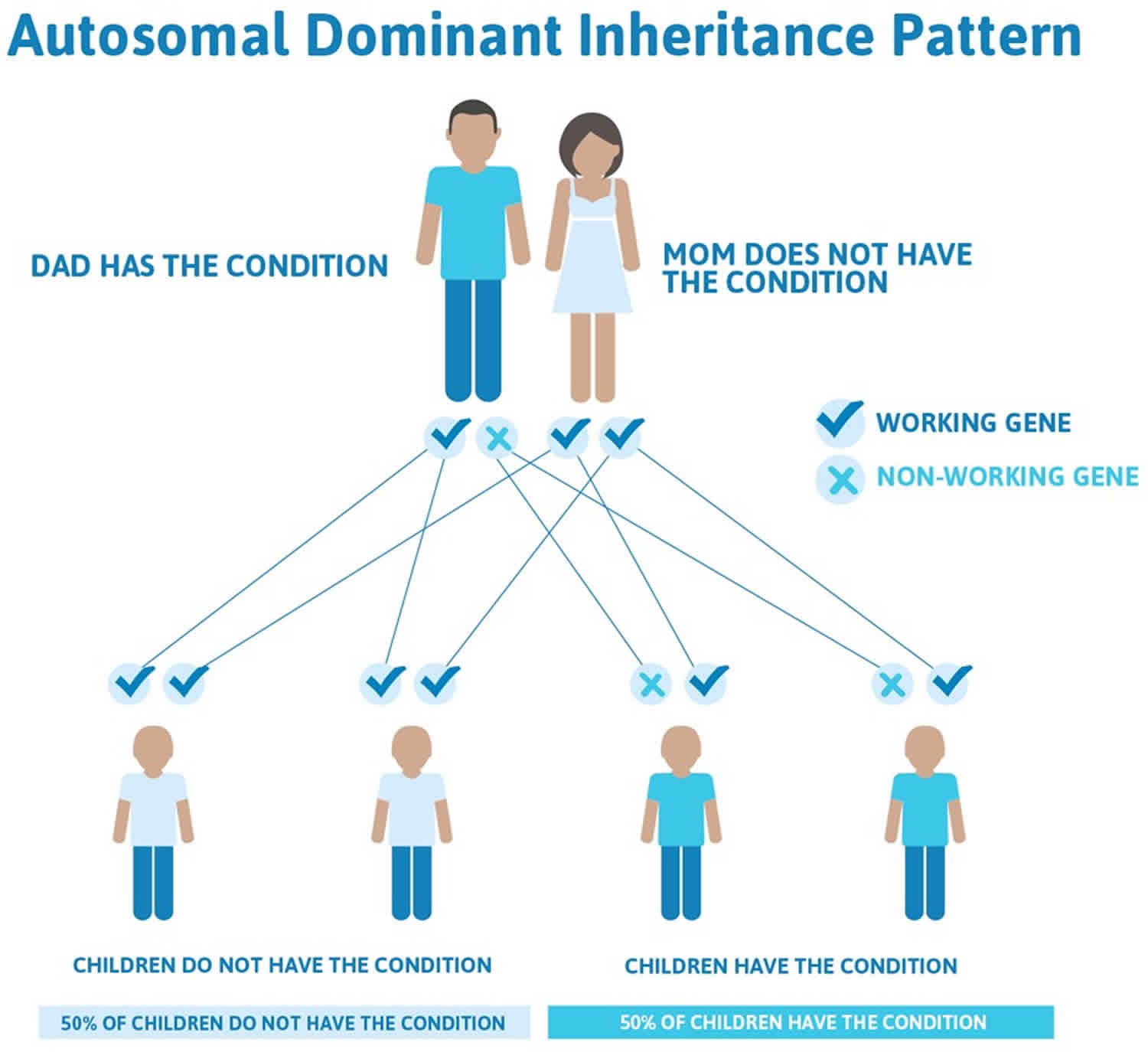
How is Crouzon syndrome inherited?
Crouzon syndrome is inherited in an autosomal dominant manner. This means that having a change (mutation) in only one copy of the responsible gene in each cell is enough to cause features of the condition.
There is nothing that either parent can do, before or during a pregnancy, to cause a child to be born with Crouzon syndrome.
In some cases, an affected person inherits the mutated gene from an affected parent. In other cases, the mutation occurs for the first time in a person with no family history of the condition. This is called a de novo mutation.
When a person with a mutation that causes an autosomal dominant condition has children, each child has a 50% (1 in 2) chance to inherit that mutation.
Figure 4. Crouzon syndrome autosomal dominant inheritance pattern
Is there a way to prevent having a child with Crouzon syndrome?
With advanced planning and appropriate testing, it may be possible to prevent having a child with Crouzon syndrome.
During a pregnancy:
If the genetic change (mutation) in an affected family member has been identified, prenatal genetic testing may be possible during pregnancy. Genetic testing may be performed on a sample obtained by chorionic villus sampling (at about 10 to 12 weeks gestation), or by amniocentesis (usually performed at about 15 to 18 weeks gestation). If the condition is confirmed in the fetus by either method, planning for an affected child and/or pregnancy management options, may be discussed with a health care provider.
Before a pregnancy:
As an alternative to prenatal diagnosis during the pregnancy, preimplantation genetic diagnosis before a pregnancy may be an option if the mutations in the family are known. Preimplantation genetic diagnosis is done after in vitro fertilization (IVF) to diagnose a genetic condition in an embryo before it is introduced into the uterus. When having preimplantation genetic diagnosis, only embryos known to be unaffected are introduced into the uterus for a possible pregnancy.
People interested in genetic testing, prenatal diagnosis, and/or preimplantation genetic diagnosis should speak with a genetic counselor or other genetics professional regarding their testing options. A genetics professional can help by:
- thoroughly evaluating the family history
- addressing questions and concerns
- assessing recurrence risks
- facilitating genetic testing if desired
- discussing reproductive options
Can an unaffected sibling of someone with Crouzon syndrome have an affected child?
Almost all people with a mutation known to cause Crouzon syndrome have features of the condition. However, in at least one family, it appeared that the responsible mutation did not affect every family member who had it 4. For this reason, Crouzon syndrome has been described by some as having variable penetrance and expressivity, even within families 5. When a condition has variable (or reduced) penetrance, it means that not every person with a mutation in the responsible gene will have apparent signs and symptoms of the condition. When a condition has variable expressivity, it means that not all people who do have features will be affected the same way – there is a range of possible features and severity. While extremely uncommon, it may be possible for family members with a mutation to “appear” unaffected, with very mild or unnoticeable features 4. Therefore, whether an unaffected sibling is at risk to have an affected child may depend on whether the sibling has a mutation in the responsible gene.
If an apparently unaffected sibling is confirmed to not have the mutation that is present in an affected family member, he/she is not considered at risk to have an affected child. In other words, a person cannot pass on a mutation that he/she does not have. Based on most reports in the literature, people without features of Crouzon syndrome typically do not carry a mutation in the responsible gene.
If an apparently unaffected sibling does have the mutation that is present in an affected family member, he/she has a 50% (1 in 2) chance to pass the mutation on to each child. In this case, even though the sibling does not have apparent features of the condition, it would not be possible to predict whether a child that inherits the mutation will be affected, or how severely he/she may be affected.
If the specific mutation causing Crouzon syndrome has been identified in an affected family member, other family members can have genetic testing to determine whether they carry the mutation and are at risk for passing the mutation on to their children.
People with personal questions about genetic risks and genetic testing are encouraged to speak with a genetic counselor or other genetics professional. A genetics professional can help by:
- thoroughly evaluating the family history
- addressing questions and concerns
- assessing recurrence risks
- facilitating genetic testing if desired
- discussing reproductive options
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counsellors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
- The National Cancer Institute provides a Cancer Genetics Services Directory (https://www.cancer.gov/about-cancer/causes-prevention/genetics/directory), which lists professionals who provide services related to cancer genetics. You can search by type of cancer or syndrome, location, and/or provider name.
If you have a health condition that has not been diagnosed, you may be interested in the Undiagnosed Diseases Network (https://undiagnosed.hms.harvard.edu/). They have information about how to apply for this multicenter research study.
Crouzon syndrome causes
Mutations in the FGFR2 gene cause Crouzon syndrome. This gene provides instructions for making a protein called fibroblast growth factor receptor 2. Among its multiple functions, this protein signals immature cells to become bone cells during embryonic development. Mutations in the FGFR2 gene probably overstimulate signaling by the FGFR2 protein, which causes the bones of the skull to fuse prematurely.
Crouzon syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. When a person with a mutation that causes an autosomal dominant condition has children, each child has a 50% (1 in 2) chance to inherit that mutation.
Crouzon syndrome symptoms
Crouzon syndrome predominantly affects the appearance of the head and face. Crouzon syndrome presents many of the same associated issues as Apert syndrome, including airway compromise, sleep apnea, hydrocephalus and eye exposure issues. In patients with Crouzon syndrome, the forehead generally appears more rounded with a more normally shaped nose, and the eyes tend to be more prominent. Another key difference is that patients with Crouzon syndrome do not have associated hand or feet anomalies present in other syndromes.
Common features:
- regressed mid-face and shallow orbits (eye sockets), which may be present at birth or become more evident; seen from the side the face has a concave appearance and the shallow orbits result in prominent eyeballs (proptosis); the arrangement of the teeth (dentition) is also affected; and
- the abnormal skull shape may result in raised intracranial pressure and require surgery to protect the restricted brain.
Crouzon syndrome may involve any combination of cranial sutures, most commonly including the coronal and sagittal sutures.
Cranial and facial malformations may vary, ranging from mild to potentially severe, including among members of the same family (kindred).
For example, the degree of cranial malformation is variable and depends on the specific cranial sutures involved as well as the order and rate of progression. In most affected individuals, there is premature fusion of the sutures (i.e., coronal and sagittal sutures) between bones forming the forehead (frontal bone) and the upper sides of the skull (parietal bones). In addition, the suture between the back and the sides of the skull (i.e., lambdoidal suture) or other sutures may be involved in some people. In most individuals with Crouzon syndrome, early sutural fusion causes the head to appear unusually short and broad (brachycephaly). In other cases, the head may appear long and narrow (scaphocephaly) or triangular (trigonocephaly). Rarely, premature closure of multiple sutures (known as Kleeblattschadel type craniosynostosis) causes the skull to be abnormally divided into three lobes (cloverleaf skull deformity). In those with Crouzon syndrome, craniosynostosis typically begins during the first year of life and progresses until approximately age two to three. However, craniosynostosis may sometimes be apparent at birth or, more rarely, may not be noted during early childhood.
In most individuals, there is unusual shallowness of the orbits or the bony cavities of the skull that accommodate the eyeballs. As a result, the eyeballs appear to protrude or bulge forward (proptosis). Due to such abnormalities, affected individuals are unusually susceptible to developing inflammation of the front, transparent regions of the eyes (i.e., exposure keratitis) as well as the membranes that line the inner surfaces of the eyelids and cover the whites of the eyes (exposure conjunctivitis). Crouzon syndrome is also often associated with additional eye abnormalities including eyes that are spaced apart wider than usual (hypertelorism) and eyes that are crossed or do not point in the same direction (strabismus). Sometimes, the various eye abnormalities can lead to a loss in vision.
Crouzon syndrome is associated with additional craniofacial abnormalities. Affected individuals often have a prominent forehead (frontal bossing); a curved nose; unusually flat or underdeveloped mid-facial regions (midface hypoplasia); and a short upper lip. In addition, a small, underdeveloped upper jaw (hypoplastic maxilla) with protrusion of the lower jaw (relative mandibular prognathism) may also occur. Clefting of the lip and/or palate (incomplete closure of the palate or an abnormal groove in the upper lip) can occur rarely. Typical dental problems include a highly arched narrow palate with crowded teeth, and upper and lower teeth that don’t meet when biting (malocclusion).
Approximately 30% of individuals with Crouzon syndrome develop hydrocephalus, a condition which is characterized by impaired flow or absorption of the fluid (i.e., cerebrospinal fluid [CSF]) that circulates through cavities (ventricles) of the brain and the spinal canal, potentially leading to increasing fluid pressure within the skull (intracranial pressure) and the brain and other associated findings.
Some affected individuals have hearing impairment due to an inability to transmit sound impulses to the brain (sensorineural hearing loss). In some infants, breathing problems may occur in infancy due to various abnormalities of the face and upper airway. In severe instances, this can lead to life-threatening breathing complications.
Crouzon syndrome diagnosis
Crouzon syndrome is usually diagnosed at birth or during infancy based upon a thorough clinical evaluation, identification of characteristic physical findings, and a variety of specialized tests. Such testing may include advanced imaging techniques, such as computerized tomography (CT) scanning or magnetic resonance imaging (MRI), or other imaging studies.
Clinical Testing and Workup
- CT scanning and MRIs are used to help detect or characterize certain abnormalities that may be associated with the disorder (e.g., craniosynostosis, other skeletal abnormalities, etc.). During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of internal structures. During MRI, a magnetic field and radio waves create detailed cross-sectional images of certain organs and tissues.
- Molecular genetic testing can confirm a diagnosis of Crouzon syndrome in some people. Molecular genetic testing can detect mutations in the FGFR2 gene known to cause the disorder, but is available only as a diagnostic service at specialized laboratories.
After a general craniofacial examination, a treatment plan is established and other specialists begin certain examinations. Photographs are also made. Basic exams include the following:
- Dental impressions.
- X-rays including a panorex for the lower jaw position, cephalograms to assess the relationship of the upper and lower jaws, CT (computed tomography) scan to assess skull growth, orbital size and jaw relationships. These scans can be converted into vivid, three-dimensional images of the skull and facial bones.
- Hearing tests when possible since patients with Crouzon syndrome tend to have ear problems.
- An eye examination.
It is important for a geneticist to meet with the family to discuss whether the condition runs in the family. It is important for the child to be evaluated by a craniofacial team to provide support and treatment for patients and their families.
Crouzon syndrome treatment
The treatment of Crouzon syndrome is dependent upon both functional and appearance-related needs, and should be addressed immediately after your child is born. Because of the complex issues that can be associated with Crouzon syndrome, your child should be treated at a medical center where he/she will have access to pediatric specialists across the many clinical areas he/she may need.
Surgery is the main form of therapy for affected children, but not all children will require surgery. Surgery is performed to create and ensure that there is enough room within the skull for the developing brain to grow; to relieve intracranial pressure (if present); and to improve the appearance of an affected child’s head.
Affected children should be seen at craniofacial clinics, which are often affiliated with major pediatric hospitals or medical centers. These clinics have a team of physicians and other healthcare providers who are experienced in treating craniofacial disorders. A team of specialists will work together to plan and carry out a child’s treatments. Such specialists include pediatricians, neurosurgeons, plastic surgeons, otolaryngologists, audiologists, ophthalmologists, dental specialists, social workers, and other healthcare professionals. Genetic counseling may be of benefit for affected individuals and their families. Psychosocial support for the entire family is essential as well.
As your child grows, he/she should also have access to psychosocial support services to address any mental, social or psychological issues that accompany these conditions.
Immediately after birth, your child should be assessed for the following:
- Respiratory function. If there is a breathing difficulty that continues after initial support in the intensive care unit then it may be required tracheostomy.
- Eyelids closure capacity. If the newborn cannot close his/her eyelids because of exophthalmos (eyes bulging out of their sockets), it may need to perform a temporary tarsorrhaphy (where stitches are carefully placed at the corners of the eyelid opening (palpebral fissure) to narrow it) so as to protect the cornea up to solve the problem permanently.
- Intracranial pressure (ICP). Rarely need to take immediate measures to confront it. The cases in which is required to perform an emergency surgery are extremely serious and nearly incompatible with life.
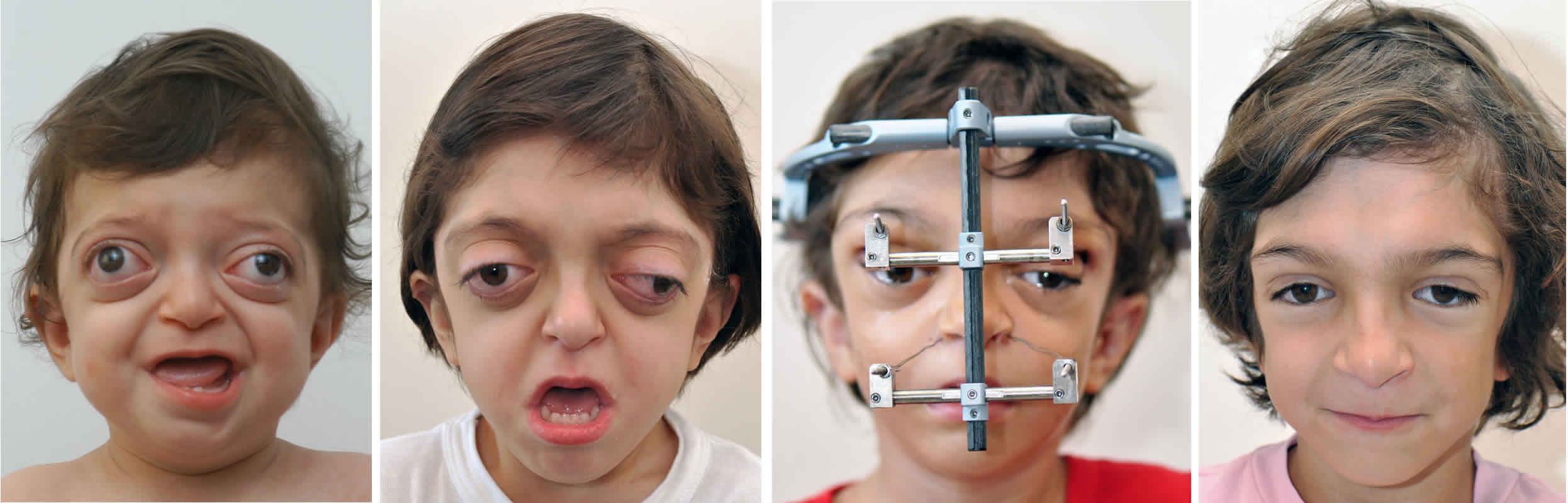
Crouzon syndrome surgery
Every patient with Crouzon syndrome has unique problems, the timing and course of surgical treatment is highly individualized. It is important to see a surgeon with expertise in pediatric plastic and reconstructive surgery who specializes in treating these rare conditions.
Some children will need ear drainage tubes inserted. In addition, if there is incomplete closure of lids causing the eyes to be severely exposed, a surgical closure may be performed by an ophthalmic surgeon. However, this is rare.
Table 1. Crouzon syndrome surgical options
| Operation | Age | Indication |
| Cranioplasty | Infancy | Skull expansion and remodelling, for cosmetic benefit and to relieve |
| Shunt surgery | Childhood | Neurosurgical operation to reduce intracranial pressure |
| Facial advancement | Childhood & Adolescence | To protect the eyes, protect against breathing difficulty, and provide cosmetic benefit. Often preceded and followed by a choanal dilation, grommet insertion and bone-anchored hearing aid |
| Choanal dilation, Grommet insertion and Bone-anchored hearing aid | Childhood | ENT procedures to improve the airway and treat chronic ear infection and hearing |
| Squint surgery and Tarsorrhaphy | Childhood | To correct ocular squint and improve vision. Tarsorrhaphy may be used to protect against exposure damage to surface of eye. |
If CT scans show the skull is not growing fast enough for brain expansion and if the soft spot is closing, the head must be enlarged. The neurosurgeon and the craniofacial surgeon perform this operation. Because almost always there is a limitation of the cranial capacity and the brain undergoes some pressure, this pressure should be alleviated in infancy with expansion (enlargement) of the cranial cavity. It is done under general anesthesia. An incision is made in the scalp in a zigzag fashion from behind one ear across the head to the other ear. This method allows the cut to be hidden in the hair.
During the surgery, the neurosurgeon removes the front part of the skull. The craniofacial surgeon removes the upper part of the orbits. Then the brain is able to expand. The two portions of the bone that are removed are then joined together with small plates. The plates will dissolve later.
The bony complex is put back in place but is advanced to increase the skull size to allow brain expansion. It is fixed in the new position again with plates which will dissolve. In some patients the front of the skull is of a strange shape and other cuts may be necessary to make this more normal. Then the scalp is closed and tubes are left in to drain the blood. If necessary, a bulky bandage is placed on the head and the child goes to the Intensive Care Unit (ICU). In most cases, normal nursing care is sufficient after a day or two, and the child will leave the hospital in three to four days. Frequently, no further skull surgery is required. If there are no indications for early correction, then the skull surgery can be done in combination with upper jaw surgery.
Usually, the initial surgery is performed on the back of the cranial area i.e. to the parietal-occipital region (Occipitoparietal Decompression). A few months later (if necessary), it follows a decompression in the frontal part of the skull (Frontal Orbital Advancement). In this surgery, it is formed the orbital area and the exophthalmos is somewhat reduced. Finally, at age of 10-12 years it is achieved a complete and permanent correction of malformation and functional problems moving the frontal part of the skull and face forward (Frontal Facial Advancement). This surgery can be performed at an earlier age, if necessary due to the functional problems that require immediate solution (obstructive apnea, increased intracranial pressure that causes vision impairment).
Although, all these problems seem to be overwhelming and it is very natural to be a source of significant anxiety for parents, however, with regular visits in a well-organized Craniofacial Center, their treatment is accomplished properly and with the minimal discomfort for the child and his/her family.
Where is the best place to have my child treated?
Crouzon syndrome is a complex condition. It requires the expert skill of several different specialists working together. Craniofacial teams experienced in the management of these patients best treat these. Centers with craniofacial teams working together have the advantage of greater experience. This definitely leads to better results and fewer complications. In addition, ongoing research at these centers offers patients the latest break-throughs in treatment. As there are only a few experienced centers in the country, it is quite common for families to travel quite some distance to get the best care. Children who are treated locally by inexperienced teams or by individual physicians not working together as a team, are more likely to have unsatisfactory results. It sometimes requires two or three additional operations to correct what has been done. Another advantage of traveling to busy centers is the opportunity to meet other families and children affected with similar problems who can offer advice. These families often share their experiences, which provides moral support.
References- Marla J. F. O’Neill. Crouzon Syndrome. http://www.omim.org/entry/123500
- Crouzon syndrome. https://ghr.nlm.nih.gov/condition/crouzon-syndrome
- Crouzon Syndrome. https://www.chop.edu/conditions-diseases/crouzon-syndrome
- Everett ET, Britto DA, Ward RE, Hartsfield JK Jr. A novel FGFR2 gene mutation in Crouzon syndrome associated with apparent nonpenetrance. Cleft Palate Craniofac J. November, 1999; 36(6):533-541.
- Helman SN, Badhey A, Kadakia S, Myers E. Revisiting Crouzon syndrome: reviewing the background and management of a multifaceted disease. Oral Maxillofac Surg. December, 2014; 18(4):373-379.





{kind=link}