
Cystinosis
Cystinosis also called cystine storage disease, is a rare multisystem genetic lysosomal storage disorder characterized by accumulation of the amino acid cystine (a building block of proteins) within cells. Excess cystine damages cells and often forms crystals that can build up and cause problems in many organs and tissues. The kidneys and eyes are especially vulnerable to damage; the muscles, thyroid, liver, pancreas, brain and testes may also be affected. Cystinosis is the most common cause of renal Fanconi syndrome in children and accounts for approximately 5 percent of all childhood cases of kidney failure.
There are three distinct types of cystinosis. In order of decreasing severity, they are:
- Nephropathic cystinosis,
- Intermediate cystinosis (also known as nephropathic juvenile cystinosis or adolescent cystinosis)
- Non-nephropathic or ocular cystinosis.
The age of onset, symptoms, and severity of cystinosis can vary greatly from one person to another. Nephropathic cystinosis presents in infancy and is the most common and severe form. Early detection and prompt treatment are critical in slowing the development and progression of symptoms associated with cystinosis. The kidneys and eyes are the two organs most often affected. Individuals with nephropathic or intermediate cystinosis ultimately require a kidney transplant. Ocular cystinosis only affects the corneas of the eyes.
Cystinosis affects approximately 1 in 100,000 to 200,000 newborns worldwide. Cystinosis affects males and females in equal numbers. The incidence is higher in the province of Brittany, France, where the disorder affects 1 in 26,000 individuals 1.
Cystinosis is caused by mutations of the CTNS gene and is inherited as an autosomal recessive disease. Cystinosis is classified as a lysosomal storage disorder. Lysosomes are membrane bound compartments within cells that break down certain nutrients such as fats, proteins and carbohydrates. Lysosomes are the primary digestive unit within cells. Some enzymes within lysosomes break down (metabolize) these nutrients, while other enzymes transport the leftover metabolic products (such as cystine) out of the lysosome. In the case of cystinosis, the lack of such a specific transporter causes cystine to accumulate in lysosomes of cells throughout the body. Cystine forms crystals (crystallizes) in many types of cells and slowly damages affected organs.
Nephropathic cystinosis
Nephropathic cystinosis is an inherited (autosomal recessive) lysosomal storage disorder caused by defective transport of the amino acid cystine out of lysosomes. The stored cystine is poorly soluble and crystallizes within the lysosomes of many cell types, leading to widespread tissue and organ damage.
Nephropathic cystinosis is the most common and severe form begins in infancy, causing poor growth and a particular type of kidney damage (renal Fanconi syndrome) in which certain molecules that should be reabsorbed into the bloodstream are instead eliminated in the urine. The kidney problems lead to the loss of important minerals, salts, fluids, and many other nutrients. The loss of nutrients impairs growth and may result in soft, bowed bones (hypophosphatemic rickets), especially in the legs. The nutrient imbalances in the body lead to increased urination, thirst, dehydration, and abnormally acidic blood (acidosis). By about the age of 2, cystine crystals may be present in the clear covering of the eye (cornea). The buildup of these crystals in the eye causes pain and an increased sensitivity to light (photophobia). Untreated children will experience complete kidney failure by about the age of 10. Other signs and symptoms that may occur in untreated people, especially after adolescence, include muscle deterioration, blindness, inability to swallow, diabetes, thyroid and nervous system problems, and an inability to father children (infertility) in affected men.
At one time, nephropathic cystinosis was fatal at a very young age. However, the development of a medication known as cysteamine (which lowers the levels of cystine in the body) and improvements in kidney transplants have transformed cystinosis from a fatal kidney disorder to a chronic, multisystem disorder with a life expectancy well into adulthood and even beyond 50 years of age.
Intermediate cystinosis
The signs and symptoms of intermediate cystinosis are the same as nephropathic cystinosis, but they occur at a later age. Intermediate cystinosis typically becomes apparent in affected individuals in adolescence. Malfunctioning kidneys and corneal crystals are the main initial features of this disorder. If intermediate cystinosis is left untreated, complete kidney failure will occur, but usually not until the late teens to mid-twenties.
Ocular cystinosis
People with non-nephropathic or ocular cystinosis typically experience photophobia due to cystine crystals in the cornea, but usually do not develop kidney malfunction or most of the other signs and symptoms of cystinosis. Due to the absence of severe symptoms, the age at which this form of cystinosis is diagnosed varies widely.
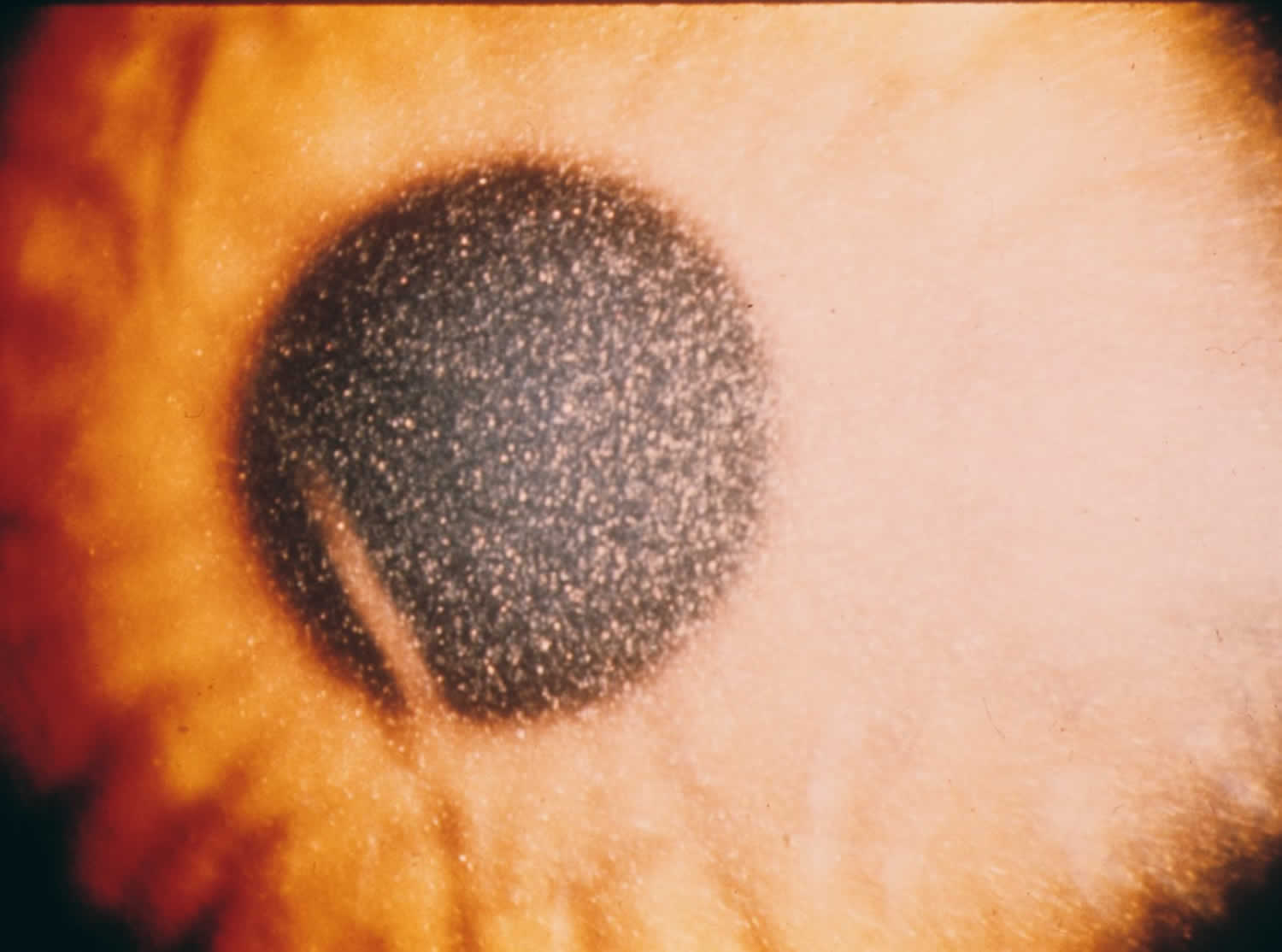
Figure 1. Ocular cystinosis
Cystinosis causes
All three types of cystinosis are caused by mutations in the CTNS gene. Mutations in CTNS gene lead to a deficiency of a transporter protein called cystinosin. Within cells, cystinosin normally moves cystine out of the lysosomes, which are compartments in the cell that digest and recycle materials. When cystinosin is defective or missing, cystine accumulates and forms crystals in the lysosomes. The buildup of cystine damages cells in the kidneys and eyes and may also affect other organs.
Investigators have determined that the CTNS gene is located on the short arm (p) of chromosome 17 (17p13). Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 17p13” refers to band 13 on the short arm of chromosome 17. The numbered bands specify the approximate location of the thousands of genes that are present on each chromosome.
Cystinosis inheritance pattern
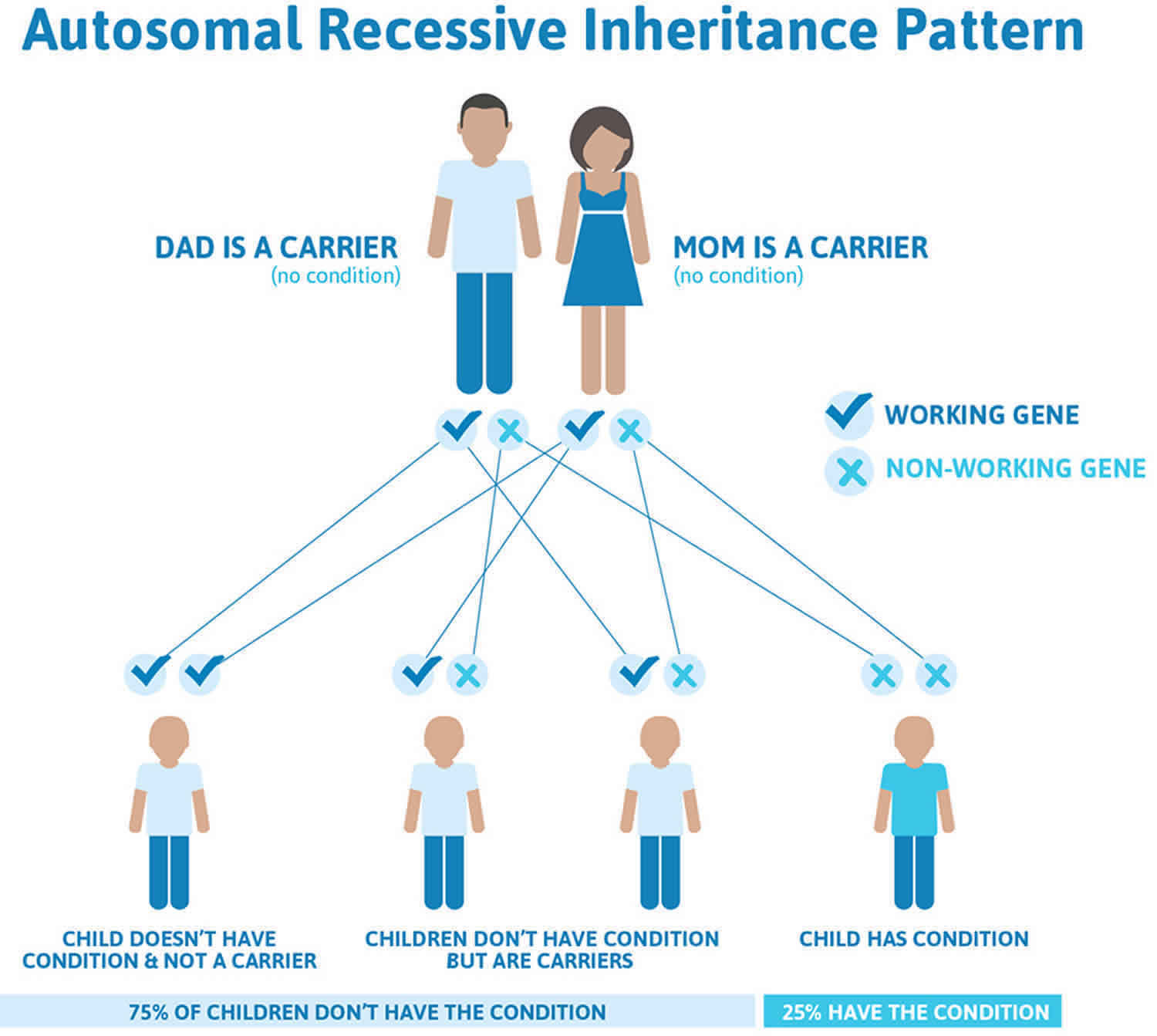
Cystinosis is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Cystinosis autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Cystinosis symptoms
The specific symptoms and severity of cystinosis vary greatly from one person to another based upon several factors including age of onset and whether the disorder is promptly diagnosed and treated. The progression of the disorder can be slowed by early diagnosis and treatment. Eventually, cystinosis can affect all tissues of the body. The age of onset for different symptoms varies greatly.
It is important to note that affected individuals may not have all of the symptoms discussed below. Affected individuals and parents of affected children should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
Nephropathic cystinosis symptoms
Nephropathic, or infantile cystinosis, is the most frequent and most severe form of cystinosis. The symptoms of nephropathic cystinosis usually become apparent within the second half of the first year of life. Specific symptoms can be mild or severe based upon each individual case and the age when treatment is started.
Growth failure and renal Fanconi syndrome are usually the first noticeable complications of the disorder. Although infants appear normal at birth, by the age of one they often fall into the third percentile for height and weight. In addition, affected infants may have episodes of vomiting, poor appetite, and feeding difficulties that contribute (along with kidney dysfunction) to nutritional deficiency and failure to gain weight and grow at the expected rate (failure to thrive). On average, growth in untreated children with cystinosis occurs at 60 percent the expected rate.
Infants with nephropathic cystinosis develop renal Fanconi syndrome, a rare disorder characterized by kidney dysfunction. The kidneys are two bean-shaped organs located just under the ribcage. The kidneys have several functions, including filtering and excreting waste products from the blood and body, creating certain hormones and helping maintain the balance of certain chemicals in the body such as potassium, sodium, chloride, calcium, magnesium, and other minerals and electrolytes. In nephropathic cystinosis, the kidney tubules fail to reabsorb a variety of needed substances, including the compounds mentioned above as well as amino acids, phosphate, calcium, glucose, carnitine, certain proteins and electrolytes. Consequently, affected individuals have abnormally low levels of many of these substances in the body.
Symptoms of renal Fanconi syndrome usually become apparent between 6 and 12 months of age and may include excessive thirst (polydipsia), excessive production and passage of urine (polyuria), electrolyte imbalances, vomiting, and dehydration with or without fever. Dehydration can be severe in some affected individuals.
The renal Fanconi syndrome can also cause hypophosphatemic rickets. In this disorder, because the kidneys are unable to reabsorb phosphorous from the urine, the body leaches phosphate from the bones to maintain blood levels of phosphorous; this results in progressive softening and weakening of the bone (rickets). Rickets can cause bone deformity and can delay walking, because it is painful. Affected children may walk gingerly. Kidney dysfunction can also cause excessive amounts of calcium to be lost from the body through the urine (hypercalciuric hypocalcemia). Low levels of calcium can cause intermittent muscle spasms (tetany) and, rarely in cystinosis, seizures.
If left untreated, the filtering function of the kidney will continue to deteriorate, eventually progressing to kidney failure at 10 years of age. Treatment with medications that lower cystine levels can slow or stop the progression of kidney disease and delay the need for a kidney transplant into the teen-age years, the 20s or later. Any existing kidney damage that occurs before diagnosis (and therefore before treatment) is irreversible.
Extrarenal symptoms
Children with nephropathic cystinosis may also develop symptoms unrelated to the kidneys (extrarenal symptoms). Again, these findings are highly variable and an affected child will not develop all of the symptoms discussed below. Specific extrarenal symptoms vary greatly depending upon the age that treatment is begun and the specific organs that become involved; those organs can include the eyes, bone marrow, liver, pancreas, spleen, intestine, brain, thyroid, muscles and testes.
At any age, children may develop an abnormal sensitivity to light (photophobia) and irritation due to the formation of cystine crystals in the cornea. The severity of photophobia can vary. In some untreated individuals, pain and recurrent corneal erosions may develop.
Around the age of 10, affected children may also develop deficiency of thyroid hormone production (hypothyroidism) due to cystine crystals accumulating in the thyroid. The thyroid is a butterfly-shaped gland located at the base of the neck. The thyroid secretes hormones into the bloodstream that influence certain activities of the body such as growth, maturation and the rate of metabolism. Symptoms of hypothyroidism are highly variable, but may include fatigue, feeling cold, dry skin, constipation and depression.
As a group, children with nephropathic cystinosis do not produce normal amounts of tears, sweat or saliva. Tear production may be diminished causing the eyes to dry out. An impaired ability to sweat can potentially cause total exhaustion or collapse due to heat (heat prostration).
Puberty may be delayed by one or two years. Untreated males experience hypogonadism, in which the testes produce reduced amounts of testosterone. Testosterone plays a key role in growth and the development of male secondary sexual characteristics during puberty.
Intelligence is usually normal, although many children experience learning disabilities. Some children may have problems with processing visual information, short-term visual memory, difficulties identifying common objects by touch (tactile recognition) and an inability to visually recognize the spatial relationship among objects (poor visuospatial skills). An example of visuospatial skills is distance and depth perception. Issues with motor speed and sustained attention have also been reported. Some affected children display behavioral and psychosocial issues, which are common in children dealing with chronic illnesses. IQ levels, while in the normal range, may be lower than would be expected based upon the IQ levels of parents and siblings.
Children with nephropathic cystinosis may have mildly altered facial features (craniofacial dysmorphology). Delayed dental development and delayed eruption of permanent teeth can also occur. Some affected individuals may develop increased pressure of cerebrospinal fluid within the brain (intracranial hypertension), which can cause headaches and swelling of the optic disc (papilledema).
Late-onset abnormalities
The increased longevity of individuals with nephropathic cystinosis has revealed that additional complications affecting organs other than the kidneys can occur later during life. These complications develop due to the chronic accumulation of cystine crystals in individuals who have not been adequately treated by cysteamine, although they have undergone a kidney transplant. These additional complications generally develop between 20 and 40 years of age.
Accumulation of cystine in muscle tissue can cause muscle disease (myopathy) leading to progressive weakness and wasting of affected muscles. Impairment of muscles in the throat can lead to swallowing and feeding difficulties. Involvement of chest muscles can result in pulmonary insufficiency.
A wide variety of gastrointestinal symptoms can develop including enlargement of the liver (hepatomegaly), high blood pressure of the main vein of the liver (portal hypertension), enlargement of the spleen (splenomegaly), gastroesophageal reflux, ulcers, inflammation of the esophagus (esophagitis), and dysfunction of the muscles of the gastrointestinal tract (dysmotility). Unusual additional symptoms include inflammatory bowel disease, tearing of the bowel causing the contents of the intestines to flow into the abdominal cavity (bowel perforation), and inflammation of the peritoneum (peritonitis), which is the membrane that lines the abdominal wall and organs.
High blood pressure (hypertension), coronary artery atherosclerosis, and blood clotting abnormalities are complications of the renal disease associated with cystinosis.
Additional findings include metabolic bone disease and an inability to properly digest food due to a lack of digestive enzymes normally produced by the pancreas (pancreatic exocrine insufficiency). Adults with cystinosis may also develop eye abnormalities including spasms of the eyelids (blepharospasm), band keratopathy and pigmentary retinopathy. Band keratopathy refers to the accumulation of calcium deposits in a band across the central surface of the cornea, which can cause pain and decreased clarity of vision (visual acuity). Pigmentary retinopathy is characterized by progressive degeneration of the retina, the thin layer of nerve cells that line the inner surface of the back of the eyes. The retina senses light and converts it to nerve signals, which are then relayed to the brain through the optic nerve. Pigmentary retinopathy can impair night and color vision and, eventually, can contribute to overall reduced clarity of vision.
Although uncommon, brain dysfunction occurs in some older adults with cystinosis. The exact reason this occurs is unknown. Specific symptoms will vary, but some affected individuals may experience a decline in both motor and mental capabilities. In very rare cases, neurological dysfunction can progress to dementia.
Individuals with nephropathic cystinosis appear to have a higher rate of diabetes than the general population because of destruction of the pancreas by cystine accumulation.
Intermediate cystinosis symptoms
Also known as nephropathic juvenile cystinosis or adolescent cystinosis, this form of cystinosis is characterized by all of the signs and symptoms of nephropathic cystinosis described above. However, onset of these symptoms does not occur until later, perhaps at 8-20 years of age. Generally, the symptoms are less severe than in the classical infantile nephropathic form and have a slower progression. If untreated, end-stage renal failure in intermediate cystinosis usually develops at some point between 15 and 25 years of age. There is a spectrum of disease severity in cystinosis, with overlap of the infantile and intermediate forms.
Ocular cystinosis symptoms
Also known as ocular or “benign” cystinosis, this form usually affects adults during middle age; it was once called adult cystinosis. Kidney disease does not occur in these individuals. The disorder appears to affect only the eyes. Untreated individuals with non-nephropathic cystinosis eventually develop photophobia due to cystine crystal accumulation in the eyes.
Cystinosis diagnosis
A diagnosis of cystinosis is based upon identification of characteristic symptoms (e.g., symptoms of renal Fanconi syndrome), a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. A prompt diagnosis of cystinosis is critical to maximize the preventive and therapeutic benefits of cystine depleting medications.
Clinical testing and work-up
A diagnosis of cystinosis can be confirmed by measuring cystine levels in certain white blood cells (“polymorphonuclear leukocytes”).
Urinary examination may reveal excess loss of nutrients including minerals, electrolytes, amino acids, carnitine and water, which is indicative of renal Fanconi syndrome.
A physician may use a special microscope called a slit lamp to view the eyes through high magnification, which can reveal cystine crystals in the cornea. This is diagnostic if performed by an experienced ophthalmologist.
A diagnosis of cystinosis can be confirmed by molecular genetic testing, which can identify the characteristic CTNS gene mutation that causes the disorder. Molecular genetic testing is available through a commercial laboratory.
Prenatal diagnosis is available for families with a known risk for having a baby with cystinosis. Cystine levels can be measured in cells obtained from the fluid that surrounds the developing fetus (amniotic fluid). A test known as chorionic villus sampling is performed at 8-9 weeks of gestation, can also be used to obtain a prenatal diagnosis of cystinosis. Chorionic villi are thin, hair-like structures found on the placenta. These cells can be examined to detect elevated levels of cystine. Another prenatal diagnosis is amniocentesis which can be performed at 14-16 weeks of gestation.
Cystinosis treatment
The treatment of cystinosis is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, kidney specialists (nephrologists), eye specialists (ophthalmologists), digestive disorder specialists (gastroenterologists), psychologists and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment.
Nephropathic and intermediate cystinosis were once progressively fatal disorders, with a lifespan for the infantile form of less than 10 years. However, the development of cystine depleting therapies along with improvements in kidney transplantation have extended the lifespan well into adulthood.
Cystine depleting therapy
In 1994, the U.S. Food and Drug Administration (FDA) approved cysteamine bitartrate (Cystagon®) for the treatment of individuals with cystinosis. In 2013, the FDA approved Procysbi®, an extended release form of cysteamine. Cysteamine is a cystine-depleting agent that can greatly lower cystine levels within cells. Therapy with cysteamine slows the development and progression of kidney damage and enhances growth in children. Cysteamine can significantly delay the need for a kidney transplant. Some individuals who underwent early, diligent treatment with cysteamine were able to delay a kidney transplant into their 20s or longer.
Therapy with cysteamine should be begun immediately after diagnosis to prevent or slow kidney damage. Cysteamine should be continued throughout life because studies have shown that long-standing treatment with cysteamine and can prevent many of the non-renal, late onset complications of cystinosis.
Cysteamine is taken orally, but oral cysteamine does not reach the cornea effectively and, therefore, fails to eliminate cystine crystals from the cornea. Cystaran® (cysteamine ophthalmic solution 0.44%) was approved by the FDA in 2012 as a treatment for corneal cystine crystal accumulation associated with cystinosis. This eye-drop solution containing cysteamine is the only FDA approved treatment for the eye complications of this disorder.
Therapy with cysteamine can cause nausea, vomiting and gastrointestinal discomfort. Cysteamine also causes excess secretion of stomach acids. Some individuals taking cysteamine may need to take proton pump inhibitors such as omeprazole to reduce the production of stomach acid, helping to improve gastrointestinal symptoms.
Cysteamine smells and tastes foul, which sometimes leads to compliance issues with affected individuals.
Symptomatic therapy
Renal Fanconi syndrome is treated with a high intake of fluids and electrolytes to prevent excessive reduction of body water (dehydration). Sodium bicarbonate, sodium citrate, magnesium and potassium may be administered to help to maintain normal electrolyte balance. Acetylcholinesterase (ACE) inhibitors are sometimes used with the hope of slowing the progression of the renal disease.
Indomethacin is an anti-inflammatory medication that is sometimes used to reduce urinary losses of water and electrolytes; sometimes it also helps to improve growth rate. If affected individuals take indomethacin, they must be closely monitored with respect to their renal function.
Phosphates and vitamin D are often given to correct impaired reabsorption of phosphate into the blood and prevent rickets. Carnitine may be prescribed for some pre-transplant individuals in order to improve muscle strength.
Proper nutrition is vitally important to ensure that affected infants and children maximize their growth potential. Growth hormone therapy has significantly improved growth in many patients. L-thyroxine is used to treat hypothyroidism, insulin is used to treat insulin-dependent diabetes and testosterone is used to treat males with underactive testicular function (hypogonadism) so that secondary sexual characteristics will develop. Infertility will not respond to testosterone treatment.
Ocular symptoms of cystinosis can be treated with avoidance of bright light, sunglasses and lubrication. In extremely rare cases, a corneal transplant may be necessary. This is usually required only for individuals with large band keratopathies or those suffering from pain due to repeated corneal erosions.
Some infants and children with cystinosis (e.g., those with dysphagia, poor nutrition and increased risk of aspiration) may require the implantation of a gastronomy tube. With this procedure, a thin tube is placed into the stomach via a small incision in the abdomen, allowing for the direct intake of food and/or medicine.
Speech and language therapy may be beneficial in some cases. Genetic counseling may be of benefit for affected individuals and their families.
Growth hormone therapy
Treatment with recombinant human growth hormone improves growth velocity. Long-term recombinant human growth hormone treatment in young children with nephropathic cystinosis prior to renal replacement therapy is safe and efficient.
Growth hormone treatment is less effective for peripubertal or adolescent patients on renal replacement therapy.
Treatment with recombinant human growth hormone does not accelerate a decline in kidney function in children with chronic kidney disease.
Renal transplantation
Despite early and prompt treatment, individuals with infantile and intermediate cystinosis eventually develop end stage renal disease (ESRD), requiring a kidney transplant. Initially, an affected individual may undergo dialysis. Dialysis is a procedure in which a machine is used to perform some of the functions of the kidney – filtering waste products from the bloodstream and helping to maintain proper levels of essential chemicals such as potassium. End stage renal disease is not reversible so individuals will eventually require a kidney transplant. The rate of progression of kidney dysfunction to end stage renal disease can vary greatly from one individual to another. Individuals with cystinosis generally respond very well to a kidney transplant, which can cure renal Fanconi syndrome because cystine does not accumulate in the donated kidney. However, cystine still accumulates in other tissues and organs of the body.
Cystinosis prognosis
In the early 1960s, nephropathic cystinosis was considered a fatal renal disease of childhood; patients died of progressive renal failure before age 10 years. The natural history of the disease has changed dramatically since the introduction of cysteamine and renal transplantation. Patients with infantile cystinosis now survive into even the fifth decade of life. Cysteamine markedly slows the progression of renal failure.
Cysteamine, introduced in the early 1980s, was shown to blunt the decline in renal function and improve linear growth in these children, despite the fact that it does not ameliorate the defect in renal tubule transport. However, the increased life expectancy afforded by the progress in medical and surgical treatment was accompanied by the development of serious complications due to the continuous accumulation of cystine in nonrenal organs, including the eye, thyroid, brain, liver, pancreas, and muscle. However, many patients survive into the third or fourth decade of life, some even survive into the fifth decade of life and are able to pursue fulfilling lifestyles.
The study results from a large European cohort showed improved survival of renal function and better patient and graft survival in children with nephropathic cystinosis who received renal transplantation compared with those who did not. The 5-year survival rate after the start of renal transplantation improved from 86.1% (prior to 1990) to 100% (since 2000) 2.
References




{kind=link}