Enterohepatic circulation
Enterohepatic circulation refers to the circulation of bile from the liver, where it is produced, to the small intestine, where it aids in digestion of fats and other substances, transported to the liver in portal venous blood, and then efficiently taken up by the hepatocyte (liver cells) and are actively secreted back into canalicular bile, completing the enterohepatic cycle 1. As a whole, the enterohepatic circulation makes each bile salt molecule available several times during a solitary digestive stage. The net effect of enterohepatic recirculation is that each bile salt molecule is reused about 20 times, often multiple times during a single digestive phase.
Enterohepatic circulation also means that some molecules which would not otherwise be very toxic can become extremely hepatotoxic as they reach unexpectedly high hepatic concentrations. Drugs may remain in the enterohepatic circulation for a prolonged period of time as a result of this recycling process.
Bile is a yellow-greenish fluid produced and secreted by the liver, consists mainly of bile salts, phospholipids, cholesterol, conjugated bilirubin, electrolytes, and water 2. Estimates are that the bile flow in humans averages 620 mL/day 2. The flow of bile is either dependent or independent on the osmotic force of bile acids. When the bile flow is dependent on the osmotic force of bile acids, it is the bile-acid dependent fraction of bile while if the flow is not dependent on the osmotic force of bile acids, it is bile acid-independent fraction of bile. Moreover, there is a linear realtionship between the amount of water that follows bile acids osmotically and the bile acids in the bile. Certain bile acids retain the capacity to induce a higher volume of bile (hypercholeresis). One classic example is ursodeoxycholic acid. There is a proposal that the hyperocholeresis might be due to the reabsorption of ursodeoxycholic acids from the epithelial cells that line the bile ducts into the bloodstream and then into the hepatic sinusoids. From the hepatic sinusoids, these hypercholeretic bile acids will be taken by the hepatocytes. In the hepatocytes, the hypercholeretic bile acids will be re-secreted into the bile canaliculi thereby increasing the bile-acid dependent fraction of bile flow and resulting in the so-called hypercholeresis. This pathway is called the cholehepatic shunt pathway. The bile acids in the bile-acid dependent fraction of bile are either newly synthesized in the liver or else they come from the enterohepatic circulation of bile acid 3. Of note, ursodeoxycholic acid (hypercholeretic bile acid) is used in primary biliary cholangitis a condition in which there is cholestasis and decreased bile secretion.
Bile travels through the liver in a series of ducts, eventually exiting through the common hepatic duct. Bile flows through this duct into the gallbladder where it is concentrated and stored 4. When stimulated by the hormone cholecystokinin (CCK), the gallbladder contracts, pushing bile through the cystic duct and into the common bile duct. Simultaneously, the sphincter of Oddi relaxes, permitting bile to enter the duodenal lumen. The hormone secretin also plays an important role in the flow of bile into the small intestine. By stimulating biliary and pancreatic ductular cells to secrete bicarbonate and water in response to the presence of acid in the duodenum, secretin effectively expands the volume of bile entering the duodenum. In the small intestine, bile acids facilitate lipid digestion and absorption. Only approximately 5% of these bile acids are eventually excreted. The majority of bile acids are efficiently reabsorbed from the ileum, secreted into the portal venous system, and returned to the liver in a process known as enterohepatic recirculation 5.
The initial driving force in enterohepatic circulation process is represented by bile acid export pump. In the intestine, bile acids absorption and translocation in venous blood is regulated by two other transporters: the apical sodium bile acid co-transporter (ASBT), present in the brush border membrane of distal ileum enterocytes, and a heterodimer of two proteins, termed organic solute transporter (OST) alpha and beta 6. These transporters are responsible for driving bile acids through basolateral membranes and into venous blood 7. The last step consists of bile acid uptake from the blood by the liver. This is mainly accomplished by the sodium/taurocholate co-transporting polypeptide (NTCP) located in the basolateral membrane of the human hepatocytes 8. In humans, nearly 2–3 grams of bile acids are present in the body, while the loss in feces is no more than 10% daily due to the high capacity of recovery by apical sodium bile acid co-transporter in the terminal ileum. The amount of bile acids left in stools is quickly replaced by liver neo-synthetic pathways. In normal circumstances, the enterohepatic circulation of a molecule of bile acid is thought to occur 3–4 times a day 9.
Intestinal bile conservation or salvage leads to the accumulation of a recycling mass of bile acid molecules, most of which is stored in the gallbladder during an overnight fast. As this mass was originally measured by an isotope dilution technique, it was termed a “pool” and this descriptor is still being used. Each biliary bile acid has its own pool and enterohepatic circulation. Biliary bile acid composition is determined by the pool sizes of individual bile acids.
The enterohepatic circulation has an input, which is the biosynthesis of bile acids from cholesterol. In the steady state, input is balanced by loss. So far as is known, renal excretion of bile acids is negligible compared to fecal excretion in all vertebrates. Therefore, input of bile acids occurs by biosynthesis in the hepatocyte, and the preponderant loss of bile acids occurs by fecal excretion. Because the steroid nucleus is considered to remain intact during colonic transit, fecal bile acid excretion is equivalent to hepatocyte bile acid biosynthesis.
Figure 1. Enterohepatic circulation
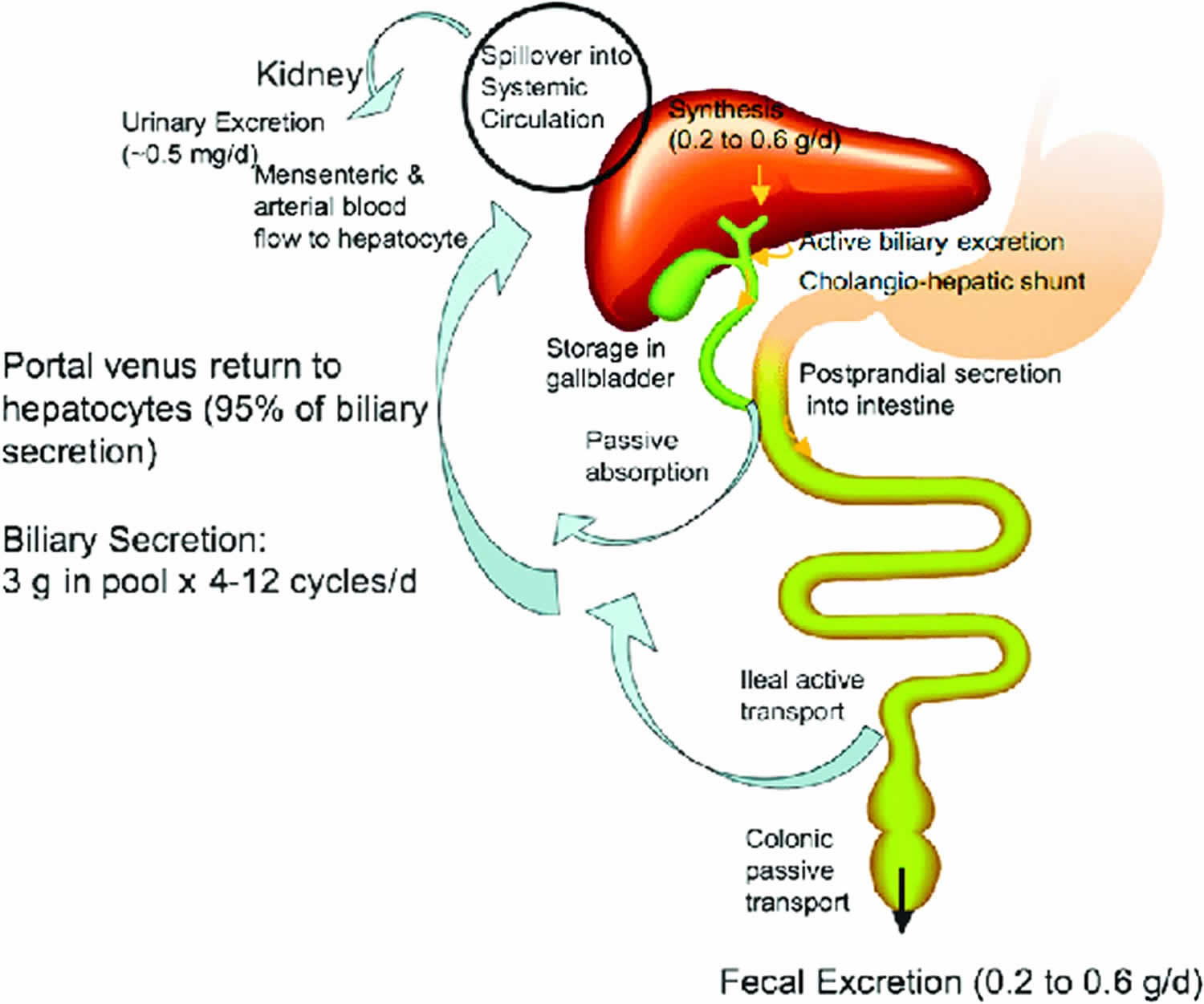
Footnote: Enterohepatic circulation of bile acids. In humans, about 0.2–0.6 g (averaging 0.5 g) bile acids are synthesized daily in human liver. Conjugated bile acids are secreted into bile and stored in the gallbladder. Some bile acids are spilled over into sinusoid blood and reabsorbed when passing through the renal tubules in the kidney and circulated back to the liver through mensenteric and arterial blood fl ow. Some bile acids secreted in the bile duct are reabsorbed in the cholangiocytes and recycled back to hepatocytes (cholangiohepatic shunt). After each meal, gallbladder contraction empties bile acids into the intestinal tract. When passing through the intestinal tract, some bile acids are reabsorbed in the upper intestine by passive diffusion, but most bile acids (95%) are reabsorbed in the ileum. Bile acids are transdiffused across the enterocyte to the basolateral membrane and excreted into portal blood circulation back to the sinusoid of hepatocytes. In the colon, deoxycholic acid is reabsorbed by passive transport and recycled with cholic acid and chenodeoxycholic acid to the liver. A bile acid pool of about 3 g is recycled 4–12 times a day. Bile acids lost in the feces (0.2–0.6 g/day) are replenished by de novo synthesis in the liver to maintain a constant bile acid pool.
[Source 10 ]Enterohepatic circulation organ systems involved
Hematologic
Bilirubin, the major pigment of bile, is an end product of heme catabolism that travels to the liver bound to albumin. Once inside the liver, the enzyme uridine diphosphate glucuronyltransferase (UDPGT) conjugates bilirubin to form bilirubin glucuronide. The water-soluble conjugated bilirubin is then secreted into bile, providing its characteristic yellow color.
Gastrointestinal and Hepatobiliary
- Liver: Site of bile formation, reuptake of bile acids and reuptake of urobilinogen
- Bile ducts: Modify and transport bile, secrete ions and water into bile
- Gallbladder: Stores and concentrates bile
- Small intestine:
- Bacteria form secondary bile acids via dehydroxylation of primary bile acids
- Bilirubin glucuronide is converted back to bilirubin
- Bacteria convert bilirubin to urobilinogen
- Duodenum: Site of lipid digestion and absorption facilitated by bile
- Ileum: Site of reabsorption of bile salts
- Portal circulation: Transports reabsorbed bile salts back to the liver
- Rectum: Urobilin and stercobilin (compounds oxidized from urobilinogen) are responsible for dark fecal pigment.
- Genitourinary: Some urobilinogen is excreted in urine.
Bile physiology
Bile formation
Bile is produced by hepatocytes, which is then modified by the cholangiocytes lining the bile ducts. The production and secretion of bile require active transport systems within hepatocytes and cholangiocytes in addition to a structurally and functionally intact biliary tree. Initially, hepatocytes produce bile by secreting conjugated bilirubin, bile salts, cholesterol, phospholipids, proteins, ions, and water into their canaliculi (thin tubules between adjacent hepatocytes that eventually join to form bile ducts). The canalicular membrane of the hepatocyte is the main bile secretory apparatus which contains the intracellular organelles, the cytoskeleton of the hepatocyte and carrier proteins. The carrier proteins in the canalicular membrane transport bile acid and ions. Transporter proteins found within the canalicular membrane use energy to secrete molecules into bile against concentration gradients. Through this active transport, osmotic and electrochemical gradients are formed. When conjugated bile salts enter the canaliculus, water follows by osmosis. The electrochemical gradient allows for the passive diffusion of inorganic ions such as sodium. The most significant promoter of bile formation is the passage of conjugated bile salts into the biliary canaliculus. The total bile flow in a day is approximately 600 ml, of which 75% is derived from the hepatocyte, and 25% is from the cholangiocytes. Approximately half of the hepatocyte component of bile flow (about 225 ml per day) is bile salt-dependent and the remaining half bile salt independent. Osmotically active solutes such as glutathione and bicarbonate promote bile salt independent bile flow 11.
Canaliculi empty bile into ductules or cholangioles or canals of Hering. The ductules connect with interlobular bile ducts, which are accompanied by branches of the portal vein and hepatic artery forming portal triads. Bile is subsequently modified by ductular epithelial cells as it passes through the biliary tree. These cells, known as cholangiocytes, dilute and alkalinize the bile through hormone-regulated absorptive and secretory processes. The cholangiocytes have receptors which modulate the bicarbonate-rich ductular bile flow, which is regulated by hormones. These receptors include receptors for secretin, somatostatin, cystic fibrosis transmembrane conductance regulator (CFTR) and chloride-bicarbonate exchanger. For example, when secretin stimulates receptors in the cholangiocyte, a cascade is initiated which activates the cystic fibrosis transmembrane conductance regulator chloride channel and allows the exchange of bicarbonate for chloride. In contrast, somatostatin inhibits the cAMP synthesis within the cholangiocytes causing the opposite effect. While bombesin, vasoactive intestinal polypeptide, acetylcholine, and secretin enhances bile flow, somatostatin, gastrin, insulin, and endothelin inhibit the flow 12.
Bile acids formation
Cholesterol catabolism by liver hepatocytes results in the synthesis of the 2 major primary bile acids, cholic acid, and chenodeoxycholic acid. These lipid soluble bile acids are conjugated mainly to glycine or taurine molecules to form water soluble primary conjugated bile acids. In humans, about 0.2–0.6 g (averaging 0.5 g) bile acids are synthesized daily in human liver 13. These bile acids travel to the gall bladder during the interdigestive phase for storage and to the second part of the duodenum via the common bile duct during digestion. 95% of the bile acids which are delivered to the duodenum will be recycled by the enterohepatic circulation.
Bile acids synthesis process involves multiple steps, with cholesterol 7 alpha-hydroxylase acting as the rate-limiting enzyme. Primary bile acids undergo dehydroxylation by bacteria in the small intestine, forming the secondary bile acids deoxycholic acid and lithocholic acid, respectively. Both primary and secondary bile acids are conjugated by the liver with an amino acid, either glycine or taurine. Conjugated bile acids are known as bile salts. Bile salts inhibit cholesterol 7alpha-hydroxylase, decreasing the synthesis of bile acids. Despite the increased water solubility of bile salts, they are amphipathic molecules overall. This critical property allows them to effectively emulsify lipids and form micelles with the products of lipid digestion. The bile acid pool is maintained via mainly the enterohepatic circulation and to a small extent (about 5%) by hepatic synthesis of bile acids, as long as the daily fecal loss of bile acids do not exceed 20% of the pool.
At the pH of the small intestine, most of the bile acids are ionized and mostly occur as their sodium salts which are then called “primary conjugated bile salts.” In the lower small intestine and colon, bacteria dehydroxylate some of the primary bile salts to form secondary conjugated bile salts (which are still water soluble). Along the proximal and distal ileum, these conjugated primary bile salts are reabsorbed actively into hepatic portal circulation. Bacteria deconjugate some of the primary and secondary conjugated bile salts back to lipid soluble bile acids, which are passively absorbed into hepatic portal circulation. Finally, the conjugated bile acids which remained un-ionized conjugated bile acids are passively absorbed.
Venous blood from the ileum goes straight into the portal vein and then into the liver sinusoids. There, hepatocytes extract bile acids very efficiently, and little escapes the healthy liver into systemic circulation. If bile does escape, jaundice may be observed.
The majority of bile acids returned in the liver in portal venous blood are conjugated bile acids that were absorbed by the ileal bile acid transport system. The minority of bile acids returned to the liver consists of a complex mixture of unconjugated bile acids. The majority of these are unconjugated bile acids formed by bacterial deconjugation of primary bile acid conjugates. In addition, there are unconjugated bile acids entering from the colon. These include the 7-deoxy bile acids deoxycholic acid and lithocholic acid as well as the 3β-hydroxy epimers of these bile acids. In animals such as rodents whose bile acids belong to the muricholic acid family, the 7-deoxy bile acids will be hyodeoxycholic (3α,6α-dihydroxy) and murideoxycholic (3α,6β-dihydroxy) acids. Other bacterial metabolites are likely to be present in trace proportions.
All of these bile acids are efficiently removed by the hepatocyte. Those not removed spill over into the systemic circulation.Thus, in healthy man, plasma bile acids are enriched in 7-deoxy metabolite of chenodeoxycholic acid conjugates as these are extracted less efficiently than conjugates of cholic acid. Bile acids escaping first pass extraction are presented to the hepatocyte once again via hepatic arterial blood flow as well as by intestinal blood flow. As a result, the half life of any plasma bile acid is < 5 minutes.
The dominant source of plasma bile acids is intestinal absorption, not hepatocyte regurgitation. In man, fasting state plasma bile acid levels average 2 μmol/liter and the concentration triples during digestion of a meal. Plasma bile acids are thus an “enterohepatic” quality, with the level fluctuating in direct proportion to the fluctuations of intestinal absorption. Because of the fluctuations in bile acid levels of peripheral venous plasma, measurement of bile acid levels has not proven to be superior valley to conventional tests (aminotransferase levels) for the detection of liver injury 14.
Bile acids not bound to albumin enter the glomerular filtrate. The amount of bile acids entering the glomerular filtrate is small because of albumin binding and the great efficiency of hepatic uptake. Those bile acids entering the glomerular filtrate are reabsorbed via apical sodium-dependent bile acid transporter in the proximal renal tubule. Urinary bile acid loss is less than 1 mg/day compared to the fecal loss of 200-600 mg/day.
Bile function
The bile has five main functions:
- Aids in the digestion of fat via fat emulsification
- Absorption of fat and fat-soluble vitamins
- Excretion of bilirubin and excess cholesterol
- Provides an alkaline fluid in the duodenum to neutralize the acidic pH of the chyme that comes from the stomach
- It provides bactericidal activity against microorganisms present in the ingested food
The main functions of bile are 2-fold:
- Facilitate lipid absorption and digestion
- Eliminate waste products from the body
Lipid Absorption and Digestion
Through the process of emulsification, bile acids break down large lipid droplets into smaller ones, increasing the surface area for digestive enzymes. Emulsification is possible due to the amphipathic property of bile salts. The hydrophilic portion of the bile salts surrounds the lipid, forcing the lipid to disperse as the negative charges repel each other. Bile salts also allow the products of lipid digestion to be transported as micelles. The core of the micelle contains monoglycerides, lysolecithin, fatty acids, and the hydrophobic portion of the bile salt. The hydrophilic portion of the bile salt surrounds the lipid core, increasing solubility. Without bile salts, the fat-soluble vitamins (A, D, E, K) cannot be absorbed.
Elimination of Waste Products
Cholesterol is eliminated through its conversion into bile acids, allowing the body to maintain cholesterol homeostasis. Bile acid sequestrants, medications intended to lower cholesterol, function by binding bile acids in the small intestine and increasing their excretion in the stool. Bilirubin is also eliminated through its secretion into bile where it eventually forms the dark pigment of feces.
Clinical significance
The biliary system pathology includes autoimmune diseases, obstructive cholestasis, non-obstructive cholestasis, infections, and malignancy. The autoimmune diseases are primary biliary cholangitis (PBC) and primary sclerosing cholangitis. In primary biliary cholangitis, there is autoimmune-mediated progressive destruction of intrahepatic small-medium sized bile ducts which results in impaired bile secretion and intrahepatic cholestasis 15. In primary sclerosing cholangitis, there is intrahepatic and extrahepatic destruction of bile ducts. Primary sclerosing cholangitis is strongly associated with ulcerative colitis. The autoimmune basis for both conditions (primary biliary cholangitis and primary sclerosing cholangitis) presents in the histologic findings as well as the presence of specific antibodies in patients with these two conditions. Moreover, both conditions are often associated with other autoimmune diseases which further supports the autoimmune basis.
The decrease or cessation of bile formation or flow is known as cholestasis. Cholestasis can result from the impaired canalicular secretion of bile, ductular disease, or from obstruction of bile flow through the biliary tree. Causes of decreased canalicular secretion include drugs, sex hormones, and inherited defects. Ductular diseases include primary biliary cirrhosis and primary sclerosing cholangitis. Bile duct obstruction is most commonly due to gallstones but is also seen with cancers of the bile duct and/or pancreas. Adenocarcinoma of the head of the pancreas is an important cause of painless jaundice secondary to obstruction of the bile duct by the enlarging head of the pancreas. Once cholestasis occurs, bilirubin starts accumulating in the body resulting in yellowish discoloration of the sclera and skin. Moreover, the accumulation of bile acids secondary to cholestasis might be responsible for the pruritus seen in these patients.
Infections of the bile ducts are not uncommon, and it might present as fever, right upper quadrant pain, and jaundice. Inflammation of the gallbladder (cholecystitis) is also common, and it is usually associated with gallstones (i.e., acute calculous cholecystitis).
Clinically, signs and symptoms of cholestasis include pruritus, dark urine, pale stools, right upper quadrant pain, fever, xanthomas, and steatorrhea (due to fat malabsorption). Like bilirubin, other substances that are normally excreted in bile such as gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP), and cholesterol accumulate in the blood. Fat malabsorption may lead to deficiencies of vitamin A, D, E, and/or K. Deficiency in vitamin K would cause bleeding while a deficiency in vitamin D may cause rickets in children and osteomalacia in adults due to decreased calcium absorption from the gut. Deficiency in vitamin A may lead to night blindness. On exam, non-tender hepatomegaly may be present. Scratch marks on the skin due to pruritus may be present. A careful history and exam with appropriate diagnostic testing are necessary to narrow the differential diagnosis of cholestasis and create the proper treatment plan 16.
Testing for bile pathology includes imaging and blood tests. Imaging tests include ultrasonography for gallbladder pathology (e.g., cholecystitis and gallstones), Magnetic resonance cholangiopancreatography (MRCP) for visualization of the biliary tree (e.g., primary sclerosing cholangitis), abdominal computed tomography (CT), abdominal magnetic resonance imaging (MRI), and hepatobiliary iminodiacetic acid (HIDA) scan. HIDA scan is particularly useful in confirming acute cholecystitis when ultrasonography is not conclusive. Absence visualization of the gallbladder on HIDA scan suggests acute cholecystitis. Blood test includes complete blood count (CBC) and liver function tests (LFT).
Complete blood count (CBC) may show leukocytosis which accompanies many hepatobiliary pathologies (e.g., cholecystitis and ascending cholangitis). Liver function test (LFT) includes alkaline phosphatase (ALP) and gamma-glutamyltransferase (GGT), aspartate aminotransferase (AST) and alanine aminotransferase (ALT), prothrombin time (INR), indirect (unconjugated bilirubin), direct (conjugated) bilirubin, total bilirubin, and serum albumin.
Marked elevation in GGT and ALP with direct hyperbilirubinemia with or without a minimal elevation in AST and ALT indicates a cholestatic pathology. Normal LFT except for high ALP would point to a bone pathology rather than a hepatobiliary pathology. Markedly elevated GGT with slightly abnormal liver function test would point to an infiltrative liver pathology (e.g., metastasis).
References- The enterohepatic circulation of bile acids in mammals: form and functions. Hofmann AF. Front Biosci (Landmark Ed). 2009 Jan 1; 14():2584-98. https://www.bioscience.org/2009/v14/af/3399/fulltext.htm
- Almajid AN, Sugumar K. Physiology, Bile. [Updated 2019 Jun 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK542254
- Esteller A. Physiology of bile secretion. World J. Gastroenterol. 2008 Oct 07;14(37):5641-9.
- Hundt M, Basit H, John S. Physiology, Bile Secretion. [Updated 2019 May 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470209
- Baiocchi L, Zhou T, Liangpunsakul S, Lenci I, Santopaolo F, Meng F, Kennedy L, Glaser S, Francis H, Alpini G. Dual Role of Bile Acids on the Biliary Epithelium: Friend or Foe? Int J Mol Sci. 2019 Apr 16;20(8).
- Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, Maeda N, Parks JS. J Biol Chem. 2003 Sep 5; 278(36):33920-7.
- The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, Ballatori N. J Biol Chem. 2005 Feb 25; 280(8):6960-8.
- Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. Hagenbuch B, Meier PJ. J Clin Invest. 1994 Mar; 93(3):1326-31.
- Intestinal bile acid physiology and pathophysiology. Martinez-Augustin O, Sanchez de Medina F. World J Gastroenterol. 2008 Oct 7; 14(37):5630-40.
- Chiang, John. (2009). Thematic Review Series: Bile Acids: Bile acids: regulation of synthesis. Journal of lipid research. 50. 1955-66. 10.1194/jlr.R900010-JLR200.
- Dosch AR, Imagawa DK, Jutric Z. Bile Metabolism and Lithogenesis: An Update. Surg. Clin. North Am. 2019 Apr;99(2):215-229.
- Chiang JYL, Ferrell JM. Bile Acids as Metabolic Regulators and Nutrient Sensors. Annu. Rev. Nutr. 2019 Apr 24
- Chiang, John. (2009). Thematic Review Series: Bile Acids: Bile acids: regulation of synthesis. Journal of lipid research. 50. 1955-66. 10.1194/jlr.R900010-JLR200.
- Cravetto, C., G. Molino, A.M. Biondi, A. Cavanna, P. Avagnina, S. Frediani: Evaluation of the diagnostic value of serum bile acid in the detection and functional assessment of liver diseases. Ann Clin Biochem 22 (Pt 6), 596-605. (1985).
- Bowlus CL, Gershwin ME. The diagnosis of primary biliary cirrhosis. Autoimmun Rev. 2014 Apr-May;13(4-5):441-4.
- Zenouzi R, Welle CL, Venkatesh SK, Schramm C, Eaton JE. Magnetic Resonance Imaging in Primary Sclerosing Cholangitis-Current State and Future Directions. Semin. Liver Dis. 2019 Apr 30





{kind=link}