Fanconi anemia
Fanconi anemia also called Fanconi’s anemia, is a rare inherited blood disorder that leads to bone marrow failure (aplastic anemia). Fanconi’s anemia is a condition that affects many parts of the body. People with Fanconi anemia may have bone marrow failure, physical abnormalities, organ defects, and an increased risk of certain cancers.
Fanconi anemia prevents your bone marrow from making enough new blood cells for your body to work normally. Fanconi anemia also can cause your bone marrow to make many faulty blood cells. This can lead to serious health problems, such as leukemia (a type of blood cancer).
Fanconi anemia is a type of aplastic anemia. In aplastic anemia, the bone marrow stops making or doesn’t make enough of all three types of blood cells. Low levels of the three types of blood cells can harm many of the body’s organs, tissues, and systems.
With too few red blood cells (anemia), your body’s tissues won’t get enough oxygen to work well. With too few white blood cells (neutropenia), your body may have problems fighting infections. This can make you sick more often and make infections worse. With too few platelets (thrombocytopenia), your blood can’t clot normally. As a result, you may have bleeding problems.
People with Fanconi anemia may also develop myelodysplastic syndrome, a condition in which immature blood cells fail to develop normally.
Individuals with Fanconi anemia have an increased risk of developing a cancer of blood-forming cells in the bone marrow called acute myeloid leukemia (AML) or tumors of the head, neck, skin, gastrointestinal system, or genital tract. The likelihood of developing one of these cancers in people with Fanconi anemia is between 10 and 30 percent. The relative risk for acute myelogenous leukemia (AML) is increased approximately 500-fold 1. In a competing risk analysis of the combined cohorts, the cumulative incidence of AML was 13% by age 50 years, with most individuals diagnosed between ages 15 and 35 years 2.
Solid tumors may be the first manifestation of Fanconi anemia in individuals who have no birth defects and have not experienced bone marrow failure.
- Head and neck squamous cell carcinomas (HNSCCs) are the most common solid tumor in individuals with Fanconi anemia. The incidence is 500- to 700-fold higher than in the general population. The head and neck squamous cell carcinomas in Fanconi anemia show distinct differences compared to head and neck squamous cell carcinomas seen in the general population. Head and neck squamous cell carcinomas:
- Occur at an earlier age (20-40 years) than in the general population;
- Are most commonly in the the oral cavity (e.g., tongue);
- Present at an advanced stage;
- Respond poorly to therapy.
- Individuals with Fanconi anemia are at increased risk for second primary cancers in the skin and genitourinary tract. The pattern of second primaries resembles that observed in HPV-associated head and neck squamous cell carcinoma in the general population 3.
- Individuals with Fanconi anemia receiving androgen treatment for bone marrow failure are also at increased risk for liver tumors.
More than half of people with Fanconi anemia have physical abnormalities. These abnormalities can involve irregular skin coloring such as unusually light-colored skin (hypopigmentation) or café-au-lait spots, which are flat patches on the skin that are darker than the surrounding area. Other possible symptoms of Fanconi anemia include malformed thumbs or forearms and other skeletal problems including short stature; malformed or absent kidneys and other defects of the urinary tract; gastrointestinal abnormalities; heart defects; eye abnormalities such as small or abnormally shaped eyes; and malformed ears and hearing loss. People with this condition may have abnormal genitalia or malformations of the reproductive system. As a result, most affected males and about half of affected females cannot have biological children are infertile. Additional signs and symptoms can include abnormalities of the brain and spinal cord (central nervous system), including increased fluid in the center of the brain (hydrocephalus) or an unusually small head size (microcephaly).
Fanconi anemia is primarily a autosomal recessive disorder: if both parents carry a defect (mutation) in the same Fanconi anemia gene, each of their children has a 25% chance of inheriting the defective gene from both parents. When this happens, the child will have Fanconi anemia. Scientists have now discovered 21 Fanconi anemia or Fanconi anemia-like genes. These genes account for over 95% of all known Fanconi anemia patients 4. Some patients do not appear to have mutations in these 21 genes, so scientists anticipate that additional Fanconi anemia genes will be discovered in the future.
Fanconi anemia occurs equally in males and females. It is found in all ethnic groups. Research has added years to the lives of people with Fanconi anemia. Decades ago, children rarely survived to adulthood. Now, there are adults with Fanconi anemia that live into their 30s and beyond. Fanconi anemia can affect all systems of the body. Many patients eventually develop acute myeloid leukemia (AML) at a very early age. Although Fanconi anemia is a blood disorder, it also can affect many of your body’s organs, tissues, and systems. Fanconi anemia also increases the risk of some cancers and other serious health problems. Fanconi anemia patients are extremely likely to develop a variety of cancers and at a much earlier age than patients in the general population. Children who inherit Fanconi anemia are at higher risk of being born with birth defects.
Patients who have had a successful bone marrow transplant and are therefore cured of the blood problem associated with Fanconi anemia still must have regular examinations to watch for signs of cancer.
Fanconi anemia is different from Fanconi syndrome. Fanconi syndrome affects the kidneys. It’s a rare and serious condition that mostly affects children.
Children who have Fanconi syndrome pass large amounts of key nutrients and chemicals through their urine. These children may have serious health and developmental problems.
Is there any data regarding life expectancy of children who underwent a bone marrow transplant?
Allogeneic hematopoietic stem cell transplantation (HCT), a type of bone marrow transplant, has long been the primary treatment method for correcting the blood defects associated with Fanconi anemia 5. In general, it has been estimated that five-year survivors of hematopoietic stem cell transplantation may have a normal to near normal life expectancy, however Fanconi anemia is a risk factor that negatively impacts survival rates. One study estimated a 58 percent 30-years survival rate for one-year survivors of hematopoietic stem cell transplantation with Fanconi anemia 5.
Does Fanconi anemia and/or allogenic hematopoietic cell transplantation affect growth?
Yes. It is not uncommon for people with Fanconi anemia to have short stature. Also, allogenic hematopoietic cell transplantation (hematopoietic stem cell transplantation) and total body irradiation can affect final height in transplanted children, particularly when the transplant is preformed prior to the age of five. Allogeneic hematopoietic stem cell transplantation may also cause a decrease in lean body mass and a decline in body mass index after transplant 6.
What other long-term health complications are childhood survivors of allogeneic hematopoietic stem cell transplantation at risk for?
There are a number of long-term health risks associated with hematopoietic stem cell transplantation (hematopoietic stem cell transplantation). Specific risks vary depending on a variety of factors, including age, gender, type of pre-transplant therapy, donor type, reason for hematopoietic stem cell transplantation, and if the person experienced early complications (e.g., graft-versus-host disease or infections). Examples of hematopoietic stem cell transplantation related health risks include, blood cancer, solid tumors, heart disease, infection, lung toxicity, and chroinc graft-versus-host-disease.
Hematopoietic stem cell transplantation related risks appear to be higher in people with Fanconi anemia. Fanconi anemia makes the body especially sensitive to radiation and chemotherapy.
Fanconi anemia causes
Fanconi anemia is an inherited disease. The term “inherited” means that the disease is passed from parents to children through genes. At least 21 faulty genes are associated with Fanconi anemia. Proteins produced from these genes are involved in a cell process known as the FA pathway. The FA pathway is turned on (activated) when the process of making new copies of DNA, called DNA replication, is blocked due to DNA damage. The FA pathway sends certain proteins to the area of damage, which trigger DNA repair so DNA replication can continue.
The FA pathway is particularly responsive to a certain type of DNA damage known as interstrand cross-links (ICLs). ICLs occur when two DNA building blocks (nucleotides) on opposite strands of DNA are abnormally attached or linked together, which stops the process of DNA replication. ICLs can be caused by a buildup of toxic substances produced in the body or by treatment with certain cancer therapy drugs.
Eight proteins associated with Fanconi anemia group together to form a complex known as the FA core complex. The FA core complex activates two proteins, called FANCD2 and FANCI. The activation of these two proteins brings DNA repair proteins to the area of the ICL so the cross-link can be removed and DNA replication can continue.
Eighty to 90 percent of cases of Fanconi anemia are due to mutations in one of three genes, FANCA, FANCC, and FANCG. These genes provide instructions for producing components of the FA core complex. Mutations in any of the many genes associated with the FA core complex will cause the complex to be nonfunctional and disrupt the entire FA pathway. As a result, DNA damage is not repaired efficiently and ICLs build up over time. The ICLs stall DNA replication, ultimately resulting in either abnormal cell death due to an inability make new DNA molecules or uncontrolled cell growth due to a lack of DNA repair processes. Cells that divide quickly, such as bone marrow cells and cells of the developing fetus, are particularly affected. The death of these cells results in the decrease in blood cells and the physical abnormalities characteristic of Fanconi anemia. When the buildup of errors in DNA leads to uncontrolled cell growth, affected individuals can develop acute myeloid leukemia or other cancers.
Fanconi anemia inheritance pattern
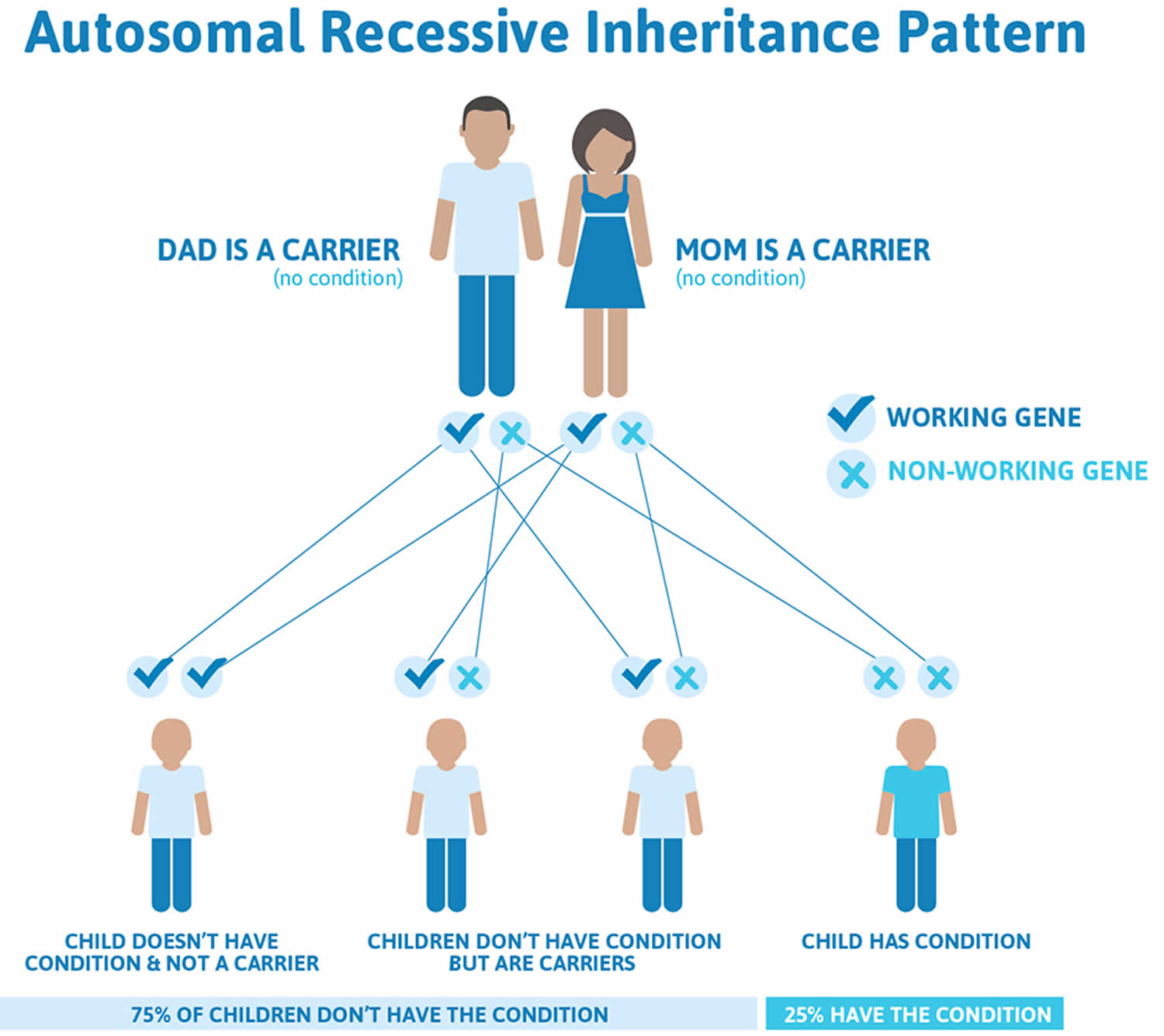
Fanconi anemia is most often inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Very rarely, Fanconi anemia is inherited in an X-linked recessive pattern. The gene associated with X-linked recessive Fanconi anemia is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
People who have only one faulty Fanconi anemia gene are Fanconi anemia “carriers.” Carriers don’t have Fanconi anemia, but they can pass the faulty gene to their children.
If both of your parents have a faulty Fanconi anemia gene, you have:
- A 25 percent chance of having Fanconi anemia
- A 25 percent chance of not having Fanconi anemia
- A 50 percent chance of being an Fanconi anemia carrier and passing the gene to any children you have
If only one of your parents has a faulty Fanconi anemia gene, you won’t have the disorder. However, you have a 50 percent chance of being an Fanconi anemia carrier and passing the gene to any children you have.
Figure 1. Fanconi anemia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Risk Factors for Fanconi anemia
Fanconi anemia occurs in all racial and ethnic groups and affects men and women equally.
In the United States, about 1 out of every 181 people is an Fanconi anemia carrier. This carrier rate leads to about 1 in 130,000 people being born with Fanconi anemia.
Two ethnic groups, Ashkenazi Jews and Afrikaners, are more likely than other groups to have Fanconi anemia or be Fanconi anemia carriers.
Ashkenazi Jews are people who are descended from the Jewish population of Eastern Europe. Afrikaners are White natives of South Africa who speak a language called Afrikaans. This ethnic group is descended from early Dutch, French, and German settlers.
In the United States, 1 out of 90 Ashkenazi Jews is an Fanconi anemia carrier, and 1 out of 30,000 is born with Fanconi anemia.
Major Risk Factors
Fanconi anemia is an inherited disease—that is, it’s passed from parents to children through genes. At least 21 faulty genes are associated with Fanconi anemia. Fanconi anemia occurs if both parents pass the same faulty Fanconi anemia gene to their child.
Children born into families with histories of Fanconi anemia are at risk of inheriting the disorder. Children whose mothers and fathers both have family histories of Fanconi anemia are at even greater risk. A family history of Fanconi anemia means that it’s possible that a parent carries a faulty gene associated with the disorder.
Children whose parents both carry the same faulty gene are at greatest risk of inheriting Fanconi anemia. Even if these children aren’t born with Fanconi anemia, they’re still at risk of being Fanconi anemia carriers.
Children who have only one parent who carries a faulty Fanconi anemia gene also are at risk of being carriers. However, they’re not at risk of having Fanconi anemia.
Screening and Prevention for Fanconi anemia
You can’t prevent Fanconi anemia because it’s an inherited disease. If a child gets two copies of the same faulty Fanconi anemia gene, he or she will have the disease.
If you’re at high risk for Fanconi anemia and are planning to have children, you may want to consider genetic counseling. A counselor can help you understand your risk of having a child who has Fanconi anemia. He or she also can explain the choices that are available to you.
If you’re already pregnant, genetic testing can show whether your child has Fanconi anemia.
In the United States, Ashkenazi Jews (Jews of Eastern European descent) are at higher risk for Fanconi anemia than other ethnic groups. For Ashkenazi Jews, it’s recommended that prospective parents get tested for Fanconi anemia-related gene mutations before getting pregnant.
Preventing Complications
If you or your child has Fanconi anemia, you can prevent some health problems related to the disorder. Pneumonia, hepatitis, and chicken pox can occur more often and more severely in people who have Fanconi anemia compared with those who don’t. Ask your doctor about vaccines for these conditions.
People who have Fanconi anemia also are at higher risk than other people for some cancers. These cancers include leukemia (a type of blood cancer), myelodysplastic syndrome (abnormal levels of all three types of blood cells), and liver cancer. Screening and early detection can help manage these life-threatening diseases.
Fanconi anemia symptoms
Major Signs and Symptoms
Your doctor may suspect you or your child has Fanconi anemia if you have signs and symptoms of:
- Anemia
- Bone marrow failure
- Birth defects
- Developmental or eating problems
Fanconi anemia is an inherited disorder—that is, it’s passed from parents to children through genes. If a child has Fanconi anemia, his or her brothers and sisters also should be tested for the disorder.
Anemia
The most common symptom of all types of anemia is fatigue (tiredness). Fatigue occurs because your body doesn’t have enough red blood cells to carry oxygen to its various parts. If you have anemia, you may not have the energy to do normal activities.
A low red blood cell count also can cause shortness of breath, dizziness, headaches, coldness in your hands and feet, pale skin, and chest pain.
Bone Marrow Failure
When your bone marrow fails, it can’t make enough red blood cells, white blood cells, and platelets. This can cause many problems that have various signs and symptoms.
With too few red blood cells, you can develop anemia. In Fanconi anemia, the size of your red blood cells also can be much larger than normal. This makes it harder for the cells to work well.
With too few white blood cells, you’re at risk for infections. Infections also may last longer and be more serious than normal.
With too few platelets, you may bleed and bruise easily, suffer from internal bleeding, or have petechiae. Petechiae are tiny red or purple spots on the skin. Bleeding in small blood vessels just below your skin causes these spots.
In some people who have Fanconi anemia, the bone marrow makes a lot of harmful, immature white blood cells called blasts. Blasts don’t work like normal blood cells. As they build up, they prevent the bone marrow from making enough normal blood cells.
A large number of blasts in the bone marrow can lead to a type of blood cancer called acute myeloid leukemia (AML).
Birth Defects
Many birth defects can be signs of Fanconi anemia. These include:
- Bone or skeletal defects. Fanconi anemia can cause missing, oddly shaped, or three or more thumbs. Arm bones, hips, legs, hands, and toes may not form fully or normally. People who have Fanconi anemia may have a curved spine, a condition called scoliosis.
- Eye and ear defects. The eyes, eyelids, and ears may not have a normal shape. Children who have Fanconi anemia also might be born deaf.
- Skin discoloration. This includes coffee-colored areas or odd-looking patches of lighter skin.
- Kidney problems. A child who has Fanconi anemia might be born with a missing kidney or kidneys that aren’t shaped normally.
- Congenital heart defects. The most common congenital heart defect linked to Fanconi anemia is a ventricular septal defect (VSD). A ventricular septal defect is a hole or defect in the lower part of the wall that separates the heart’s left and right chambers.
Developmental Problems
Other signs and symptoms of Fanconi anemia are related to physical and mental development. They include:
- Low birth weight
- Poor appetite
- Delayed growth
- Below-average height
- Small head size
- Mental retardation or learning disabilities
Signs and Symptoms of Fanconi Anemia in Adults
Some signs and symptoms of Fanconi anemia may develop as you or your child gets older. Women who have Fanconi anemia may have some or all of the following:
- Sex organs that are less developed than normal
- Menstruating later than women who don’t have Fanconi anemia
- Starting menopause earlier than women who don’t have Fanconi anemia
- Problems getting pregnant and carrying a pregnancy to full term
Men who have Fanconi anemia may have sex organs that are less developed than normal. They also may be less fertile than men who don’t have the disease.
Fanconi anemia diagnosis
People who have Fanconi anemia are born with the disorder. They may or may not show signs or symptoms of it at birth. For this reason, Fanconi anemia isn’t always diagnosed when a person is born. In fact, most people who have the disorder are diagnosed between the ages of 2 and 15 years.
The tests used to diagnose Fanconi anemia depend on a person’s age and symptoms. In all cases, medical and family histories are an important part of diagnosing Fanconi anemia. However, because Fanconi anemia has many of the same signs and symptoms as other diseases, only genetic testing can confirm its diagnosis.
Specialists Involved
A geneticist is a doctor or scientist who studies how genes work and how diseases and traits are passed from parents to children through genes.
Geneticists do genetic testing for Fanconi anemia. They also can provide counseling about how Fanconi anemia is inherited and the types of prenatal (before birth) testing used to diagnose it.
An obstetrician may detect birth defects linked to Fanconi anemia before your child is born. An obstetrician is a doctor who specializes in providing care for pregnant women.
After your child is born, a pediatrician also can help find out whether your child has Fanconi anemia. A pediatrician is a doctor who specializes in treating children and teens.
A hematologist (blood disease specialist) also may help diagnose Fanconi anemia.
Family and Medical Histories
Fanconi anemia is an inherited disease. Some parents are aware that their family has a medical history of Fanconi anemia, even if they don’t have the disease.
Other parents, especially if they’re Fanconi anemia carriers, may not be aware of a family history of Fanconi anemia. Many parents may not know that Fanconi anemia can be passed from parents to children.
Knowing your family medical history can help your doctor diagnose whether you or your child has Fanconi anemia or another condition with similar symptoms.
If your doctor thinks that you, your siblings, or your children have Fanconi anemia, he or she may ask you detailed questions about:
- Any personal or family history of anemia
- Any surgeries you’ve had related to the digestive system
- Any personal or family history of immune disorders
- Your appetite, eating habits, and any medicines you take
If you know your family has a history of Fanconi anemia, or if your answers to your doctor’s questions suggest a possible diagnosis of Fanconi anemia, your doctor will recommend further testing.
Diagnostic Tests and Procedures
The signs and symptoms of Fanconi anemia aren’t unique to the disease. They’re also linked to many other diseases and conditions, such as aplastic anemia. For this reason, genetic testing is needed to confirm a diagnosis of Fanconi anemia. Genetic tests for Fanconi anemia include the following.
Chromosome Breakage Test
This is the most common test for Fanconi anemia. It’s available only in special laboratories (labs). It shows whether your chromosomes (long chains of genes) break more easily than normal.
Skin cells sometimes are used for the test. Usually, though, a small amount of blood is taken from a vein in your arm using a needle. A technician combines some of the blood cells with certain chemicals.
If you have Fanconi anemia, the chromosomes in your blood sample break and rearrange when mixed with the test chemicals. This doesn’t happen in the cells of people who don’t have Fanconi anemia.
Cytometric Flow Analysis
Cytometric flow analysis, is done in a lab. This test examines how chemicals affect your chromosomes as your cells grow and divide. Skin cells are used for this test.
A technician mixes the skin cells with chemicals that can cause the chromosomes in the cells to act abnormally. If you have Fanconi anemia, your cells are much more sensitive to these chemicals.
The chromosomes in your skin cells will break at a high rate during the test. This doesn’t happen in the cells of people who don’t have Fanconi anemia.
Mutation Screening
A mutation is an abnormal change in a gene or genes. Geneticists and other specialists can examine your genes, usually using a sample of your skin cells. With special equipment and lab processes, they can look for gene mutations that are linked to Fanconi anemia.
Diagnosing Different Age Groups
Before Birth (Prenatal)
If your family has a history of Fanconi anemia and you get pregnant, your doctor may want to test you or your fetus for Fanconi anemia.
Two tests can be used to diagnose Fanconi anemia in a developing fetus:
- amniocentesis and
- chorionic villus sampling (CVS).
Both tests are done in a doctor’s office or hospital.
Amniocentesis is done 15 to 18 weeks after a pregnant woman’s last period. A doctor uses a needle to remove a small amount of fluid from the sac around the fetus. A technician tests chromosomes (chains of genes) from the fluid sample to see whether they have faulty genes associated with Fanconi anemia.
Chorionic villus sampling is done 10 to 12 weeks after a pregnant woman’s last period. A doctor inserts a thin tube through the vagina and cervix to the placenta (the temporary organ that connects the fetus to the mother).
The doctor removes a tissue sample from the placenta using gentle suction. The tissue sample is sent to a lab to be tested for genetic defects associated with Fanconi anemia.
At Birth
Three out of four people who inherit Fanconi anemia are born with birth defects. If your baby is born with certain birth defects, your doctor may recommend genetic testing to confirm a diagnosis of Fanconi anemia.
Childhood and Later
Some people who have Fanconi anemia are not born with birth defects. Doctors may not diagnose them with the disorder until signs of bone marrow failure or cancer occur. This usually happens within the first 10 years of life.
Signs of bone marrow failure most often begin between the ages of 3 and 12 years, with 7 to 8 years as the most common ages. However, 10 percent of children who have Fanconi anemia aren’t diagnosed until after 16 years of age.
If your bone marrow is failing, you may have signs of aplastic anemia. Fanconi anemia is one type of aplastic anemia.
In aplastic anemia, your bone marrow stops making or doesn’t make enough of all three types of blood cells: red blood cells, white blood cells, and platelets.
Aplastic anemia can be inherited or acquired after birth through exposure to chemicals, radiation, or medicines.
Doctors diagnose aplastic anemia using:
- Family and medical histories and a physical exam.
- A complete blood count (CBC) to check the number, size, and condition of your red blood cells. The CBC also checks numbers of white blood cells and platelets.
- A reticulocyte count. This test counts the number of new red blood cells in your blood to see whether your bone marrow is making red blood cells at the proper rate.
- Bone marrow tests. For a bone marrow aspiration, a small amount of liquid bone marrow is removed and tested to see whether it’s making enough blood cells. For a bone marrow biopsy, a small amount of bone marrow tissue is removed and tested to see whether it’s making enough blood cells.
If you or your child is diagnosed with aplastic anemia, your doctor will want to find the cause. If your doctor suspects you have Fanconi anemia, he or she may recommend genetic testing.
Fanconi anemia treatment
At the present time, stem cell transplantation is the only long-term cure for the blood defects in Fanconi anemia. Stem cells can be taken from a donor’s marrow or peripheral blood, or can be obtained through cord blood harvested at the time of a baby’s birth. To prepare for transplant, the patient’s own bone marrow is destroyed, making space for the new, healthy stem cells to engraft. Donor stem cells can be matched or partially mismatched to the patient’s tissue type. The closer the match, the less likely that the new stem cells will recognize the patient’s cells as foreign and attack them, a complication know as graft-versus-host disease.
Doctors decide how to treat Fanconi anemia based on a person’s age and how well the person’s bone marrow is making new blood cells.
Goals of Treatment
Long-term treatments for Fanconi anemia can:
- Cure the anemia. Damaged bone marrow cells are replaced with healthy ones that can make enough of all three types of blood cells on their own.
—Or—
- Treat the symptoms without curing the cause. This is done using medicines and other substances that can help your body make more blood cells for a limited time.
Screening and Short-Term Treatment
Even if you or your child has Fanconi anemia, your bone marrow might still be able to make enough new blood cells. If so, your doctor might suggest frequent blood count checks so he or she can watch your condition.
Your doctor will probably want you to have bone marrow tests once a year. He or she also will screen you for any signs of cancer or tumors.
If your blood counts begin to drop sharply and stay low, your bone marrow might be failing. Your doctor may prescribe antibiotics to help your body fight infections. In the short term, he or she also may want to give you blood transfusions to increase your blood cell counts to normal levels.
However, long-term use of blood transfusions can reduce the chance that other treatments will work.
Long-Term Treatment
The four main types of long-term treatment for Fanconi anemia are:
- Blood and marrow stem cell transplant
- Androgen therapy
- Synthetic growth factors
- Gene therapy
Blood and Marrow Stem Cell Transplant
A blood and marrow stem cell transplant is the current standard treatment for patients who have Fanconi anemia that’s causing major bone marrow failure. Healthy stem cells from another person, called a donor, are used to replace the faulty cells in your bone marrow.
If you’re going to receive stem cells from another person, your doctor will want to find a donor whose stem cells match yours as closely as possible.
When the healthy stem cells come from you, the procedure is called an autologous transplant. When the stem cells come from another person, called a donor, it is an allogeneic transplant. Blood or bone marrow transplants most commonly are used to treat blood cancers or other kinds of blood diseases that decrease the number of healthy blood cells in the body. These transplants also may be used to treat other disorders.
For allogeneic transplants, your doctor will try to find a donor whose blood cells are the best match for you. Your doctor will consider using cells from your close family members, from people who are not related to you and who have registered with the National Marrow Donor Program, or from publicly stored umbilical cord blood. Although it is best to find a donor who is an exact match to you, new transplant procedures are making it possible to use donors who are not an exact match.
Blood or bone marrow transplants are usually performed in a hospital. Often, you must stay in the hospital for one to two weeks before the transplant to prepare. During this time, you will have a narrow tube placed in one of your large veins. You may be given medicine to make you sleepy for this procedure. You also will receive special medicines and possibly radiation to destroy your abnormal stem cells and to weaken your immune system so that it won’t reject the donor cells after the transplant.
On the day of the transplant, you will be awake and may get medicine to relax you during the procedure. The stem cells will be given to you through the narrow tube in your vein. The stem cells will travel through your blood to your bone marrow, where they will begin making new healthy blood cells.
After the transplant, your doctor will check your blood counts every day to see if new blood cells have started to grow in your bone marrow. Depending on the type of transplant, you may be able to leave, but stay near the hospital, or you may need to remain in the hospital for weeks or months. The length of time will depend on how your immune system is recovering and whether or not the transplanted cells stay in your body. Before you leave the hospital, the doctors will give you detailed instructions that you must follow to prevent infection and other complications. Your doctor will keep monitoring your recovery, possibly for up to one year.
Although blood or bone marrow transplant is an effective treatment for some conditions, the procedure can cause early or late complications. The required medicines and radiation can cause nausea, vomiting, diarrhea, tiredness, mouth sores, skin rashes, hair loss, or liver damage. These treatments also can weaken your immune system and increase your risk for infection. Some people may experience a serious complication called graft-versus-host disease if the donated stem cells attack the body. Other people may reject the donor stem cells after the transplant, which can be an extremely serious complication.
Stem cell transplants are most successful in younger people who:
- Have few or no serious health problems
- Receive stem cells from a brother or sister who is a good donor match
- Have had few or no previous blood transfusions
During the transplant, you’ll get donated stem cells in a procedure that’s like a blood transfusion. Once the new stem cells are in your body, they travel to your bone marrow and begin making new blood cells.
A successful stem cell transplant will allow your body to make enough of all three types of blood cells.
Even if you’ve had a stem cell transplant to treat Fanconi anemia, you’re still at risk for some types of blood cancer and cancerous solid tumors. Your doctor will check your health regularly after the procedure.
Androgen Therapy
Before improvements made stem cell transplants more effective, androgen therapy was the standard treatment for people who had Fanconi anemia. Androgens are man-made male hormones that can help your body make more blood cells for long periods.
Androgens increase your red blood cell and platelet counts. They don’t work as well at raising your white blood cell count.
Unlike a stem cell transplant, androgens don’t allow your bone marrow to make enough of all three types of blood cells on its own. You may need ongoing treatment with androgens to control the effects of Fanconi anemia.
Also, over time, androgens lose their ability to help your body make more blood cells, which means you’ll need other treatments.
Androgen therapy can have serious side effects, such as liver disease. This treatment also can’t prevent you from developing leukemia (a type of blood cancer).
Synthetic Growth Factors
Your doctor may choose to treat your Fanconi anemia with growth factors. These are substances found in your body, but they also can be man-made.
Growth factors help your body make more red and white blood cells. Growth factors that help your body make more platelets still are being studied.
More research is needed on growth factor treatment for Fanconi anemia. Early results suggest that growth factors may have fewer and less serious side effects than androgens.
Gene Therapy
Researchers are looking for ways to replace faulty Fanconi anemia genes with normal, healthy genes. They hope these genes will make proteins that can repair and protect your bone marrow cells. Early results of this therapy hold promise, but more research is needed.
Surgery
Fanconi anemia can cause birth defects that affect the arms, thumbs, hips, legs, and other parts of the body. Doctors may recommend surgery to repair some defects.
For example, your child might be born with a ventricular septal defect—a hole or defect in the wall that separates the lower chambers of the heart. His or her doctor may recommend surgery to close the hole so the heart can work properly.
Children who have Fanconi anemia also may need surgery to correct digestive system problems that can harm their nutrition, growth, and survival.
One of the most common problems is an Fanconi anemia-related birth defect in which the trachea (windpipe), which carries air to the lungs, is connected to the esophagus, which carries food to the stomach.
This can cause serious breathing, swallowing, and eating problems and can lead to lung infections. Surgery is needed to separate the two organs and allow normal eating and breathing.
Living with Fanconi anemia
Improvements in blood and marrow stem cell transplants have increased the chances of living longer with Fanconi anemia. Also, researchers are studying new and promising treatments for Fanconi anemia. However, the disorder still presents serious challenges to patients and their families.
Fanconi anemia is a life-threatening illness. If you or your child is diagnosed with Fanconi anemia, you and your family members may feel shock, anger, grief, and depression. If you’re the parent or grandparent of a child who has Fanconi anemia, you may blame yourself for causing the disease.
Your doctor will want to test all of your children for Fanconi anemia if one of your children is born with the disorder. If you’re diagnosed with Fanconi anemia as an adult, your doctor may suggest testing your brothers and sisters for the disorder.
All of these things can create stress and anxiety for your entire family. Family counseling for Fanconi anemia may give you and other relatives important support, comfort, and advice.
One of the hardest issues to deal with is telling children that they have Fanconi anemia and what effect it will have on their lives.
Most Fanconi anemia support groups believe that parents need to give children information about the disorder in terms they can understand. These groups recommend answering questions honestly and directly, stressing the positive developments in treatment and survival.
If your child becomes upset or begins to act out after learning that he or she has Fanconi anemia, you may want to seek counseling.
Special Concerns and Needs
Many people who have Fanconi anemia survive to adulthood. If you have Fanconi anemia, you’ll need ongoing medical care. Your blood counts will need to be checked regularly.
Even if you have a blood and marrow stem cell transplant, you remain at risk for many cancers. You’ll need to be screened for cancer more often than people who don’t have Fanconi anemia.
If Fanconi anemia has left you with a very low platelet count, your doctor may advise you to avoid contact sports and other activities that can lead to injuries.
If your child has Fanconi anemia, he or she may have problems eating or keeping food down. Your doctor may recommend additional, special feedings to support growth and good health.
Support Groups
You or your family members may find it helpful to know about resources that can give you emotional support and helpful information about Fanconi anemia and its treatments.
Your doctor or hospital social worker may have information about counseling and support services. They also may be able to refer you to support groups that offer help with financial planning (treatment for Fanconi anemia can be costly).
Fanconi anemia prognosis
Treatment of aplastic anemia with medications, supportive use of blood products, and stem cell transplantation increases the life expectancy beyond the projected median of approximately age 30 years 7.
Cancer prevention, in particular the avoidance of smoking, and screening to identify early malignancies may reduce the mortality rate from cancer. With regard to the first serious adverse event, patients with a large number of birth defects are at higher risk of early-onset severe aplastic anemia, while those with fewer anomalies are more likely to develop leukemia or a solid tumor as young adults.
Although many patients with Fanconi anemia are short and have skeletal anomalies, intelligence is usually normal, and education and career planning should be encouraged.
Mortality and morbidity
Regarding mortality and morbidity, major adverse events for patients with Fanconi anemia are aplastic anemia (usually severe), leukemia, and solid tumors 8. The projected median survival from all causes for more than 2000 cases reported in the literature has improved in the past decade; from 1927-1999 and 2000-2009, median survivals are age 21 years and 29 years, respectively 7.
Bone marrow failure usually presents in childhood, with petechiae, bruising, and hemorrhages due to thrombocytopenia; pallor and fatigue from anemia; and infections due to neutropenia. The annual hazard rate for severe aplastic anemia reached 5% per year by age 10 years and was less than 1% per year in adults, with a cumulative incidence of 50% by age 50 years.
Leukemia usually presents primarily in teens and young adults, reaching a hazard rate of 1% per year, with a cumulative incidence of 10% by age 50 years. About one third of the cases of Fanconi anemia and leukemia in the literature did not have a prior diagnosis of Fanconi anemia, as well as a preceding phase of aplastic anemia. More than 100 cases in the literature were reported to have myelodysplastic syndrome (MDS).
The hazard rate for solid tumors rises steadily to greater than 10% per year by age 45 years, with a cumulative incidence of 25% by age 50 years, often without prior hematologic disease. As for acute myelogenous leukemia (AML), about one third of reported cases presented with a tumor and were subsequently diagnosed as Fanconi anemia.
A positive correlation between absent or abnormal radii and other congenital anomalies and bone marrow failure has been noted. The relative hazard of bone marrow failure and leukemia is higher in FANCG, compared with FANCA, and in FANCC, compared with FANCA. Patients with homozygous null mutations in FANCA have a higher risk of leukemia than those with allelic mutations, leading to an abnormal protein. Patients with biallelic mutations in BRCA2/FANCD1 have an extraordinarily high risk of acute myeloid leukemia, brain tumors (medulloblastoma), and Wilms tumors, with an approximately 95% chance of developing one of these tumors by age 5 years. Genetic background (Japanese vs Ashkenazi Jewish) and specific allelic mutations in FANCC can modulate the phenotype.
The risk of liver tumors is increased 400-fold, the risk of leukemia is about 500-fold, and head and neck cancers are increased approximately 600-fold. The risk of esophageal cancer is increased 2000-fold, and the risk of vulvar/vaginal cancer is increased 3000-fold. In competing risk analyses, the cumulative incidence of solid tumors reaches 30% by age 45 years and does not level off. Although bone marrow failure and leukemia, which may be treated or prevented by hematopoietic stem cell transplantation or gene therapy, are the concerns in treating children and adolescents, solid tumors remain the major threat to older patients with Fanconi anemia.
In a retrospective analysis of 145 patients with Fanconi anemia, 9 patients evolved to leukemia and 14 developed 18 solid tumors 9. The ratio of observed-to-expected cancers for all cancer diagnoses or for solid tumors was 40, and the ratio was 600 for leukemia. The cumulative incidence of leukemia, death from marrow failure, death from a solid tumor, and having a stem cell transplant (not necessarily a favorable outcome) was 10%, 11%, 29%, and 43%, respectively. Note that the risk of head and neck squamous cell carcinomas appeared to be higher in patients who had received a bone marrow transplantation 10.
A study by Sauter et al 11 suggested that the prevalence of oral human papillomavirus (HPV) is greater in persons with Fanconi anemia. The study found the oral HPV rate to be 11.1% in 126 patients with Fanconi anemia, versus 2.5% in 162 unaffected first-degree family members. More specifically, the oral HPV rate in sexually active persons with Fanconi anemia was 17.7%, versus 2.4% in family members, while in sexually inactive individuals with Fanconi anemia the prevalence of HPV was 8.7%, versus 2.9% in siblings 11.
A study by Sathyanarayana et al 12 suggested that in patients with Fanconi anemia, greater age is positively correlated with the incidence of chronic kidney disease.
Fanconi anemia life expectancy
People who have Fanconi anemia have a greater risk than other people for some cancers. About 10 percent of people who have Fanconi anemia develop leukemia.
People who have Fanconi anemia and survive to adulthood are much more likely than others to develop cancerous solid tumors.
The risk of solid tumors increases with age in people who have Fanconi anemia. These tumors can develop in the mouth, tongue, throat, or esophagus. The esophagus is the passage leading from the mouth to the stomach.
Women who have Fanconi anemia are at much greater risk than other women of developing tumors in the reproductive organs.
Fanconi anemia is an unpredictable disease. The average lifespan for people who have Fanconi anemia is between 20 and 30 years. The most common causes of death related to Fanconi anemia are bone marrow failure, leukemia, and solid tumors.
Advances in care and treatment have improved the chances of surviving longer with Fanconi anemia. Blood and marrow stem cell transplant is the major advance in treatment. However, even with this treatment, the risk of some cancers is greater in people who have Fanconi anemia.
References- Tamary H, Nishri D, Yacobovich J, Zilber R, Dgany O, Krasnov T, Aviner S, Stepensky P, Ravel-Vilk S, Bitan M, Kaplinsky C, Ben Barak A, Elhasid R, Kapelusnik J, Koren A, Levin C, Attias D, Laor R, Yaniv I, Rosenberg PS, Alter BP. Frequency and natural history of inherited bone marrow failure syndromes: the Israeli Inherited Bone Marrow Failure Registry. Haematologica. 2010;95:1300–7.
- Mehta PA, Tolar J. Fanconi Anemia. 2002 Feb 14 [Updated 2018 Mar 8]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1401
- Morris LG, Sikora AG, Patel SG, Hayes RB, Ganly I. Second primary cancers after an index head and neck cancer: subsite-specific trends in the era of human papillomavirus-associated oropharyngeal cancer. J Clin Oncol. 2011;29:739–46.
- Fanconi anemia. http://fanconi.org/index.php/learn_more
- Sanders JE, Woolfrey AE, Carpenter PA, et al. Late effects among pediatric patients followed for nearly 4 decades after transplantation for severe aplastic anemia. Blood. 2011;118(5):1421‐1428. doi:10.1182/blood-2011-02-334953
- Freycon F, Trombert-Paviot B, Casagranda L, et al. Final height and body mass index after fractionated total body irradiation and allogeneic stem cell transplantation in childhood leukemia. Pediatr Hematol Oncol. 2012;29(4):313‐321. doi:10.3109/08880018.2012.666781
- Fanconi anemia prognosis. https://emedicine.medscape.com/article/960401-overview#a6
- Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010 May. 24(3):101-22.
- Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003 Feb 1. 101(3):822-6.
- Rosenberg PS, Alter BP, Ebell W. Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica. 2008 Apr. 93(4):511-7.
- Sauter SL, Wells SI, Zhang X, et al. Oral human papillomavirus is common in individuals with Fanconi anemia. Cancer Epidemiol Biomarkers Prev. 2015 May. 24 (5):864-72.
- Sathyanarayana V, Lee B, Wright NB, et al. Patterns and frequency of renal abnormalities in Fanconi anaemia: implications for long-term management. Pediatr Nephrol. 2018 Apr 12.




{kind=link}