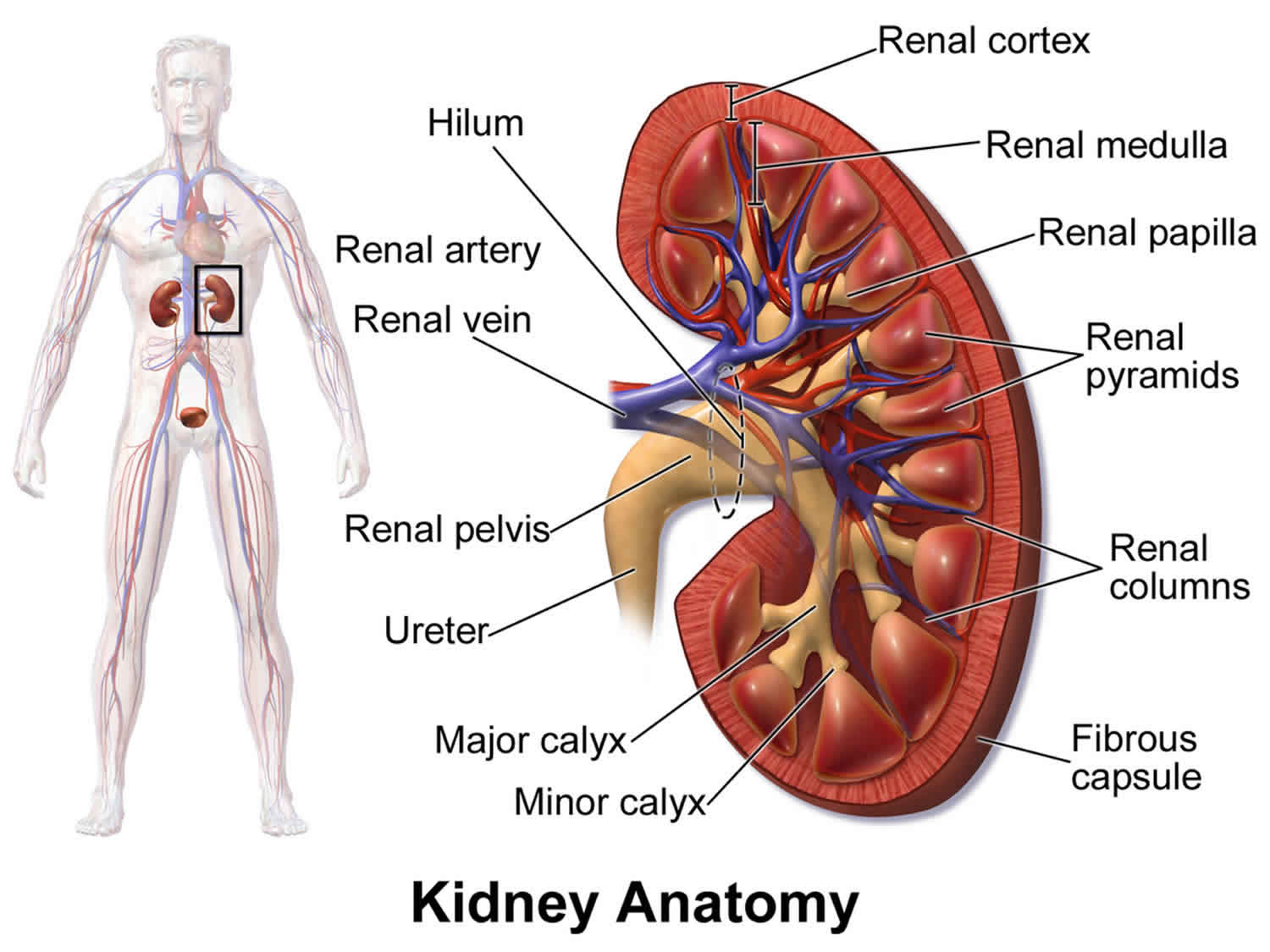
What is Fanconi syndrome
Fanconi syndrome is a rare disorder of the kidney tubule function that results in excess amounts of glucose, bicarbonate, phosphates (phosphorus salts), uric acid, potassium, and certain amino acids being excreted in the urine 1.
Fanconi syndrome is unrelated to and should not be confused with Fanconi anemia.
Fanconi syndrome can be:
- Hereditary Fanconi syndrome
- Acquired Fanconi syndrome
Hereditary Fanconi syndrome
This disorder usually accompanies another genetic disorder, particularly cystinosis. Cystinosis is an inherited (autosomal recessive) metabolic disorder in which cystine accumulates within cells and tissues (and is not excreted to excess in the urine as occurs in cystinuria). Besides renal tubular dysfunction, other complications of cystinosis include eye disorders, hepatomegaly, hypothyroidism, and other manifestations.
Fanconi syndrome may also accompany Wilson disease, hereditary fructose intolerance, galactosemia, oculocerebrorenal syndrome (Lowe syndrome), mitochondrial cytopathies, and tyrosinemia. Inheritance patterns vary with the associated disorder.
Acquired Fanconi syndrome
This disorder may be caused by various drugs, including certain cancer chemotherapy drugs (e.g, ifosfamide, streptozocin), antiretrovirals (eg, didanosine, cidofovir), and outdated tetracycline. All of these drugs are nephrotoxic. Acquired Fanconi syndrome also may occur after renal transplantation and in patients with multiple myeloma, amyloidosis, intoxication with heavy metals or other chemicals, or vitamin D deficiency.
Acquired Fanconi syndrome may caused by:
- Exposure to certain drugs (including some chemotherapy and antiretroviral drugs)
- Exposure to heavy metals or other chemicals
- Vitamin D deficiency
- Kidney transplantation
- Multiple myeloma
- Amyloidosis
Fanconi syndrome usually occurs with another hereditary disorder, such as cystinosis. Cystinosis is an inherited disorder of amino acid metabolism characterized by abnormal deposits of the amino acid cystine throughout the body and abnormal concentrations of cystine in the urine. Abnormal cystine deposits cause eye disorders, an enlarged liver, and an underactive thyroid gland.
The prognosis of Fanconi bickel syndrome depends on the underlying disease.
Key Points
- Multiple defects impair proximal tubular reabsorption of glucose, phosphate, amino acids, bicarbonate, uric acid, water, potassium, and sodium.
- Fanconi syndrome is usually caused by a drug or accompanies another genetic disorder.
- In hereditary Fanconi syndrome, proximal tubular acidosis, hypophosphatemic rickets, hypokalemia, polyuria, and polydipsia usually appear in infancy.
- Test urine for glucosuria (particularly in the presence of normal serum glucose), phosphaturia, and aminoaciduria.
- Treat by giving combinations as needed of potassium or sodium with either bicarbonate or citrate, or sometimes with just a supplemental potassium salt.
Fanconi syndrome symptoms
In hereditary Fanconi syndrome, symptoms of excessive drinking and excessive urination usually begin during infancy.
A child with Fanconi syndrome and cystinosis may have failure to thrive, slowed growth, and chronic kidney disease. Interstitial nephritis develops, leading to progressive renal failure that may be fatal before adolescence. Kidney failure may require a kidney transplant during childhood. The retinas show patchy depigmentation.
In acquired Fanconi syndrome, adults, symptoms may not develop until the disorder has been present for some time. The most common symptoms in adults include muscle weakness and bone pain due to osteomalacia and fractures due to bone weakness.
Most often, some damage to bones or kidney tissue has occurred before the diagnosis is made.
Fanconi syndrome causes
Fanconi syndrome can be caused by faulty genes, or it may result later in life due to kidney damage. Sometimes the cause of Fanconi syndrome is unknown.
Common causes of Fanconi syndrome in children are genetic defects that affect the body’s ability to break down certain compounds such as:
- Cystine (cystinosis)
- Fructose (fructose intolerance)
- Galactose (galactosemia)
- Glycogen (glycogen storage disease)
Cystinosis is the most common cause of Fanconi syndrome in children.
Other causes of Fanconi syndrome in children include:
- Exposure to heavy metals such as lead, mercury, or cadmium
- Lowe syndrome, a rare genetic disorder of the eyes, brain, and kidneys
- Wilson disease
- Dent disease, a rare genetic disorder of the kidneys
In adults, Fanconi syndrome can be caused by various things that damage the kidneys, including:
- Certain medicines, including azathioprine, cidofovir, gentamicin, and tetracycline
- Kidney transplant
- Light chain deposition disease
- Multiple myeloma
- Primary amyloidosis
Fanconi syndrome diagnosis
Laboratory tests may show that too much of the following substances may be lost in the urine:
- Amino acids
- Bicarbonate
- Glucose
- Magnesium
- Phosphate
- Potassium
- Sodium
- Uric acid
Diagnosis is made by showing the abnormalities of renal function, particularly high levels of glucose in urine (in the presence of normal serum glucose), phosphaturia, and aminoaciduria. In cystinosis, slit-lamp examination may show cystine crystals in the cornea.
Loss of these substances can lead to a variety of problems. Further tests and a physical exam may show signs of:
- Dehydration due to excess urination
- Growth failure
- Osteomalacia
- Rickets
- Type 2 renal tubular acidosis
Fanconi syndrome treatment
Many different diseases can cause Fanconi syndrome. The underlying cause and its symptoms should be treated as appropriate.
- Sometimes sodium bicarbonate or potassium bicarbonate or sodium citrate or potassium citrate
- Sometimes potassium supplementation
Other than removing the offending nephrotoxin, there is no specific treatment.
Acidosis may be lessened by taking tablets or solutions of sodium bicarbonate or potassium bicarbonate or sodium citrate or potassium citrate, e.g, Shohl’s solution (sodium citrate and citric acid; 1 mL is equivalent to 1 mmol of bicarbonate) given 1 mEq/kg body weight twice daily to three times daily or 5 to 15 mL after meals and at bedtime.
Potassium depletion may require replacement therapy with a potassium-containing salt.
Hypophosphatemic rickets can be treated.
Kidney transplantation has been successful in treating renal failure. However, when cystinosis is the underlying disease, progressive damage may continue in other organs and eventually result in death.
What is Fanconi Bickel syndrome
Fanconi Bickel syndrome is a rare condition characterized by the accumulation of a substance called glycogen in different parts of the body. Glycogen is created when the body needs to store glucose (sugar). When the body needs sugar again, glycogen is transformed back into glucose for use 2. People with Fanconi Bickel syndrome do not store the appropriate amount of glycogen. Therefore, Fanconi Bickel syndrome is known as a glycogen storage disease. Specifically, glycogen accumulates in the liver and kidneys. Signs and symptoms begin in the first few months of life and include growing slower than expected (failure to thrive), excessive urination (polyuria), and weakened bones (rickets). Later in life, children may have short stature and a swollen liver and spleen (hepatosplenomegaly) 3.
Fanconi Bickel syndrome is caused by mutations to the SLC2A2 gene and is inherited in an autosomal recessive manner. Diagnosis of Fanconi Bickel syndrome is based on a clinical examination that shows signs of Fanconi Bickel syndrome. The condition can be confirmed by genetic testing. Treatment is focused on relieving symptoms of the condition, particularly the symptoms that affect the kidneys 4.
Fanconi Bickel syndrome symptoms
Fanconi Bickel syndrome is characterized by the accumulation of glycogen in the liver and kidneys. This causes symptoms such as having weakened bones (rickets), being very small for one’s age (failure to thrive), and a specific type of kidney malfunction called renal tubular dysfunction. The accumulation of glycogen can also cause swelling of the liver and spleen (hepatosplenomegaly) 5. Between meals, affected individuals may have low blood sugar (hypoglycemia). People with Fanconi Bickel syndrome typically enter puberty later than expected. As adults, the weakened bones may result in osteopenia or osteoporosis 2. Adults with Fanconi Bickel syndrome may be shorter than other people, and they may have bowed legs 6.
Fanconi Bickel syndrome may have the following symptoms:
- Abdominal bloating
- Chronic acidosis
- Elevated alkaline phosphatase
Fanconi Bickel syndrome causes
Fanconi Bickel syndrome is caused by a mutation in the SLC2A2 gene. This gene tells the body how to make a protein called glucose-transporter protein 2 (GLUT2). This protein is responsible for transporting glucose through different cells in the body. When GLUT2 is not working properly because of a mutation in SLC2A2, the body cannot transport glucose. Therefore, glucose builds up in the liver and kidneys. This glucose is stored as glycogen, and the buildup of glycogen in these body parts cause the symptoms of Fanconi Bickel syndrome 7.
Fanconi Bickel syndrome inheritance pattern
Fanconi Bickel syndrome is inherited in an autosomal recessive manner 3. This means that a person affected with Fanconi Bickel syndrome has changed (mutated) copies of both SLC2A2 genes. We inherit one copy of each gene from our mother and the other from our father. A person with only one changed copy of SLC2A2 typically does not have symptoms and is known as a carrier. The children of any two carriers have a 3:
- 25% chance of being unaffected
- 50% chance of being a carrier like both parents
- 25% chance of having Fanconi Bickel syndrome.
Fanconi Bickel syndrome diagnosis
Fanconi Bickel syndrome is diagnosed by a clinical examination that is consistent with symptoms of the condition. This clinical evaluation may reveal findings such as rickets (weakened bones) and high levels of glucose, protein, and phosphate in the urine. People with Fanconi Bickel syndrome may also have low levels of phosphate and high levels of cholesterol in the blood. People with Fanconi Bickel syndrome tend to have low blood sugar (hypoglycemia) between meals. If a diagnosis of Fanconi Bickel syndrome is suspected, it can be confirmed with genetic testing of the SLC2A2 gene 3.
Fanconi Bickel syndrome treatment
Management of Fanconi Bickel syndrome generally focuses on treating the signs and symptoms of the condition. For treatment of the symptoms of kidney disease, a doctor may focus on replacing the water and electrolytes that are lost from the kidneys. Additionally, vitamin D and phosphate supplements can help prevent bone weakening (rickets) 7.
It is recommended that people with Fanconi Bickel syndrome follow a galactose-restricted diet. Galactose is a substance that is broken down into glucose. Because people with Fanconi Bickel syndrome have trouble moving glucose throughout the body, limiting galactose prevents the buildup of glucose and glycogen in the liver and kidneys. Galactose is found in food such as milk, cheese, yogurt, and legumes. People with Fanconi Bickel syndrome should primarily eat fructose as their main carbohydrate. In addition, people with Fanconi Bickel syndrome should eat small, frequent meals in order to avoid developing low blood sugar between meals. Finally, cornstarch may be used as a substitute to provide necessary sugars to the body 7.
Fanconi Bickel syndrome prognosis
The treatments mentioned above may alleviate some of the signs and symptoms of the condition. The symptoms of kidney disease and the swelling of the liver and spleen typically resolve before adulthood. However, dietary treatments and supplements typically do not improve growth, which can result in short stature during adulthood 8.
Some of the symptoms of Fanconi Bickel syndrome such as rickets can have a large effect on people’s lives. However, with the proper treatment, some of the symptoms of Fanconi Bickel syndrome can resolve with age. People with this condition have been known to reach adulthood, and there have been reports that affected individuals can have children of their own 8.
References- Fanconi Syndrome. https://www.merckmanuals.com/home/kidney-and-urinary-tract-disorders/disorders-of-kidney-tubules/fanconi-syndrome
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th edition. New York: W H Freeman; 2002. Chapter 21, Glycogen Metabolism. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21190
- Glycogen storage disease due to GLUT2 deficiency. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=2088
- Kehar M, Bijarnia S, Ellard S, Houghton J, Saxena R, Verma IC, and Wadhwa N. Fanconi Bickel syndrome – mutation in SLC2A2 gene. Indian Journal of Pediatrics. November 2014; 81(11):1237-1239. https://www.ncbi.nlm.nih.gov/pubmed/24912437
- Grünert SC, Schwab KO, Pohl M, Sass JO, Santer R. Fanconi-Bickel syndrome: GLUT2 mutations associated with a mild phenotype. Mol Genet Metab. March 2012; 105(3):433-437.
- Mohandas Nair K, Sakamoto O, Jagadeesh S, Nampoothiri S. Fanconi-Bickel syndrome. Indian Journal of Pediatrics. January 2012; 79(1):112-114. https://www.ncbi.nlm.nih.gov/pubmed/21327337
- Fanconi bickel syndrome. https://www.omim.org/entry/227810
- Pena L and Charrow J. Fanconi-Bickel syndrome: report of life history and successful pregnancy in an affected patient. American Journal of Medical Genetics. February 2011; 155A(2):415-417. https://www.ncbi.nlm.nih.gov/pubmed/21271664





{kind=link}