What is G6PD deficiency
G6PD deficiency is short for glucose 6 phosphate dehydrogenase deficiency, is a group of hereditary abnormalities that occurs almost exclusively in males in which the activity of the red blood cells glucose 6 phosphate dehydrogenase enzyme is markedly diminished 1. G6PD deficiency mainly affects red blood cells, which carry oxygen from your lungs to tissues throughout your body. In affected individuals, a defect in an enzyme called glucose-6-phosphate dehydrogenase causes red blood cells to break down prematurely. This destruction of red blood cells is called hemolysis. The red blood cells glucose 6 phosphate dehydrogenase enzyme deficiency may result in hemolytic anemia, particularly when the body is exposed to certain foods, drugs, infections or stress, possibly during diabetic acidosis, and in the neonatal period. The enzyme glucose-6-phosphate dehydrogenase helps red blood cells work properly and is essential for assuring a normal life span for red blood cells, and for oxidizing processes.
The most common medical problem associated with glucose-6-phosphate dehydrogenase deficiency (G6PD deficiency) is hemolytic anemia, which occurs when red blood cells are destroyed faster than the body can replace them. Hemolytic anemia leads to paleness, yellowing of the skin and whites of the eyes (jaundice), dark urine, fatigue, shortness of breath, and a rapid heart rate. In people with G6PD deficiency, hemolytic anemia is most often triggered by bacterial or viral infections or by certain drugs (such as some antibiotics and medications used to treat malaria). Hemolytic anemia can also occur after eating fava beans or inhaling pollen from fava plants (a reaction called favism).
Glucose-6-phosphate dehydrogenase deficiency is also a significant cause of mild to severe jaundice in newborns. Many people with this disorder, however, never experience any signs or symptoms and are unaware that they have the condition.
G6PD deficiency is inherited in an X-linked recessive manner and symptoms are more common in males (particularly African Americans and those from certain parts of Africa, Asia, and the Mediterranean). About 1 in 10 African American males in the United States has G6PD deficiency, but that it could be found in virtually any population. An estimated 400 million people worldwide have G6PD deficiency (glucose-6-phosphate dehydrogenase deficiency). G6PD deficiency occurs most frequently in certain parts of Africa, Asia, the Mediterranean, and the Middle East.
G6PD deficiency is caused by mutations in the G6PD gene. Treatment may involve medicines to treat infection, stopping drugs that are causing red blood cell destruction, and/or transfusions, in some cases 2.
G6PD deficiency mainly affects red blood cells, which carry oxygen from the lungs to tissues throughout the body. The most common medical problem it can cause is hemolytic anemia. That happens when red blood cells are destroyed faster than the body can replace them.
If you have G6PD deficiency, you may not have symptoms. Symptoms happen if your red blood cells are exposed to certain chemicals in food or medicine, certain bacterial or viral infections, or stress. They may include:
- Paleness
- Jaundice
- Dark urine
- Fatigue
- Shortness of breath
- Enlarged spleen
- Rapid heart rate
You are more likely to develop G6PD deficiency if you:
- Are African American
- Are of Middle Eastern decent, particularly Kurdish or Sephardic Jewish
- Are male
- Have a family history of the deficiency
A form of G6PD deficiency is common in whites of Mediterranean descent. This form is also associated with acute episodes of hemolysis. Episodes are longer and more severe than in the other types of the disorder.
A blood test can tell if you have it. Treatments include medicines to treat infection, avoiding substances that cause the problem with red blood cells, and sometimes transfusions.
G6PD deficiency prognosis
In most G6PD deficiency cases, hemolytic episodes go away on their own. In rare case, kidney failure or death may occur following a severe hemolytic event.
Can a person get G6PD deficiency from a blood transfusion?
No. A person cannot acquire G6PD deficiency from a blood transfusion from a donor with G6PD. G6PD is a genetic disorder and is inherited in an X-linked recessive manner.
Due to many unanswered questions, there is controversy about whether G6PD-deficient people should become blood donors and about the quality of their blood during processing and storage. Risk factors associated with the use of G6PD-deficient blood in transfusion have not been well established. There is little evidence that G6PD-deficient people should be excluded from donating red blood cells. But, most studies on newborns and children have recommended routine screening for G6PD deficiency because their immature hepatic (liver) function potentially lessens their ability to handle any excess bilirubin. Transfusions of G6PD-deficient blood also may potentially have negative effects on premature newborns, or people who need repeated transfusions. For these recipients, screening for G6PD deficiency may be appropriate. The potential effects may differ depending on the region, the specifics of the recipient, the quantity transfused, and the type of G6PD deficiency 3.
The World Health Organization (WHO) guidelines recommend that blood be accepted from “individuals with G6PD deficiency or other inherited red cell membrane defects, without a history of hemolysis; however, their blood is not suitable for intrauterine transfusion, neonatal exchange transfusion or for patients with G6PD deficiency” 3.
Can women have symptoms of glucose-6-phosphate dehydrogenase (G6PD) deficiency?
Females can have symptoms of G6PD deficiency. Females can be affected if they have a mutation in both copies of the G6PD gene, or in some cases, if they have only one mutated G6PD gene.
Depending on how mutations affect G6PD enzyme activity, symptoms in any individual can range from mild to severe. Females with mutations in both of their G6PD gene copies can experience the full range of symptoms similar to affected males (who have one mutation). Females who have one G6PD mutation can also have mild to severe symptoms as a result of a phenomenon called skewed lyonization. This phenomenon is also referred to as skewed X-inactivation.
Because females have two X chromosomes and males have one X chromosome, one copy of the X chromosome in each of a female’s cells is “inactivated.” This is to prevent females from having twice as much genetic information coming from the X chromosome. Typically, the “choice” of which X chromosome to inactivate is random, which theoretically would lead to each X chromosome being inactivated around 50% of the time. However, in come cases, the activation is skewed, leading to one X chromosome being inactivated significantly more than the other. If a female has a G6PD mutation on the X chromosome that is significantly more active, she can have mild to severe symptoms of G6PD deficiency. The severity depends on how the mutation affects G6PD activity, and the extent to which X-chromosome inactivation is skewed.
G6PD deficiency triggers
Red blood cell destruction can be triggered by infections, certain foods (such as fava beans), and certain medicines, including:
- Antimalarial medicines such as quinine
- Aspirin (high doses)
- Nonsteroidal anti-inflammatory drugs (NSAIDs)
- Quinidine
- Sulfa drugs
- Antibiotics such as quinolones, nitrofurantoin
Other chemicals, such as those in mothballs, can also trigger an episode.
G6PD deficiency foods to avoid
Fava beans. Acute hemolytic anemia from eating fava beans (favism) can be rapid. Favism can occur at any age, but occurs more often and more severely in children. A child may have a slightly elevated temperature within 24-48 hours and can become irritable and unruly, or subdued and lethargic. Nausea, abdominal pain and diarrhea may develop. Vomiting only occurs rarely. Within 6 to 24 hours, urine may become noticeably dark and can appear red, brown or even black. Affected children may become pale and their resting heartrate may be high (tachycardia). Jaundice can also develop and the liver and spleen may become enlarged. In severe cases, evidence may be seen of hypovolemic shock, in which blood and fluid loss is so severe that the heart cannot pump enough blood to the body, or, less likely, heart failure.
G6PD deficiency medications to avoid
Drugs and chemicals that should be avoided by persons with G6PD deficiency 4:
- Acetanilid
- Doxorubicin
- Furazolidone (Furoxone)
- Methylene Blue
- Nalidixic acid (NeGram)
- Naphthalene
- Niridazole (Ambilhar)
- Nitrofurantoin (Furadantin)
- Phenazopyridine (Pyridium)
- Phenylhydrazine
- Primaquine
- Primaquine, the only drug licensed for radical cure of vivax and ovale malaria, and the only licenced antimalarial with specific gametocytocidal activity against Plasmodium falciparum, causes dose-dependent haemolysis in G6PD-deficient individuals 5.
- World Health Organization (WHO) recommendation on the use of primaquine. The G6PD status of patients should be used to guide administration of primaquine for preventing malaria relapse. 6:
- To prevent relapse, treat P. vivax or P. ovale malaria children and adults (except pregnant women, infants aged < 6 months, women breastfeeding infants aged < 6 months, women breastfeeding older infants unless they are known not to be G6PD deficient and people with G6PD deficiency) with a 14-day course of primaquine at 0.25–0.5 mg base/kg body weight daily in all transmission settings.
- In people with G6PD deficiency, consider preventing relapse by giving primaquine at 0.75 mg base/kg body weight once a week for 8 weeks, with close medical supervision for potential primaquine-induced haemolysis.
- When a patient’s G6PD status is unknown and G6PD testing is not available, a decision to prescribe primaquine must be based on an assessment of the risks and benefits of adding primaquine.
- For women who are pregnant or breastfeeding, consider weekly chemoprophylaxis with chloroquine until delivery and breastfeeding are completed; then, on the basis of the woman’s G6PD status, treat with primaquine to prevent future relapse 7.
- Sulfacetamide
- Sulfamethoxazole (Gantanol)
- Sulfanilamide
- Sulfapyridine
- Thiazolesulfone
- Toluidine blue
- Trinitrotoluene (TNT)
For a complete list of medications and drugs that individuals with G6PD deficiency should avoid can be found here: https://www.g6pd.org/en/G6PDDeficiency/SafeUnsafe.aspx
G6PD deficiency symptoms
People with G6PD deficiency generally do not have symptoms unless their red blood cells are exposed to certain chemicals in food or medicine, certain bacterial or viral infections, or to stress. Many people with G6PD deficiency never experience symptoms 8. The most common medical problem associated with G6PD deficiency is hemolytic anemia, which occurs when red blood cells are destroyed faster than the body can replace them. This type of anemia leads to paleness, yellowing of the skin and whites of the eyes (jaundice), dark urine, fatigue, shortness of breath, enlarged spleen, and a rapid heart rate. Some patients have a history of chronic hemolytic anemia. Skin ulcers are uncommon but may occur in people with severe G6PD deficiency.
Because G6PD deficiency is inherited in an X-linked recessive manner, it is more common for males to have symptoms. This is because males have only one copy of the G6PD gene. If this one copy has a mutation, they will definitely have G6PD deficiency. However, while females have two copies of the G6PD gene, some females are as severely affected as males. This can be the case in females who have a mutation in both copies of the G6PD gene, or even in females who have only one mutation. Females with one mutation may have lower G6PD activity than would normally be expected due to a phenomenon called skewed lyonization.
Symptoms are more common in men and may include:
- Dark urine
- Fever
- Pain in abdomen
- Enlarged spleen and liver
- Fatigue
- Pallor
- Rapid heart rate
- Shortness of breath
- Yellow skin color (jaundice)
G6PD deficiency causes
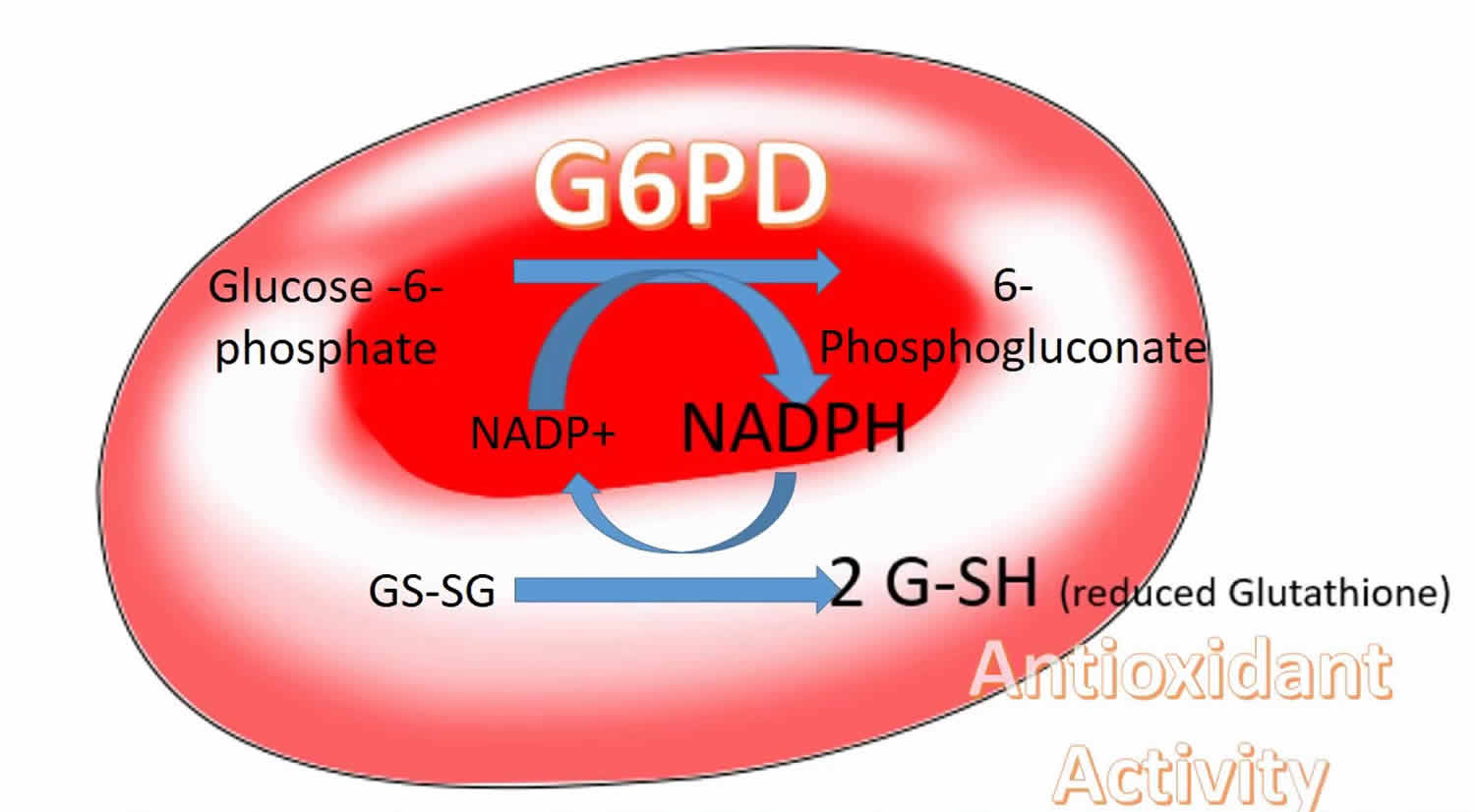
G6PD deficiency (glucose-6-phosphate dehydrogenase deficiency) results from mutations in the G6PD gene. This gene gives the body instructions for making an enzyme called glucose-6-phosphate dehydrogenase. The glucose-6-phosphate dehydrogenase enzyme is involved in the normal processing of carbohydrates. It also protects red blood cells from the effects of potentially harmful molecules called reactive oxygen species (ROS), which are byproducts of normal cellular functions. Chemical reactions involving glucose-6-phosphate dehydrogenase enzyme produce compounds that prevent reactive oxygen species from building up to toxic levels within red blood cells.
If mutations in the G6PD gene reduce the amount of glucose-6-phosphate dehydrogenase enzyme or alter its structure, this enzyme can no longer play its protective role. As a result, reactive oxygen species can accumulate and damage red blood cells. Factors such as infections, certain drugs, or ingesting fava beans can increase the levels of reactive oxygen species, causing red blood cells to be destroyed faster than the body can replace them. A reduction in the number of red blood cells causes the signs and symptoms of hemolytic anemia.
As part of a chemical reaction, glucose-6-phosphate dehydrogenase enzyme brings about (catalyzes) the coenzyme NADPH, which protects cells from oxidative damage. A mutation in the G6PD gene results in low levels of functional glucose-6-phosphate dehydrogenase, which in turn leads to low levels of NADPH and a depletion of an antioxidant known as glutathione, which is necessary to protect the cell’s hemoglobin and its cell wall (red cell membrane) from highly reactive oxygen radicals (oxidative stress). Normally, the amount of NADPH, although reduced, is adequate for the health of a red blood cell. However, this reduction in NADPH makes red blood cells more susceptible to destruction from oxidative stress than other cells, resulting in their premature break down when in the presence of triggering factors. G6PD is a housecleaning enzyme that is expressed in all cells of the body. However, the body can compensate for the effects of G6PD deficiency in cells other than red blood cells.
Researchers believe that people who have a G6PD mutation may be partially protected against malaria, an infectious disease carried by a certain type of mosquito. A reduction in the amount of functional glucose-6-phosphate dehydrogenase appears to make it more difficult for this parasite to invade red blood cells. G6PD deficiency (glucose-6-phosphate dehydrogenase deficiency) occurs most frequently in areas of the world where malaria is common.
More than 400 different mutations have been found in individuals with G6PD deficiency. Mutations, with the exception of the G6PD A mutation, are associated with more or less enzyme deficiency, but never with complete enzyme deficiency, which is not compatible with life. The disorder has been classified into variants based upon the degree of deficiency and associated clinical symptoms.
In many cases, a mutation occurs as a new (sporadic or de novo) mutation, which means that in these cases the gene mutation has occurred at the time of the formation of the egg or sperm for that child only, and no other family member will have the mutation. In cases with a family history, the G6PD gene mutation is inherited in an X-linked manner.
G6PD deficiency inheritance
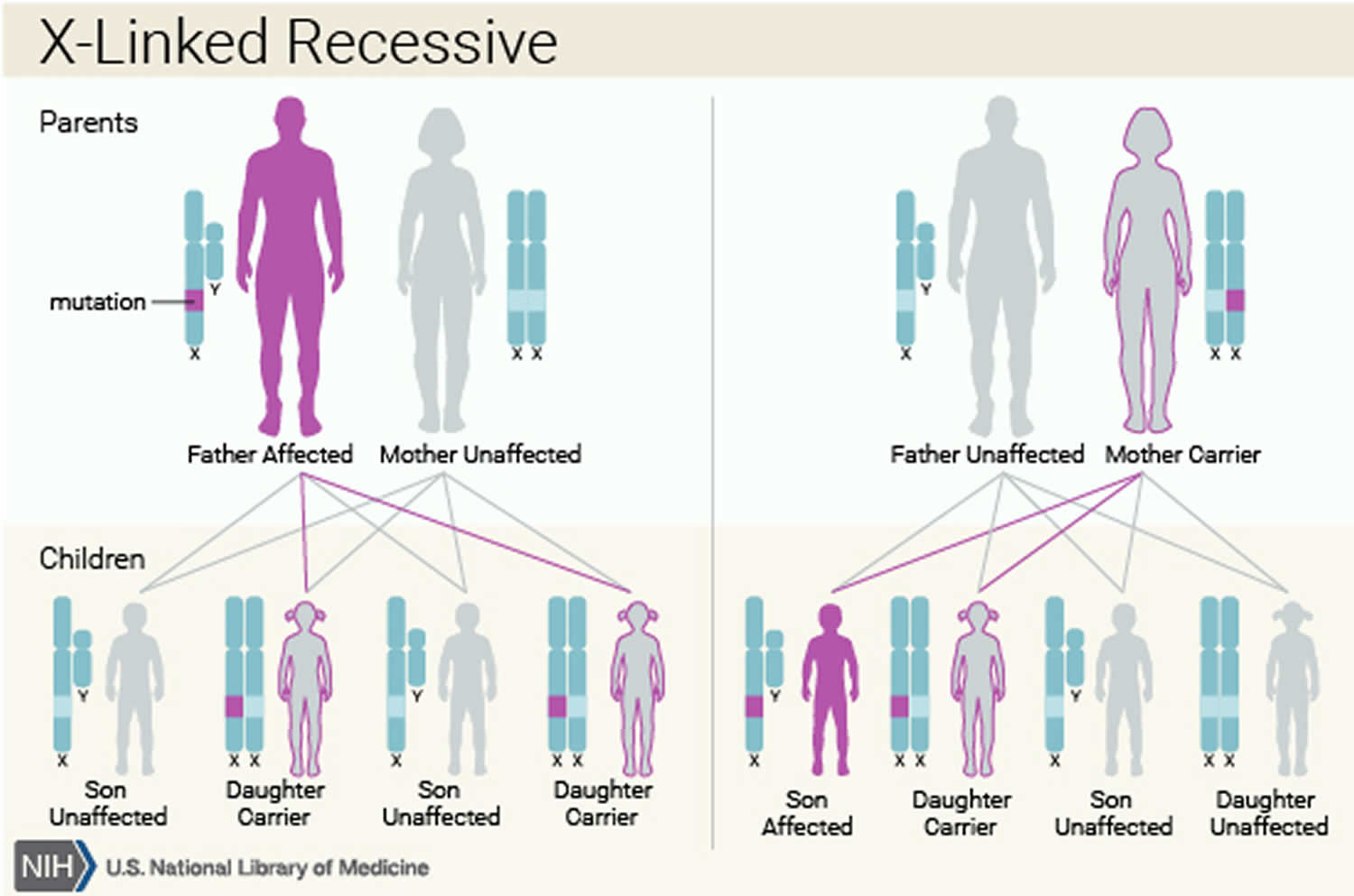
G6PD deficiency is inherited in an X-linked recessive manner. The G6PD gene associated with G6PD deficiency is located on the X chromosome, which is one of the two sex chromosomes. X-linked recessive conditions are much more common in males, who have only one X chromosome (and one Y chromosome). Females have two X chromosomes, so if they have a mutation on one of them, they still have one X chromosome without the mutation. Females with one X chromosome mutation are known as carriers and are usually unaffected. However, females can be affected if they have a mutation in both copies of the G6PD gene, or in some cases, if they have only one mutation. Females with one mutation may have lower G6PD activity than would normally be expected due to a phenomenon called skewed lyonization 1.
If a mother is a carrier of an X-linked recessive condition and the father is not, the risk to each child depends on whether the child is male or female.
- Each son has a 50% chance to be unaffected, and a 50% chance to be affected
- Each daughter has a 50% chance to be unaffected, and a 50% chance to be a carrier
A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons. If a father has G6PD deficiency and the mother is not a carrier, all sons will be unaffected, and all daughters will be carriers.
There is nothing either parent can do, before or during a pregnancy, to cause a child to have this condition.
Figure 1. G6PD deficiency X-linked recessive inheritance
G6PD deficiency diagnosis
A diagnosis is based upon the identification of characteristic physical findings and symptoms, a thorough clinical evaluation, a detailed patient history, and/or specialized tests. If a person experiences symptoms, e.g. blood in the urine, and spontaneously reports eating fava beans and comes from an area or a population where G6PD deficiency is common, suspicion of the disorder should be high.
If doctors suspect a person has G6PD deficiency, they will conduct a variety of blood tests to confirm a diagnosis and rule out other conditions that cause similar conditions. The diagnosis depends upon demonstrating decreased activity of the G6PD enzyme through either a quantitative assay or a screening test such as fluorescent spot test
Molecular genetic testing can detect mutations in the specific gene known to cause G6PD, but is available only as diagnostic service at specialized laboratories.
Diagnosis of G6PD deficiency depends on the demonstration of decreased enzyme activity through either a quantitative assay or a screening test. Assay of the enzyme is generally carried out by measuring the rate of reduction of NADP+ to NADPH in an ultraviolet spectrophotometer. Several acceptable visual screening tests have been described, and the prepared reagents for carrying out some of these procedures are commercially available. Although detection of enzyme deficiency in the healthy, fully affected (hemizygous) male presents no problem and can be achieved readily through either assay or screening tests, difficulties arise when a patient with G6PD deficiency of the A– type has undergone a hemolytic episode. As the older, more enzyme-deficient cells are removed from the circulation and are replaced by young cells, the level of the enzyme begins to increase toward normal. Under such circumstances, suspicion that the patient may be G6PD-deficient should be raised by the fact that enzyme activity is not increased, even though the reticulocyte count is elevated. Centrifugation of the blood followed by testing of the most dense (oldest) red cells has been employed as a means for the detection of G6PD deficiency in persons with the A– defect who have recently undergone hemolysis. It is helpful to carry out family studies or to wait until the circulating red cells have aged sufficiently to betray their lack of enzyme.
Even greater difficulties are encountered in attempting to diagnose the heterozygous enzyme deficiency state: The presence of a population of normal red cells coexisting with the deficient cells may mask the enzyme deficiency when screening tests are used. Even enzyme assays on heterozygous females may frequently be in the normal range. Here methods that depend upon histochemical demonstration of individual red cell enzyme activity may be useful. In addition, the ascorbate-cyanide test, in which screening is carried out on a whole cell population rather than on a lysate, may be more sensitive than the other screening procedures.
The identification of specific G6PD variants requires the use of relatively sophisticated biochemical techniques. The enzyme must be partially purified and then its Km for NADP+ and glucose 6-phosphate, utilization of substrate analogs, pH optima, and electrophoretic mobility must be determined in standard systems. Detailed characterization of G6PD variants is of value chiefly in appraising the role of G6PD deficiency in the etiology of nonspherocytic hemolytic anemia. For example, if a black male with chronic hemolysis is found to have a common A– variant of G6PD, it is necessary to seek elsewhere for a source of hemolysis. On the other hand, if an unusual thermolabile variant is found, it is more likely that the G6PD deficiency plays an etiologic role in the hemolytic process.
G6PD deficiency treatment
Most affected individuals do not require treatment. The most important aspect of management for G6PD deficiency is to avoid agents that might trigger an attack.
Some adults may need short-term treatment with fluids to prevent hemodynamic shock (in which there is inadequate supply of blood to the organs) or, in severe cases where the rate of hemolysis is very rapid, even blood transfusions or an exchange transfusion may be required. Blood transfusions are more likely to be indicated in children than adults, and in children with favism can prove life-saving. In areas where G6PD deficiency is common, care must be taken to avoid giving G6PD deficient blood to the patient.
Neonatal jaundice is treated by placing the infant under special lights (bili lights) that alleviate the jaundice. In more severe cases, an exchange transfusion may be necessary. This procedure involves removing an affected infant’s blood and replacing it with fresh donor blood or plasma.
Genetic counseling may be of benefit for patients and their families.
References- Glucose-6-phosphate dehydrogenase deficiency. https://rarediseases.info.nih.gov/diseases/6520/glucose-6-phosphate-dehydrogenase-deficiency
- Glucose-6-phosphate dehydrogenase deficiency. https://medlineplus.gov/ency/article/000528.htm
- Andre M.N. Renzaho, Eliette Husser, Michael Polonsky. Should Blood Donors Be Routinely Screened for Glucose-6-Phosphate Dehydrogenase Deficiency? A Systematic Review of Clinical Studies Focusing on Patients Transfused With Glucose-6-Phosphate Dehydrogenase–Deficient Red Cells. Transfusion Medicine Reviews. January, 2014; 28(1):7-17.
- Erythrocyte disorders: Anemias due to increased destruction of erythrocytes with enzyme deficiencies. https://www.g6pd.org/en/G6PDDeficiency/ResearchPapers/Beutler_01.aspx
- Recht J, Ashley EA, White NJ. Use of primaquine and glucose-6-phosphate dehydrogenase deficiency testing: Divergent policies and practices in malaria endemic countries. Sinnis P, ed. PLoS Neglected Tropical Diseases. 2018;12(4):e0006230. doi:10.1371/journal.pntd.0006230. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5908060
- Testing for G6PD deficiency for safe use of primaquine in radical cure of P. vivax and P. ovale. October 2016 http://apps.who.int/iris/bitstream/handle/10665/250297/WHO-HTM-GMP-2016.9-eng.pdf;jsessionid=2ACF1C68330D74D5493330A5E8837728?sequence=1
- Guidelines for the treatment of malaria, 3rd edition. Geneva: World Health Organization; 2015. http://apps.who.int/iris/bitstream/handle/10665/162441/9789241549127_eng.pdf?sequence=1
- Glucose-6-phosphate dehydrogenase deficiency. https://ghr.nlm.nih.gov/condition/glucose-6-phosphate-dehydrogenase-deficiency





{kind=link}