What is Gardner syndrome
Gardner syndrome is a subtype of familial adenomatous polyposis, an autosomal dominant genetic disease, that is characterized by combined presence of multiple intestinal polyps (colorectal polyps) and various types of tumors, both benign (noncancerous) and malignant (cancerous) and extraintestinal manifestations 1. The extraintestinal manifestations include multiple osteomas, desmoids, epidermoid cysts, connective tissue tumors, thyroid carcinomas, and hypertrophy of the pigmented epithelium of the retina 2. Osteoma is a benign neoplasm of bone tissue characterized by slow continuous growth that usually affects the long bones and cranial bones and is a major symptom for Gardner syndrome.
The prevalence of Gardner syndrome is 1 per one million people within the United States, with an incidence of 1 in 8000 individuals 3. Most commonly, Gardner syndrome patients present with epidermoid cysts.
Clinical features of Gardner syndrome can be divided into two types: cutaneous and non-cutaneous.
The most noticeable cutaneous feature of Gardner syndrome is the appearance of epidermoid cysts. These cysts can be differentiated from ordinary epidermoid cysts by the following factors:
- Epidermoid inclusion cysts of Gardner syndrome (50–65%) occur at an earlier age (around puberty) than ordinary cysts
- Epidermoid cysts occur in less common locations such as the face, scalp and extremities compared to ordinary cysts
- Cysts tend to be multiple in over half of the patients with Gardner syndrome
- As with ordinary epidermoid cysts, cysts in Gardner syndrome are usually asymptomatic (without symptoms), however in some cases they may be pruritic (itchy) and/or inflamed, and they may rupture.
- Sometimes the cysts have hybrid features with pilomatricoma-like histopathology
Other cutaneous features include desmoid cysts, fibromas, lipomas, leiomyomas, neurofibromas and pigmented skin lesions.
Non-cutaneous features include:
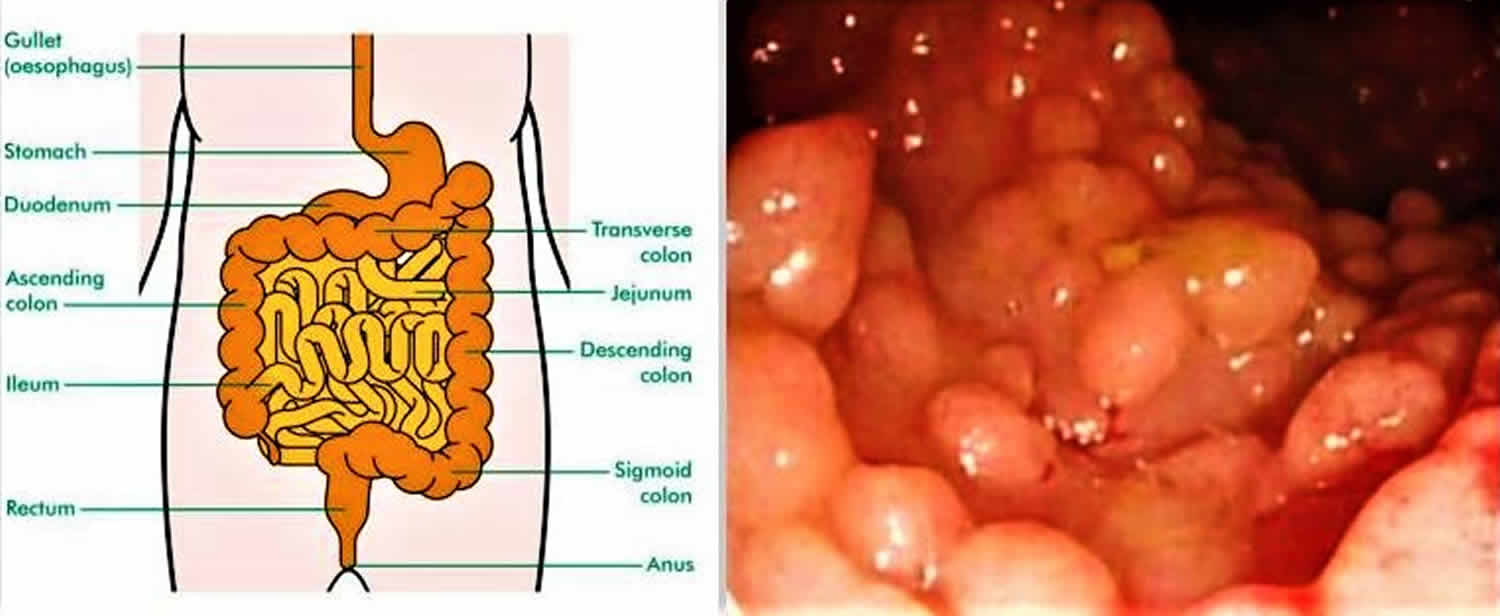
- Gastrointestinal polyps that nearly always transform into colonic adenocarcinomas (colon cancer).
- Osteomas – these benign bone tumours are essential in making the diagnosis of Gardner syndrome. They occur most commonly in the mandible (jawbone) but may also grow in the skull and long bones.
- Dental abnormalities – as well as osteomas in the jaw there may be other dental abnormalities such as unerupted extra teeth and caries
- Multifocal pigmented lesions of the fundus in the eye – seen in 80% of patients. These lesions may be present shortly after birth and can be the first marker of the disease.
Typically, patients with Gardner syndrome may present with osteomas of the mandible and skull, epidermal cysts, or fibromatosis 3. These manifestations are often found to be asymptomatic but may present with pruritus, inflammation, and rupture. Various non-cutaneous manifestations also exist with Gardner syndrome. A characteristic finding is bilateral, multiple, pigmented, ocular fundus lesions, known as congenital hypertrophy of the retinal epithelium. The development of intestinal polyposis and colorectal adenocarcinoma are key features of Gardner syndrome. Other neoplasms have also been found, such as duodenal carcinomas around the ampulla of Vater, hepatoblastoma, adrenal adenomas, papillary or follicular thyroid cancer.
People affected by Gardner syndrome have a high risk of developing colorectal cancer at an early age. They are also at an increased risk of developing other familial adenomatous polyposis-related cancers, such as those of the small bowel, stomach, pancreas, thyroid, central nervous system, liver, bile ducts, and/or adrenal gland. Other signs and symptoms of Gardner syndrome include dental abnormalities; osteomas (benign bone growths); various skin abnormalities such as epidermoid cysts, fibromas (a benign tumor of the connective tissue), and lipomas; and desmoid tumors. It is caused by changes (mutations) in the APC gene and inherited in an autosomal dominant manner. Although there is no cure for Gardner syndrome, management options are available to reduce the risk of cancer. These may include high risk screening, prophylactic surgeries and/or certain types of medications 4.
Figure 1. Gardner syndrome teeth
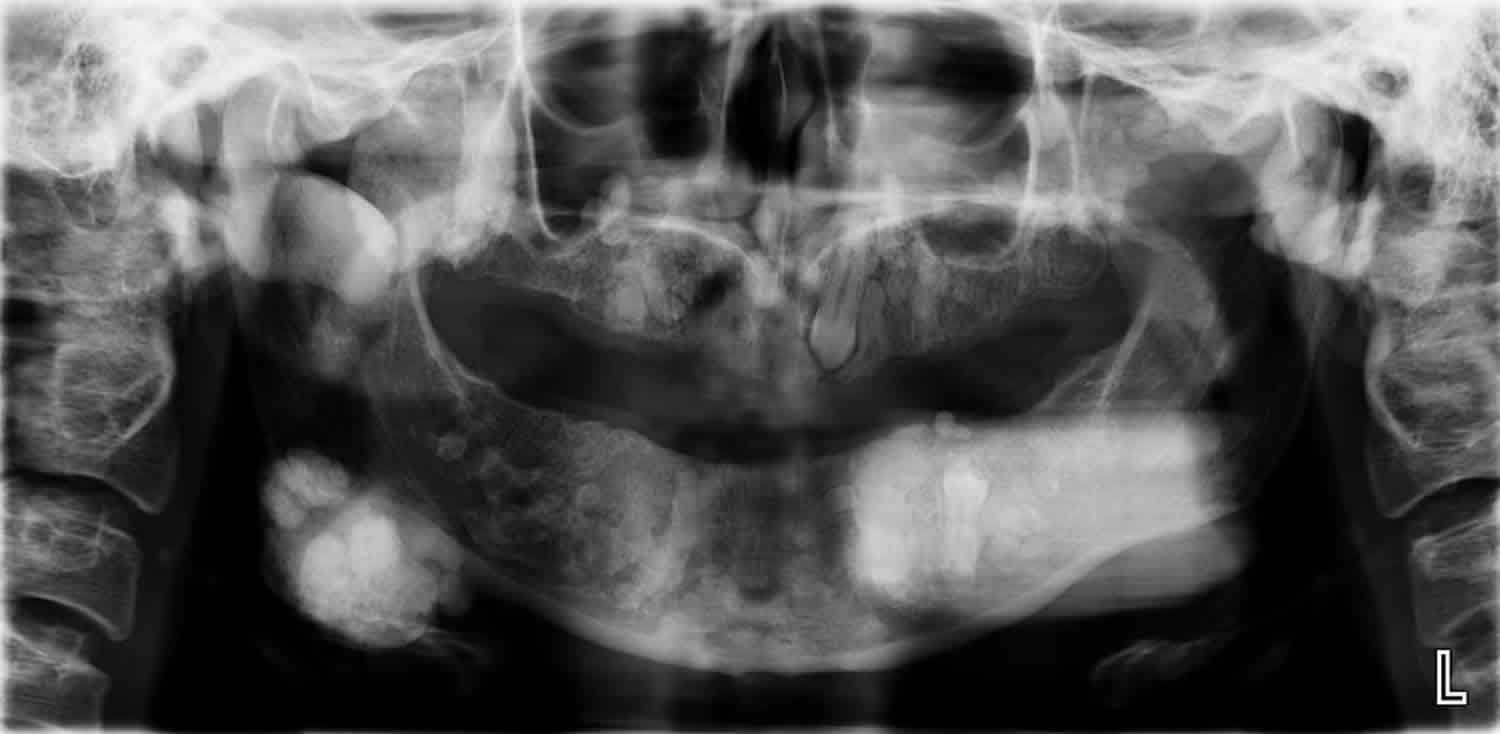
Footnote: There are unerupted teeth present with impacted left maxilla tooth (possibly canine) and left mandibular premolar. There are innumerable sclerotic lesions in the mandible and maxilla bilaterally, including pedunculated lesions in the right maxilla peripherally and right posterior body/ angle of mandible. Larger lesions are also seen in the left body of mandible and right mandibular ramus. Findings are suggestive of multiple osteomas. Malignant degeneration cannot be excluded and if this is a clinical concern then cross sectional imaging could be performed
[Source 5 ]Figure 2. Gardner syndrome with multiple osteomas of the mandible, facial and skull bones
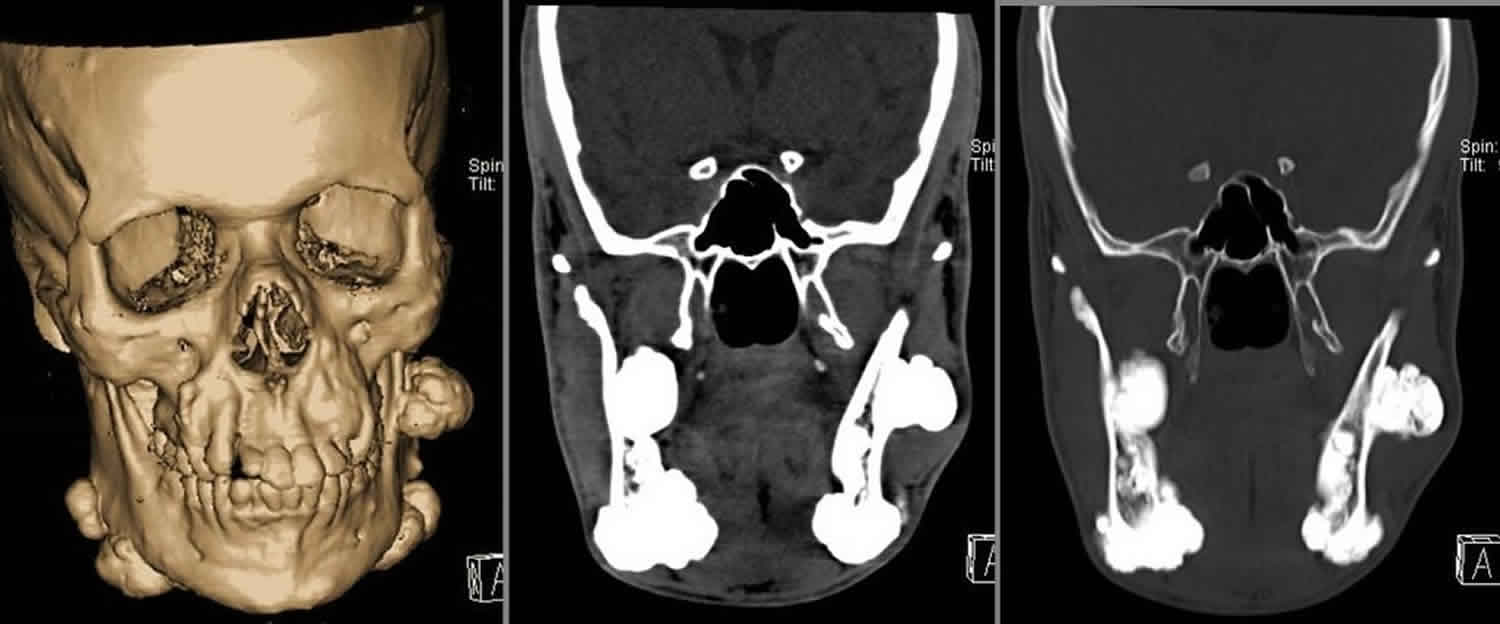
Footnote: Multiple variably sized osteomas are arising from the frontal and pareital bones (more numerous on the right) and the facial bones, namely the maxillae and zygomata, the orbital roofs (larger on the left) and the mandible. The largest osteomas are arising from the mandible. Multiple osteomas are seen within ethmoidal air cells (more numerous on the left). Multiple maxillary and mandibular supernumerary teeth are noted.
[Source 6 ]What is Gardner Diamond syndrome?
Gardner-Diamond syndrome is unrelated to the Gardner syndrome. Gardner-Diamond syndrome is a rare condition characterized by episodes of unexplained, painful bruising that mostly occurs on the arms, legs, and/or face 7. Gardner-Diamond syndrome is most common in Caucasian women who have mental illness or emotional stress. Symptoms typically include the formation of multiple, small, purple bruises that may be associated with burning, redness and swelling. Most affected people report that the bruising occurs either spontaneously, or some time after trauma or surgery at other sites of the body. The cause of Gardner-Diamond syndrome is poorly understood. Management typically involves psychiatric treatment 8.
Is genetic testing available for Gardner syndrome?
Genetic testing is available for APC, the gene known to cause Gardner syndrome. Carrier testing for at-risk relatives and prenatal testing are possible if the disease-causing mutation in the family is known. Because colon screening for those at risk for Gardner syndrome begins as early as age ten years, genetic testing is generally offered to children by this age.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counsellors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
- The National Cancer Institute provides a Cancer Genetics Services Directory (https://www.cancer.gov/about-cancer/causes-prevention/genetics/directory), which lists professionals who provide services related to cancer genetics. You can search by type of cancer or syndrome, location, and/or provider name.
Is Gardner syndrome associated with joint issues?
Joint issues are not considered a feature of Gardner syndrome. However, several studies have found that joint symptoms sometimes develop in people who have undergone a restorative proctocolectomy. Restorative proctocolectomy is often performed in people affected by ulcerative colitis or familial adenomatous polyposis (FAP). Gardner syndrome is considered a form of familial adenomatous polyposis (FAP) 9.
Does Gardner syndrome cause exhaustion?
Exhaustion is not considered a direct feature of Gardner syndrome. However, treatment of Gardner syndrome includes complete removal of the colon, which causes a change in bowel and dietary habits. This change can alter the absorption of nutrients and potentially lead to vitamin deficiencies that may impact energy levels 10.
I have Gardner syndrome with a bony growth in my neck that is completely surrounding a nerve. Where can I find information and help about this?
The bony growths associated with Gardner syndrome are called osteomas. They usually develop in the skull or jaw bone (the mandible), but can occur in any bone of the body. They do not spread to other parts of the body, which means they are considered a benign tumor (not malignant). Osteomas usually do not cause symptoms or health problems. However, an osteoma can be removed by surgery for cosmetic reasons or if it begins to interfere with normal movement or function 4.
Gardner syndrome causes
Gardner syndrome is caused by changes (mutations) in the APC gene located on chromosome 5, which is called a “tumor suppressor gene.” Tumor suppressor genes encode the APC proteins that are part of the system that controls cell division, regulation, and growth. APC gene is specifically involved in controlling how often a cell divides and the manner of attachment of the cell to other cells within the tissue and the cell polarization and the morphology of some of its structures. APC gene also serves as a determinant of the cell’s mobility and direction in terms of chromosomal movement during cell division. Specifically, the protein beta-catenin, controlled by the APC gene, prevents those proteins responsible for stimulating cell division from being overly activated, thereby, preventing cell overgrowth and proliferation. Frequently it is the inactivation of the APC gene and the subsequent, inactivation of beta-catenin that is observed in cancerous cells.
Mutations in the APC gene lead to uncontrolled cell growth which results in the development of the colon polyps, tumors and cancers that can be associated with Gardner syndrome. The symptoms found in each person and the severity of the condition depend on which part of the APC gene is mutated.
In addition to such genetic mutations, a loss of DNA methylation, a mutation of the RAS gene on chromosome 12, a deletion of the colon cancer gene on chromosome 18, as well as, a mutation in TP53 gene located on chromosome 17 have also been linked as a possible cause of Gardner syndrome 3.
Inheritance pattern
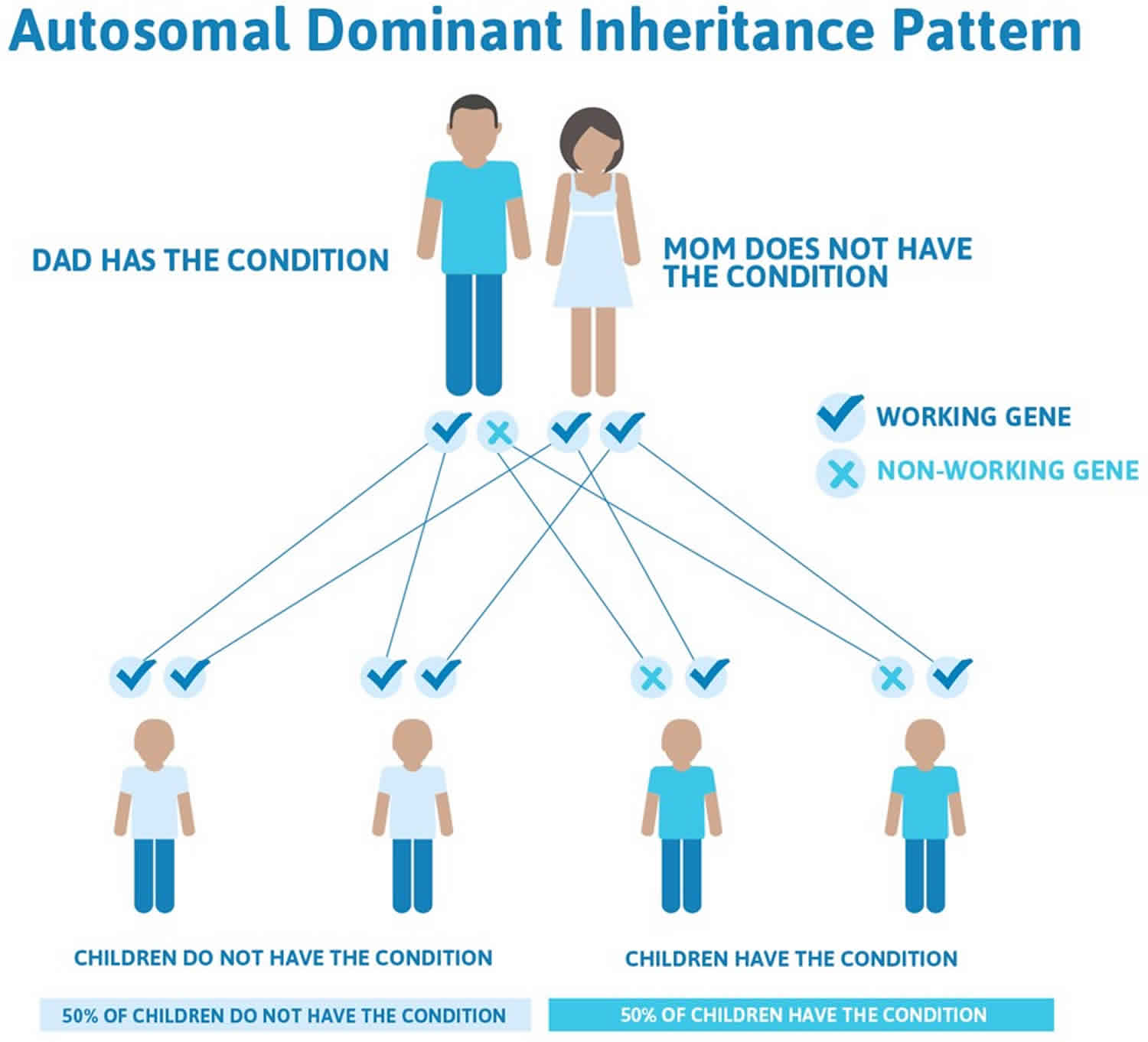
Gardner syndrome is inherited in an autosomal dominant manner 11. This means that to be affected, a person only needs a change (mutation) in one copy of the responsible gene in each cell. In some cases, an affected person inherits the mutation from an affected parent. Other cases may result from new (de novo) mutations in the gene. These cases occur in people with no history of the disorder in their family. A person with Gardner syndrome has a 50% chance with each pregnancy of passing along the altered gene to his or her child.
Figure 3. Gardner syndrome autosomal dominant inheritance pattern
Gardner syndrome symptoms
The signs and symptoms of Gardner syndrome vary from person to person. Gardner syndrome is a form of familial adenomatous polyposis (FAP), which is characterized primarily by hundreds to thousands of noncancerous (benign) polyps in the colon that begin to appear at an average age of 16 years. Unless the colon is removed, these polyps will become malignant (cancerous), leading to early-onset colorectal cancer at an average age of 39 years 4.
The following are symptoms commonly associated with Gardner syndrome: patients may be asymptomatic or self-report cramping, diarrhea, rectal bleeding, constipation secondary to the obstruction, and/or vomiting 3. In addition, multiple gastrointestinal polyps (typically presenting in the colon) may be observed, developing extra teeth, cysts may develop under the skin, bony tumors on bones, such as the skull, as well as, fibromas. The risk of developing colon cancer is also much greater in a Gardner syndrome patient.
Other features of Gardner syndrome may include:
- Dental abnormalities
- Fundic gland or adenomatous polyps of the stomach
- Adenomatous polyps of the small intestines
- Osteomas (benign bone growths)
- Congenital hypertrophy of the retinal pigment epithelium (a flat, pigmented spot within the outer layer of the retina)
- Benign skin abnormalities such as epidermoid cysts, fibromas (a benign tumor of the connective tissue), and lipomas
- Adrenal masses
- Desmoid tumors
- Other types of cancer (small bowel, stomach, pancreas, thyroid, central nervous system, liver, bile ducts, and/or adrenal gland)
Gardner syndrome prognosis
The long-term outlook (prognosis) for people with Gardner syndrome depends on the signs and symptoms present in each person and the age of diagnosis. By age 35 years, 95% of affected people have polyps. Once they appear, the polyps rapidly increase in number. Without colectomy, hundreds to thousands of colon polyps are typically observed and colon cancer is inevitable. The average age of colon cancer diagnosis in untreated individuals is 39 years (range 34-43 years) 4. However, with early diagnosis and high-risk management, the prognosis is good. In fact, the 5-year survival rate for patients who undergo protocolectomy is nearly 100%. If alternative surgeries are performed that remove the colon but not the rectum, the recurrence rate is 30% in 20 years and 45% in 30 years; continued surveillance of the remaining rectum is, therefore, imperative 12.
The skin abnormalities (epidermoid cysts, fibromas, and lipomas) associated with Gardner syndrome are mainly of cosmetic concern, as they do not appear to become malignant (cancerous). Osteomas (bony growths) do not usually cause medical problems and do not become malignant. Although desmoid tumors are typically benign (noncancerous), they have a tendency to invade surrounding tissues and/or compress nearby organs and can be very difficult to remove 4.
Gardner syndrome diagnosis
Gardner syndrome can be diagnosed via a medical examination and discussion of medical and familial history, self-reported symptoms, endoscopy of the lower gastrointestinal (GI) tract, and through the use of molecular testing of the blood for screening of genetic mutations known to be linked to the disease. Pre-natal genetic testing can also be performed, and if positive, endoscopic screenings for polyps can begin as early as 10 years of age.
Radiological studies are essential for patients and family members with suspected Gardner syndrome.
- Images of the long bone may show up osteomas.
- Images of the mandible at an early age may show up subtle defects.
- Eye exams at an early age can detect pigmented lesions of the fundus.
- Colonoscopy and other invasive tests to check for polyp involvement every 1-2 years.
Gardner syndrome treatment
Although there is no cure for Gardner syndrome, prevention, treatment and management options are available to reduce the risk of cancer. Prevention is the most common approach to treatment for those individuals who are aware of the respective familial inheritance or once this is discovered. This type of treatment protocol can include a healthy diet, the use of nonsteroidal anti-inflammatory drug (NSAIDs) such as sulindac, or a COX2 inhibitor like celecoxib which can aid in the stifling of the growth of polyps in the colon, especially since it is well known that Gardner syndrome patients are at a greater risk for developing colon cancer. Surveillance via lower gastrointestinal endoscopies is also recommended to monitor polyp growth and development and to ascertain their transformation into malignant tumors. If more than 20 or more polyps are present, removal of the colon is recommended to reduce the risk for colon cancer development.
Affected people typically undergo regular screening for the various polyps and tumors associated with Gardner syndrome to permit early diagnosis and treatment. This screening regimen may include 13, 4:
- Sigmoidoscopy or colonoscopy every one to two years, beginning at age ten to 12 years. Once polyps are detected, colonoscopy is recommended annually until colectomy (removal of colon).
- Esophagogastroduodenoscopy beginning by age 25 and repeated every one to three years.
- Annual physical examination, including a thorough thyroid evaluation beginning in the late teenage years.
- Screening for desmoid tumors and hepatoblastoma (a specific type of liver cancer that is diagnosed in young children) may also be recommended in some people.
A colectomy is usually recommended when more than 20 or 30 polyps and/or multiple advanced polyps are identified. Sulindac, a nonsteroidal anti-inflammatory drug (NSAIDs), is sometimes prescribed in people with Gardner syndrome who have had a colectomy to treat polyps in the remaining rectum 14.
Treatment for desmoid tumors varies depending on the size and location of the tumor, but may include watchful waiting, surgery, NSAIDS, anti-estrogen medications, chemotherapy and/or radiation therapy 4. Osteomas (bony growths) may be removed for cosmetic reasons – if they are severely deforming or if they are a nuisance 4. Treatment of epidermoid cysts in Gardner syndrome is similar to that used for ordinary cysts and involves excision 15. Occasionally intralesional steroid injections may be used if the cysts are inflamed.
References- Gardner syndrome. https://rarediseases.info.nih.gov/diseases/6482/gardner-syndrome
- Gardner syndrome: presurgical planning and surgical management of craniomaxillofacial osteomas. J Craniofac Surg. 2011 May;22(3):946-8. doi: 10.1097/SCS.0b013e31821028a6.https://www.ncbi.nlm.nih.gov/pubmed/21558892
- Charifa A, Zhang X. Gardner Syndrome. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482342
- Jasperson KW, Patel SG, Ahnen DJ. APC-Associated Polyposis Conditions. 1998 Dec 18 [Updated 2017 Feb 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1345
- Gardner syndrome. https://radiopaedia.org/cases/gardner-syndrome
- Gardner syndrome. https://radiopaedia.org/cases/gardner-syndrome-2
- Gardner-Diamond syndrome. https://rarediseases.info.nih.gov/diseases/6481/gardner-diamond-syndrome
- Benjamin P Geisler, Bruce J Dezube. Psychogenic purpura (Gardner-Diamond syndrome). UpToDate. Waltham, MA: UpToDate; December, 2015
- Thomas PD, Keat AC, Forbes A, Ciclitira PJ, Nicholls RJ.. Extraintestinal manifestations of ulcerative colitis following restorative proctocolectomy. Eur J Gastroenterol Hepatol. 1999 Sep;11(9):1001-5.. September 1999; 11(9):1001-1005.
- Almendingen K, Fausa O, Høstmark AT, Bratlie J, Mørkerid L, Aabakken L, Vatn MH. Serum nutrients and habitual dietary intake in colectomized FAP patients in Norway. European Journal of Nutrition. 2009; 48:129-136.
- Jasperson KW, Patel SG, Ahnen DJ. APC-Associated Polyposis Conditions. 1998 Dec 18 [Updated 2017 Feb 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1345/
- Gardner syndrome. https://emedicine.medscape.com/article/190486-overview
- New NCCN Guidelines® Now Available for Genetic/Familial High-Risk Assessment: Colorectal. https://www.nccn.org/about/news/ebulletin/ebulletindetail.aspx?ebulletinid=294
- Familial Adenomatous Polyposis. https://rarediseases.org/rare-diseases/familial-adenomatous-polyposis/
- Gardner syndrome. https://www.dermnetnz.org/topics/gardner-syndrome/





{kind=link}