What are glucocorticoids
Glucocorticoids are corticosteroid hormones, which are a class of essential steroid hormones, secreted from the adrenal glands, that are essential for life and are important in the regulation of adaptation to stress, carbohydrate-, protein-, fat-, calcium- and bone-metabolism, immune function, growth and behavioral regulation by acting as end products of the stress-responsive hypothalamic-pituitary-adrenal axis 1. Glucocorticoids are essential to life and after removal of both adrenals humans will not survive for long without glucocorticoid replacement. Naturally occurring glucocorticoids are part of the feedback mechanism your body utilizes to reduce immune activity (inflammation). Exogenous glucocorticoids (synthetic or natural) are used to treat diseases caused by an overactive immune system (e.g. allergies, asthma, autoimmune diseases & sepsis). Glucocorticoids are distinguished from mineralocorticoids and sex steroids that have different receptors, target cells and effects.
When the hypothalamus produces corticotrophin-releasing hormone (CRH), it stimulates the pituitary gland to release adrenal corticotrophic hormone (ACTH). These hormones, in turn, alert the adrenal glands to produce glucocorticoids (corticosteroid hormones).
Glucocorticoids released by the adrenal cortex include:
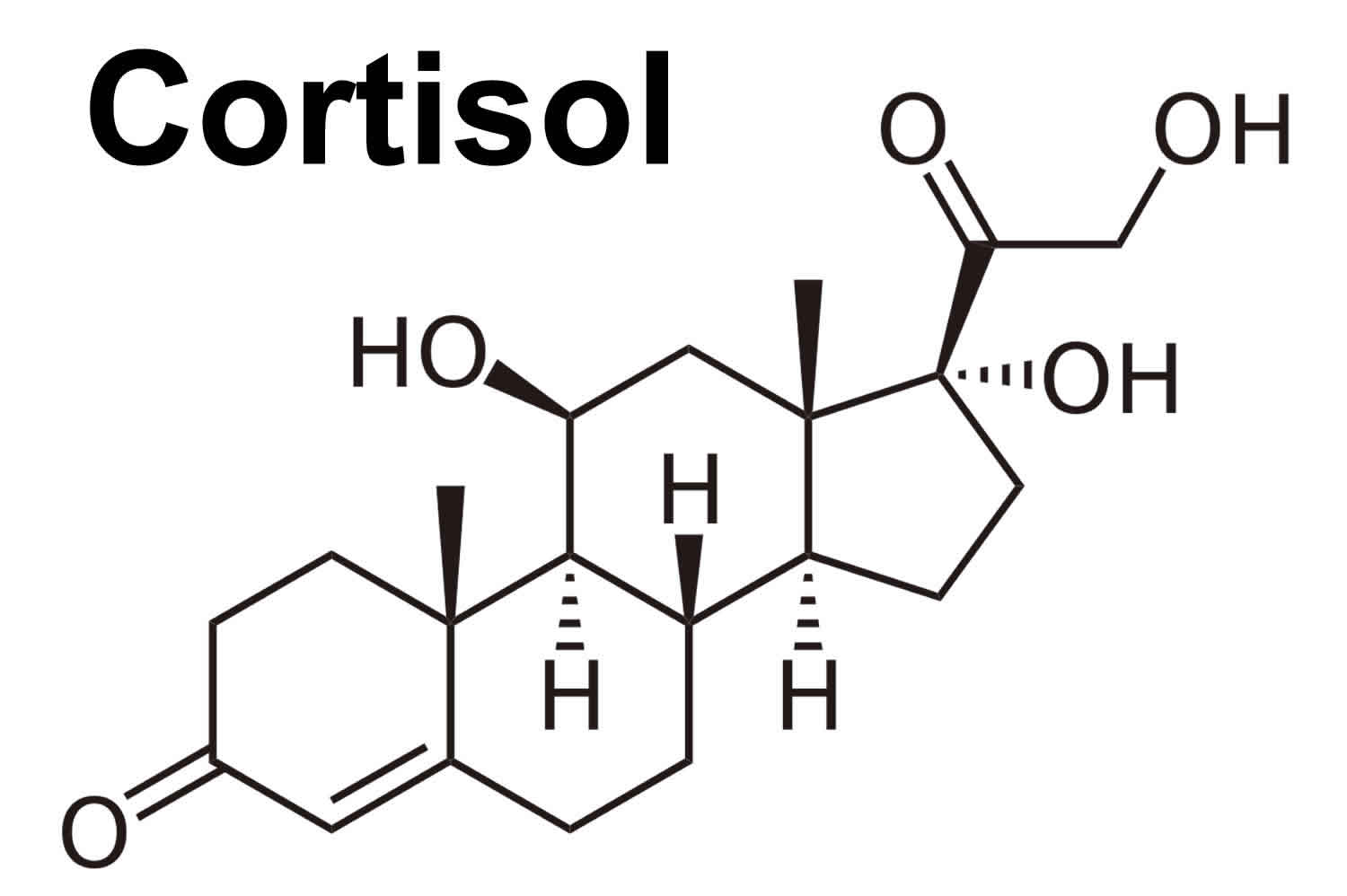
- Cortisol: When used as a medication, it is known as hydrocortisone, cortisol regulates how your body converts fats, proteins, and carbohydrates to energy. Cortisol also helps regulate blood pressure and cardiovascular function. Cortisol is produced in humans by the zona fasciculata of the adrenal cortex within the adrenal gland. Cortisol is released in response to stress and low blood-glucose concentration. Cortisol functions to increase blood sugar through gluconeogenesis, to suppress the immune system, and to aid in the metabolism of fat, protein, and carbohydrates 2. Cortisol also decreases bone formation 3.
- Corticosterone (another glucocorticoid that is only released in small amounts in human) is then converted into aldosterone (mineralocorticoid).
Figure 1. Mechanism of action of glucocorticoids
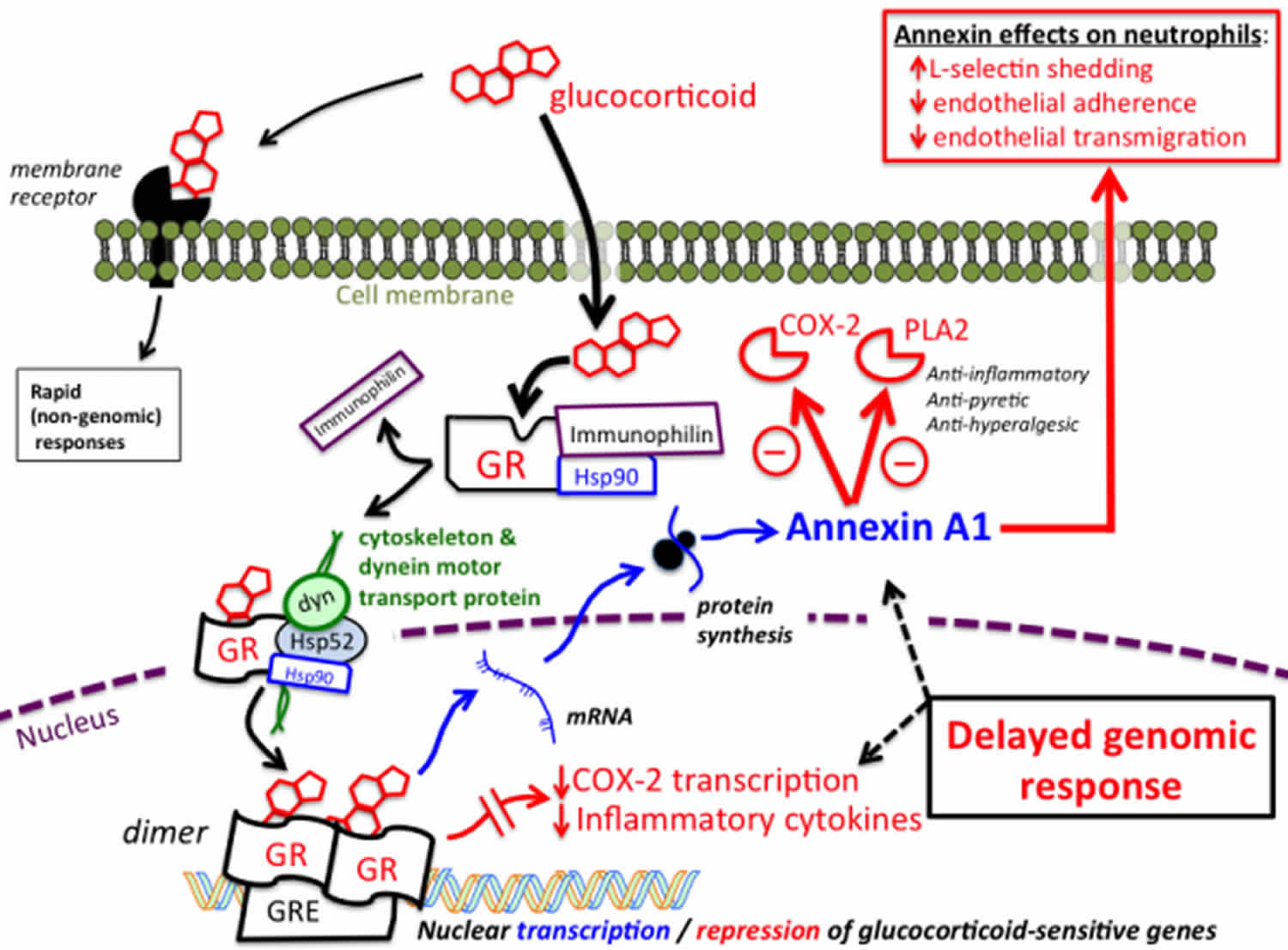
Footnote: The majority of effects produced by glucocorticoids result from initial steroid binding to intracellular glucocorticoid receptors followed by translocation to the nucleus and changes in gene transcription. In their steroid-free (unbound) state, intracellular glucocorticoid receptors (GR) are bound to stabilizing proteins that include heat-shock protein 90 (Hsp90) and immunophilin. The unbound form of the receptor is not capable of affecting gene transcription. Binding of steroid initiates a conformational change that results in an exchange of chaperone proteins, which permits attachment of the steroid-GR complex to the dynein protein trafficking pathway. This results in translocation of the steroid-glucocorticoid receptor complex from the cytoplasm into the nucleus. Once in the nucleus, the steroid-glucocorticoid receptor complex dimerizes and binds to glucocorticoid response elements (GRE) associated with the regulatory region of glucocorticoid-sensitive genes. Binding of the glucocorticoid-glucocorticoid receptor dimer either represses, or stimulates the transcription of sensitive genes, resulting in changes in synthesis of mRNA, followed by changes in protein synthesis. These steps are necessary for producing most cellular responses to glucocorticoids, and typically take hours to days to develop. Both a reduction in the synthesis of inflammatory cytokines, as well as upregulation in the synthesis of annexin A1 are known to play important roles in mediating the antiinflammatory and immunomodulatory effects of glucocorticoids. As illustrated at the top left, in some situations glucocorticoids are able to produce more rapid responses by binding to membrane-associated receptors and exerting effects that do not involve changes in gene regulation. These non-genomic mechanisms remain poorly understood.
Abbreviations: COX = Cyclooxygenase; GR = Glucocorticoid receptor; Hsp = Heat Shock Protein(s); PLA2 = Phospholipase A2.
[Source 4 ]Glucocorticoids molecules reach all tissues, including the brain, readily penetrate the cell membrane, and interact with ubiquitous cytoplasmic/nuclear glucocorticoid receptors, through which they exert markedly diverse actions 5. In pharmacologic doses, glucocorticoids are used as potent immunosuppressive agents in the management of many inflammatory, autoimmune and lympho-proliferative diseases 6. At the cellular level, actions of glucocorticoids are mediated by an intracellular receptor protein, the glucocorticoid receptor (its gene name is “nuclear receptor subfamily 3, group C, member 1: NR3C1”), which belongs to the steroid/sterol/thyroid/retinoid/orphan receptor superfamily of nuclear transactivating factors with over 200 members in general and over 40 in mammals currently cloned and characterized across species 7. Human glucocorticoid receptor consists of 777 amino acid residues 7. Glucocorticoid receptor is ubiquitously expressed in almost all human tissues and organs including neural stem cells 8. Glucocorticoid receptor functions as a hormone-dependent transcription factor that regulates the expression of glucocorticoid-responsive genes, which probably represent 3-10% of the human genome and can be influenced by the ligand-activated glucocorticoid receptor directly or indirectly 9.
The more important actions of cortisol include:
- Inhibition of protein synthesis in tissues, increasing the blood concentration of amino acids.
- Promotion of fatty acid release from adipose tissue, increasing the utilization of fatty acids and decreasing the use of glucose as energy sources.
- Stimulation of liver cells to synthesize glucose from noncarbohydrates, such as circulating amino acids and glycerol, increasing the blood glucose concentration.
These actions of cortisol help keep blood glucose concentration within the normal range between meals. This control is important, because a few hours without food can exhaust the supply of liver glycogen, a major source of glucose.
Negative feedback controls cortisol release (see Figure 5). This is much like control of thyroid hormones, involving the hypothalamus, anterior pituitary gland, and adrenal cortex. The hypothalamus secretes corticotropin-releasing hormone (CRH) into the pituitary gland portal veins, which carry CRH to the anterior pituitary, stimulating it to secrete adrenal corticotrophic hormone (ACTH). In turn, ACTH stimulates the adrenal cortex to release cortisol. Cortisol inhibits the release of corticotropin-releasing hormone (CRH) and adrenal corticotrophic hormone (ACTH), and as concentrations of these fall, cortisol production drops.
The set point of the feedback mechanism controlling cortisol secretion may change to meet the demands of changing conditions. For example, under stress—as from injury, disease, or emotional upset—information concerning the stressful condition reaches the brain. In response, brain centers signal the hypothalamus to release more corticotropin-releasing hormone (CRH), elevating the blood cortisol concentration until the stress subsides.
Is cortisol a glucocorticoid?
Yes.
Is prednisone a glucocorticoid?
Yes.
Figure 2. The pituitary gland location
Figure 3. The hypothalamus and pituitary gland (anterior and posterior) endocrine pathways and target organs
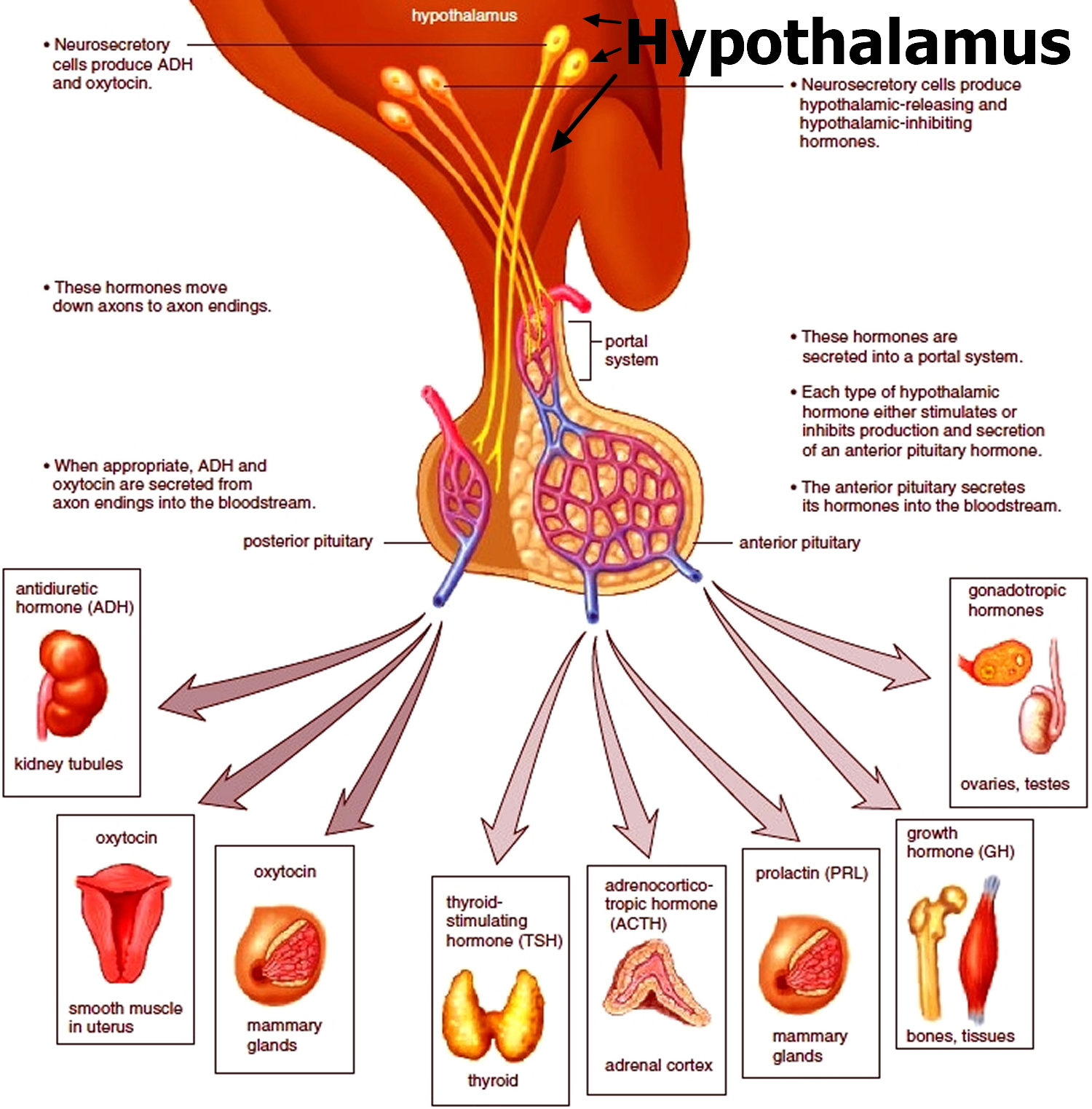
Figure 4. Adrenal gland anatomy
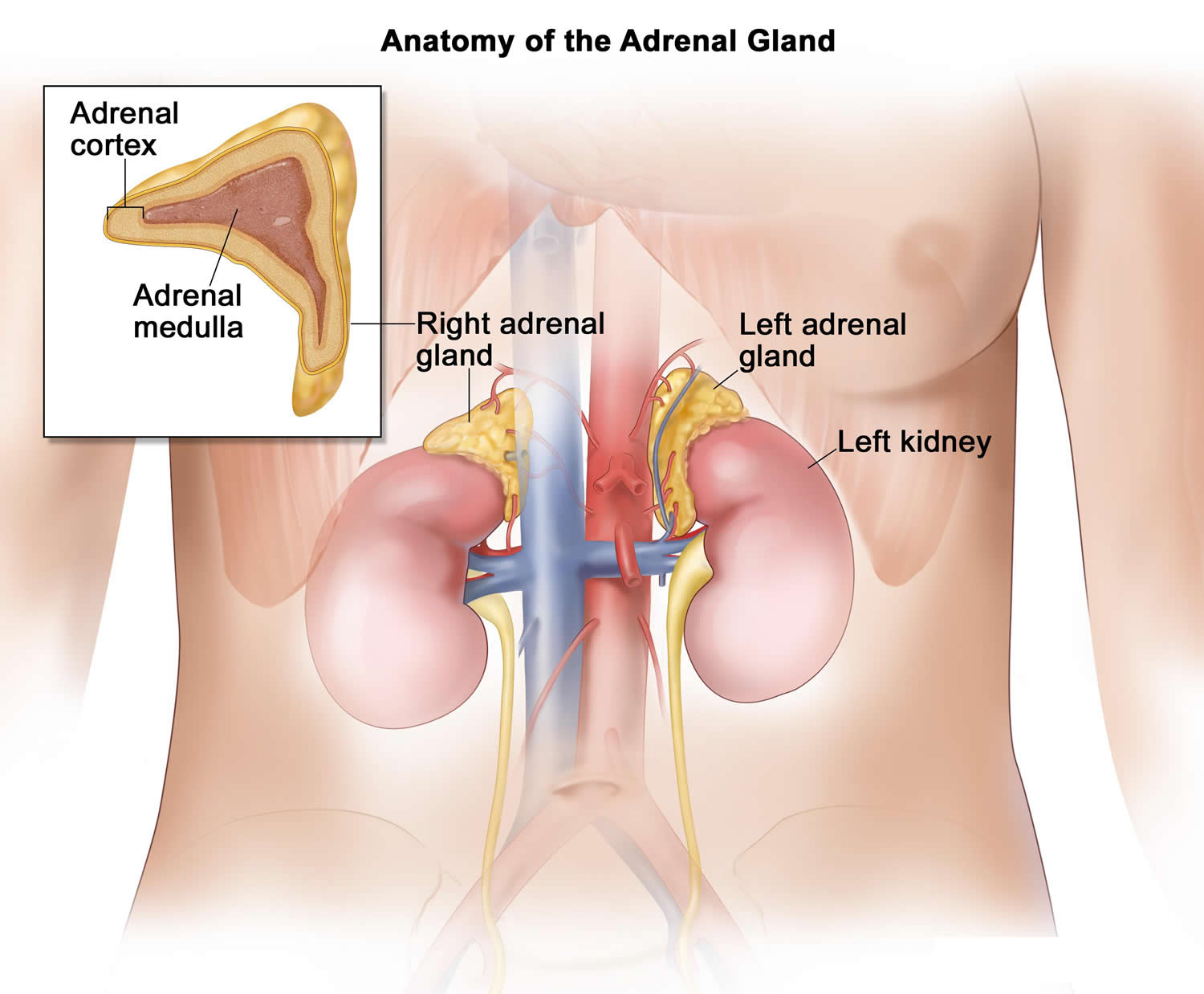
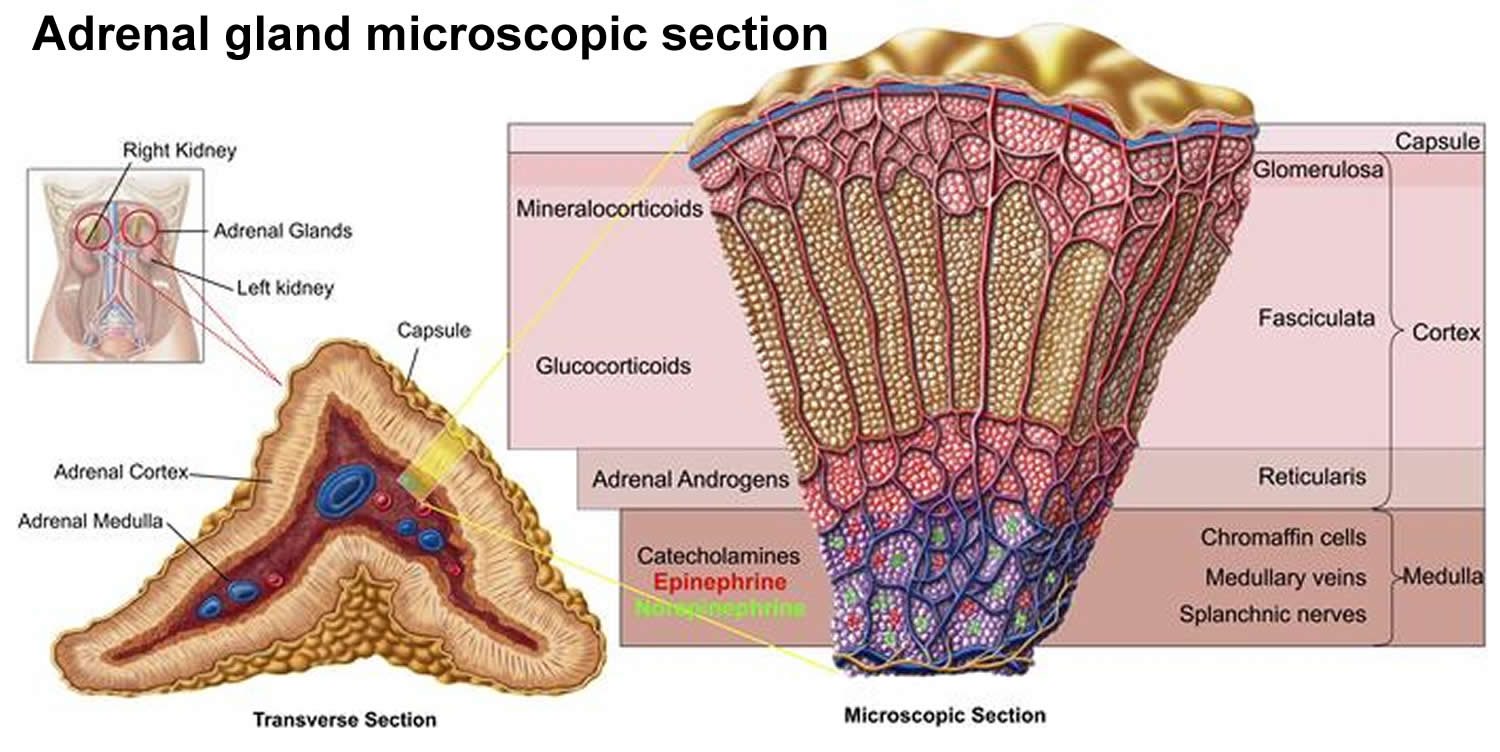
Figure 5. Negative feedback regulates cortisol secretion
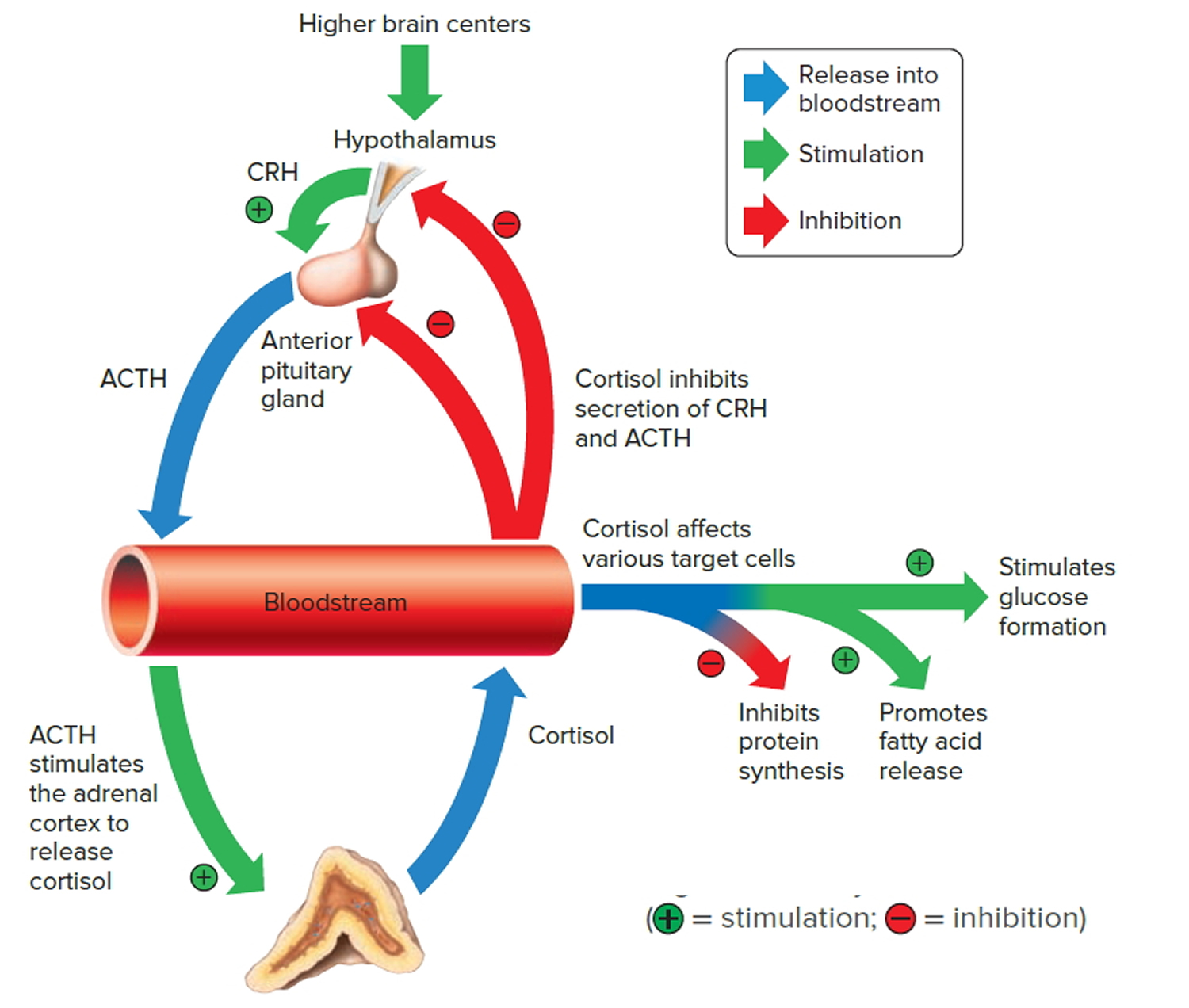 Abbreviations: CRH = corticotropin-releasing hormone; ACTH = adrenal corticotrophic hormone
Abbreviations: CRH = corticotropin-releasing hormone; ACTH = adrenal corticotrophic hormoneTable 1. Expected clinical manifestations in tissue hypersensitivity or resistance to glucocorticoids
| Affected area | Glucocorticoid excess | Glucocorticoid deficiency |
|---|---|---|
| Glucocorticoid hypersensitivity | Glucocorticoid resistance | |
| Central nervous system | Insomnia, anxiety, depression, defective cognition | Fatigue, somnolence, malaise, defective cognition |
| Liver | +Gluconeogenesis,+liposynthesis | Hypoglycemia |
| Fat | Accumulation of visceral fat (metabolic syndrome) | Loss of weight |
| Blood vessels | Hypertension | Hypotension |
| Bone | Stunted growth, osteoporosis | |
| Inflammation/immunity | Immune suppression, antiinflammation, vulnerability to certain infections and tumors | +Inflammation, +autoimmunity |
Table 2. Reported pathological states associated with a change in tissue sensitivity to glucocorticoids
| Resistance | Hypersensitivity |
|---|---|
| Familial/sporadic glucocorticoid resistance syndrome | Visceral-type obesity-related hypertension and insulin resistance |
| Bronchial asthma | |
| Rheumatoid arthritis | AIDS (HIV type-1 infection) |
| Osteoarthritis | |
| Systemic lupus erythematosus | |
| Crohn’s disease | |
| Ulcerative colitis | |
| Septic shock/acute respiratory distress syndrome |
Glucocorticoid function
Anti-inflammatory Effects and Mechanisms
Glucocorticoids reduce inflammation through a combination of both inhibition & upregulation of gene transcription, including:
- INHIBITION of genes regulating expression of:
- UPREGULATION of the expression of Annexin A1, which in turn:
Neutrophil migration through the vasculature to sites of inflammation is markedly reduced. This effect, combined with an enhanced release of cells from the bone marrow, and reduced neutrophil apoptosis, causes the white blood cell count to increase 11.
Metabolic Effects and Mechanisms
Carbohydrate and Protein Metabolism
Cortisol is released during times of stress to supply glucose as an energy substrate to organs facing stressful conditions. Stress responses that increase cortisol release include vigorous exercise, psychological stress or fear, acute trauma, surgery, pain, severe infection and hypoglycemia. Cortisol’s effects to elevate blood glucose are important in maintaining energy homeostasis during the stress response, and ensure that critical organs (such as the brain) continue to receive nutrients at a time when they are most needed for survival.
The mechanisms involved in elevating blood glucose include:
- Increased gluconeogenesis (glucose formation)
- the enzymes involved in amino acid metabolism & gluconeogenesis are upregulated
- the liver forms glucose from amino acids & fatty acids
- Reduce glucose uptake & utilization by peripheral tissues
- cortisol antagonizes the effect of insulin on peripheral tissues in order to increase blood glucose
- cortisol stimulates the translocation of glucose transporters away from the plasma membrane into the cell interior
- Increase protein breakdown (to provide amino acids for gluconeogenesis)
- protein breakdown affects multiple tissues including muscle and skin 14
- Activate lipolysis (to provide fatty acids for gluconeogenesis)
- the effects of growth hormone on hormone-sensitive lipase in adipocytes are stimulated, resulting in the release of free fatty acids.
- increased free fatty acids further increase insulin resistance.
Clinical outcomes:
- atrophy of lymphoid tissue
- decreased muscle mass
- thinning of the skin (cigarette-paper-like consistency, fragile, easy to bruise)
- hyperglycemia, worsening of diabetes
Lipid Metabolism
Chronically elevated glucocorticoids can produce a dramatic redistribution of body fat. The redistribution of fat is believed to result from different sensitivities of peripheral vs truncal fat cells to:
- the lipolytic effects of glucocorticoids themselves
- lipolytic effects of insulin that is released in response to glucocorticoid-induced hyperglycemia
Clinical outcomes:
- increased fat in the back of the neck (buffalo hump)
- increased facial fat (moon face)
- loss of fat in body extremities
Central Nervous System
Glucocorticoids can cause diverse neurological effects and behavioral changes that may reflect steroid effects on neuronal excitability. The mechanisms remain poorly understood.
Formed Elements of Blood
Administration of glucocorticoids results in diverse changes to different types of white blood cells. The number of circulating eosinophils (which makes up 2-4% of white blood cells in the blood) is typically reduced by glucocorticoids, due to increased apoptosis and sequestration of eosinophils in extravascular sites (possibly due to upregulation of the CXCR4 chemokine receptor). In direct contrast, the number of circulating neutrophils (which normally comprise 60-70% of circulating white blood cells) is increased by glucocorticoids due to a combination of decreased neutrophil tissue migration, inhibition of neutrophil apoptosis, and enhanced release of cells from the bone marrow. The effect on neutrophils typically results in a net (increased white blood cell count) 15. Certain lymphoid malignancies are also destroyed, most likely due to activation of programmed cell death 16.
Glucocorticoid resistance
Glucocorticoid resistance also named Chrousos syndrome, is a rare, sporadic or familial condition caused by mutation of the NR3C1 gene encoding the glucocorticoid receptor 17. The glucocorticoid receptor gene (NR3C1) is located on chromosome 5q31 and contains 9 exons. Glucocorticoid resistance is characterized by biochemically proven hypercortisolism without the clinical stigmata of Cushing syndrome, and by partial or generalized insensitivity to glucocorticoids 17.
Figure 5. Glucocorticoid resistance syndrome (Chrousos syndrome)
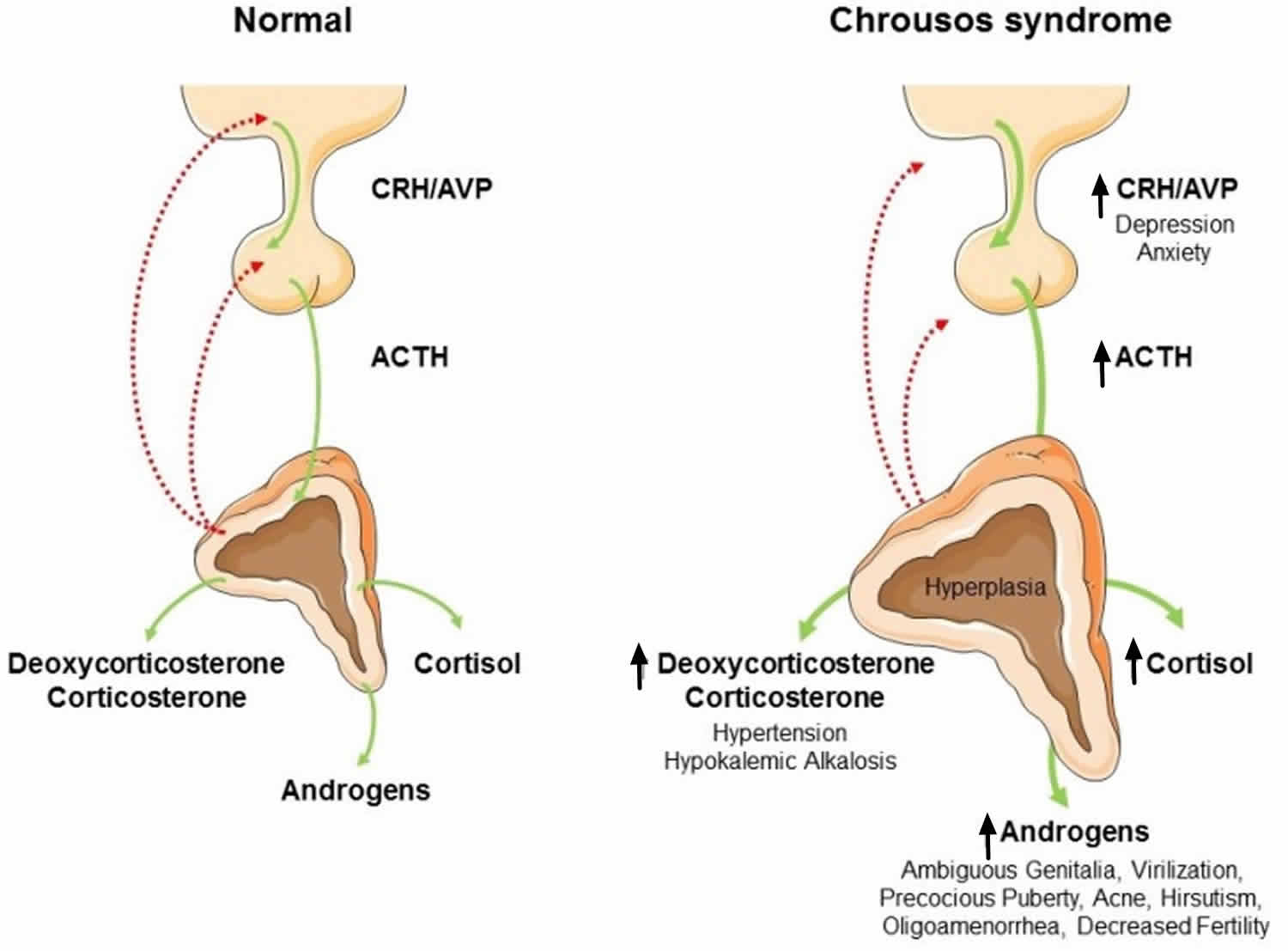
Abbreviations: CRH = corticotropin-releasing hormone; ACTH = adrenocorticotropic hormone; AVP = arginine-vasopressin
[Source 18 ]Due to this insensitivity, and thereby inadequate negative feedback, serum ACTH, and therefore cortisol production were compensatory stimulated. The chronic excess of ACTH results in an overstimulated steroid biosynthesis, including increased production of adrenal steroids with androgenic and/or mineralocorticoid activity 19. The clinical spectrum ranges from a completely asymptomatic form 20, to severe, life threatening conditions such as severe hypokalaemia, alkalosis or hypoglycemia. In addition, hyperandrogenism (acne, hirsutism, infertility, oligo-amenorrhea in females, oligospermia and infertility in males, precocious puberty in children) 21 and mineralocorticoid excess (hypertension and hypokalemic alkalosis) 22 can also be observed. Fatigue is the most common sign of the disease 20. The diagnosis is based on a detailed evaluation of the hypothalamic-pituitary-adrenal axis. Measurement of serum cortisol levels in samples collected in the morning under fasting conditions, at midnight and after dexamethasone administration, together with evaluation of 24 hour urinary-free cortisol excretion, are mandatory investigations for diagnosis. Serum cortisol and 24 hour urinary free cortisol excretion remain elevated after administration of low dose dexamethasone 23. Contrary to Cushing’s syndrome, in patients with Chrousos syndrome, the hypothalamic-pituitary-adrenal axis preserves its circadian rhythm 23.
To date more than 15 different mutations of the glucocorticoid receptor that cause glucocorticoid resistance have been identified 17. It has been shown that the mutant receptors may exert a dominant negative effect on the wild-type receptor, or may decrease the receptor’s affinity to the ligand. In addition, a mislocalization of the mutant receptor, delayed or failed translocation to the nucleus or decreased transcriptional activity due to decreased binding through glucocorticoid response element 21 can lead to glucocorticoid resistance.
Treatment of glucocorticoid resistance includes administration of a high dose of glucorcorticoids in order to suppress the excessive ACTH-stimulated secretion of mineralocorticoids and androgens 24. However, our patient had no clinical symptoms of mineralocorticoid or androgen excess, and it is not known whether a high dose of glucocorticoids could offer an option for the treatment of infertility with an acceptable maternal and fetal risk, and whether glucocorticoids should be continued during pregnancy. Because the Arg714Glu variant of the glucocorticoid receptor gene may cause both mild and severe phenotypes, a high dose of glucocorticoids may be of value to prevent fetal androgen and mineralocorticoid excess in an affected fetus predisposed to a severe phenotype 17. Detailed genetic counseling is indicated and prenatal genetic testing possible in order to determine the fetus genotype.
Glucocorticoid deficiency
Adrenal insufficiency is a serious pathologic condition characterized by decreased production or action of glucocorticoids and/or mineralocorticoids and adrenal androgens 25. Adrenal insufficiency is a life-threatening disorder that may be classified as primary, secondary or tertiary adrenal insufficiency, resulting from diseases affecting the adrenal cortex, the anterior pituitary gland or the hypothalamus, respectively (see Figure 6). The clinical signs and symptoms of adrenal insufficiency include anorexia, abdominal pain, weakness, weight loss, fatigue, hypotension, salt craving and hyperpigmentation of the skin in case of primary adrenal insufficiency. The diagnosis of adrenal insufficiency can be confirmed by demonstrating inappropriately low cortisol secretion, determining whether the cortisol deficiency is secondary or primary, and defining the cause of the disorder. Treatment with glucocorticoid and/or mineralocorticoid replacement should be initiated when glucocorticoid and or mineralocorticoid deficiency is suspected.
Figure 6. Adrenal insufficiency
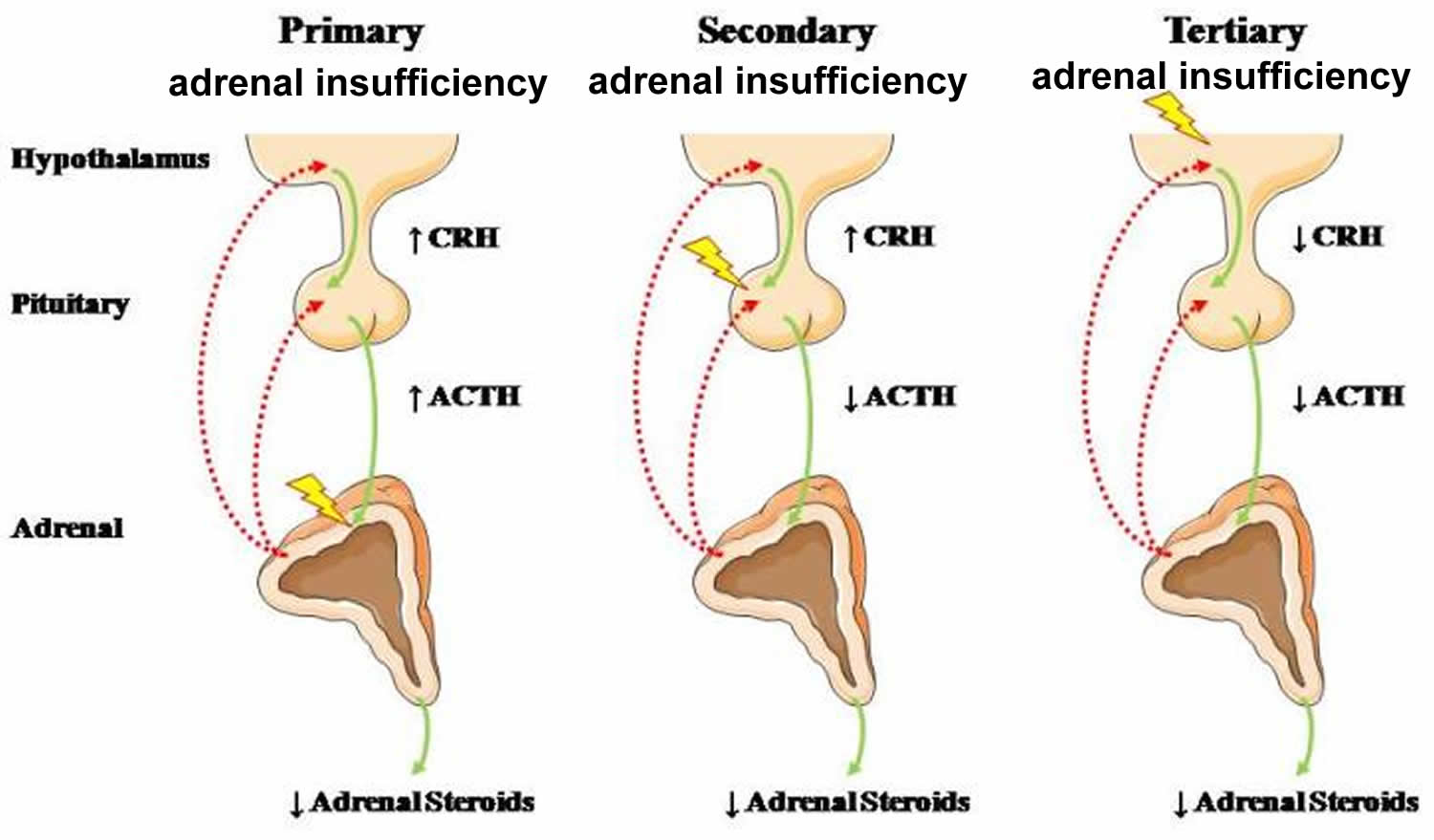 Abbreviations: CRH = corticotropin-releasing hormone; ACTH = adrenocorticotropic hormone
[Source 25 ]
Abbreviations: CRH = corticotropin-releasing hormone; ACTH = adrenocorticotropic hormone
[Source 25 ]Primary adrenal insufficiency affects more frequently women, and clinical manifestations can present at any age, although most often between 30 and 50 years 26. In primary adrenal insufficiency, although the above mentioned causes lead to gradual destruction of the adrenal cortex, the symptoms and signs of the disease appear when the loss of adrenocortical tissue is higher than 90% 27. At the molecular and cellular level, a viral infection, even subclinical, or an excessive tissue response to inflammatory signals may potentially lead to apoptosis or necrosis of adrenocortical cells. In the initial phase of chronic gradual destruction, the adrenal reserve is decreased and although the basal steroid secretion is normal, the secretion in response to stress is suboptimal. Consequently, any major or even minor stressor can precipitate an acute adrenal crisis. With further loss of adrenocortical tissue, even basal steroid secretion is decreased, leading to the clinical manifestations of the disease. Low plasma cortisol concentrations result in the increase of production and secretion of ACTH (adrenocorticotropic hormone) due to decreased negative feedback inhibition 27. The elevated plasma ACTH concentrations are responsible for the well-recognized hyperpigmentation observed in these patients.
Secondary adrenal insufficiency occurs more frequently than primary adrenal insufficiency 28. Secondary adrenal insufficiency estimated prevalence is 150–280 per million and is more common in women than men 29. Affected patients are mostly diagnosed in the sixth decade of life 30.
The most common cause of tertiary adrenal insufficiency is chronic exogenous administration of synthetic glucocorticoids, which causes prolonged suppression of hypothalamic corticotropin-releasing hormone (CRH) secretion through negative feedback mechanisms 31.
In secondary or tertiary adrenal insufficiency, the resultant ACTH deficiency leads to decreased secretion of cortisol and adrenal androgens, while mineralocorticoid production remains normal. In the early stages, basal ACTH secretion is normal, while stress-induced ACTH secretion is impaired 27. With further loss of basal ACTH secretion, there is atrophy of zonae fasciculata and reticularis of the adrenal cortex. Therefore, basal cortisol secretion is decreased, but aldosterone secretion by the zona glomerulosa is preserved.
Causes of Primary Adrenal Insufficiency
The cause of primary adrenal insufficiency has changed over time. Prior to 1920, the most common cause of primary adrenal insufficiency was tuberculosis, while since 1950, the majority of cases (80-90%) have been ascribed to autoimmune adrenalitis, which can be isolated (40%) or in the context of an autoimmune polyendocrinopathy syndrome (60%) 32.
Autoimmune adrenalitis (Addison’s disease)
This condition is characterized by destruction of the adrenal cortex by cell-mediated immune mechanisms. Antibodies that react against steroid 21-hydroxylase are detected in approximately 90% of patients with autoimmune Addison’s disease 33, but only rarely in patients with other causes of adrenal insufficiency or normal subjects 34. Considerable progress has been made in identifying genetic factors that predispose to the development of autoimmune adrenal insufficiency 32. In addition to the major histocompatibility complex (MHC) haplotypes DR3-DQ2 and DR4-DQ8, other genetic factors, such as protein tyrosine phosphatase non-receptor type 22 (PTPN22), cytotoxic T lymphocyte antigen 4 (CTLA-4), and the major histocompatibility complex class II transactivator (CIITA) have been associated with this condition 35.
Primary adrenal insufficiency may also present as part of autoimmune polyendocrinopathy syndromes. Patients with autoimmune polyendocrinopathy syndrome type 1 (APS1) or Autoimmune Polyendocrinopathy, Candidiasis, Ectodermal Dystrophy syndrome may present with chronic mucocutaneous candidiasis, adrenal insufficiency, hypoparathyroidism, hypoplasia of the dental enamel and nail dystrophy, while type 1 diabetes mellitus or pernicious anemia, may develop later in life 36. Clinical manifestations of autoimmune polyendocrinopathy syndrome type 2 (APS2) include autoimmune adrenal insufficiency, autoimmune thyroid disease and/or type 1 diabetes, whereas autoimmune polyendocrinopathy syndrome type 4 (APS4) is characterized by autoimmune adrenal insufficiency and one or more other autoimmune diseases, such as atrophic gastritis, hypogonadism, pernicious anemia, celiac disease, myasthenia gravis, vitiligo, alopecia and hypophysitis, but without any autoimmune disorders of APS1 or APS2 35.
Adrenoleukodystrophy
This is an X-linked recessive disorder affecting 1 in 20.000 males 32. The molecular basis of this condition has been ascribed to mutations in the ABCD1 gene, which result in defective beta oxidation of very long chain fatty acids within peroxisomes. The abnormally high concentrations of very long chain fatty acids in affected organs, including the adrenal cortex, result in the clinical manifestations of this disorder, which include neurological impairment due to white-matter demyelination and primary adrenal insufficiency, with the latter presenting in infancy or childhood 37.
Hemorrhagic infarction
Bilateral adrenal infarction caused by hemorrhage or adrenal vein thrombosis may also lead to adrenal insufficiency 38. The diagnosis is usually made in critically ill patients in whom a computed tomography (CT) scan of the abdomen shows bilateral adrenal enlargement. Several coagulopathies and the heparin-induced thrombocytopenia syndrome have been associated with adrenal vein thrombosis and hemorrhage, while the primary antiphospholipid syndrome has been recognized as a major cause of adrenal hemorrhage 27. Adrenal hemorrhage has been mostly associated with meningococcemia (Waterhouse-Friderichsen syndrome) and Pseudomonas aeruginosa infection 39.
Infectious adrenalitis
Many infectious agents may attack the adrenal gland and result in adrenal insufficiency, including tuberculosis (tuberculous adrenalitis), disseminated fungal infections and HIV-associated infections, such as adrenalitis due to cytomegalovirus and mycobacterium avium complex 40.
Drug-induced adrenal insufficiency
Drugs that may cause adrenal insufficiency by inhibiting cortisol biosynthesis, particularly in individuals with limited pituitary and/or adrenal reserve, include aminoglutethimide (antiepileptic), etomidate (anesthetic-sedative) 41, ketoconazole (antimycotic) 42 and metyrapone 43. Drugs that accelerate the metabolism of cortisol and most synthetic glucocorticoids by inducing hepatic mixed-function oxygenase enzymes, such as phenytoin, barbiturates, and rifampicin can also cause adrenal insufficiency in patients with limited pituitary or adrenal reserve, as well as those who are on replacement therapy with glucocorticoids 44. Furthermore, some of novel tyrosine kinase-targeting drugs (e.g. sunitinib) have been shown in animal studies to cause adrenal dysfunction and hemorrhage 45.
Other causes of primary adrenal insufficiency are listed in Table 3.
Table 3. Causes of Primary Adrenal Insufficiency
| Disease | Pathogenetic Mechanism |
| Autoimmune adrenalitis | |
| Isolated | Associations with HLA-DR3-DQ2, HLADR4-DQ8, MICA, CTLA-4, PTPN22, CIITA, CLEC16A, Vitamin D receptor |
| APS type 1 (APECED) | AIRE gene mutations |
| APS type 2 | Associations with HLA-DR3, HLA-DR4, CTLA-4 |
| APS type 4 | Associations with HLA-DR3, CTLA-4 |
| Infectious adrenalitis | |
| Tuberculous adrenalitis | Tuberculosis |
| AIDS | HIV-1, cytomegalovirus |
| Fungal adrenalitis | Histoplasmosis, cryptococcosis, coccidiodomycosis |
| Syphilis | Treponema pallidum |
| African Trypanosomiasis | Trypanosoma brucei |
| Bilateral adrenal hemorrhage | Meningococcal sepsis (Waterhouse- Friderichsen syndrome), primary antiphospholipid syndrome |
| Bilateral adrenal metastases | Primarily lung, stomach, breast and colon cancer |
| Bilateral adrenal infiltration | Primary adrenal lymphoma, amyloidosis, haemochromatosis |
| Bilateral adrenalectomy | Unresolved Cushing’s syndrome, bilateral adrenal masses, bilateral pheochromocytoma |
| Drug-induced adrenal insufficiency | |
| Anticoagulants (heparin, warfarin), tyrosine kinase inhibitors (sunitinib) | Hemorrhage |
| Aminoglutethimide | Inhibition of P450 aromatase (CYP19A1) |
| Trilostane | Inhibition of 3β-hydroxysteroid dehydrogenase type 2 (HSD3B2) |
| Ketoconazole, fluconazole, etomidate | Inhibition of mitochondrial cytochrome P450-dependent enzymes (e.g. CYP11A1, CYP11B1) |
| Phenobarbital | Induction of P450-cytochrome enzymes (CYP2B1, CYP2B2), which enhance cortisol metabolism |
| Phenytoin, rifampin, troglitazone | Induction of P450-cytochrome enzymes (primarily CYP3A4), which enhance cortisol metabolism |
| Genetic disorders | |
| Adrenoleukodystrophy or adrenomyeloneuropathy | ABCD1 and ABCD2 gene mutations |
| Congenital adrenal hyperplasia | |
| 21-Hydroxylase deficiency | CYP21A2 gene mutations |
| 11β-Hydroxylase deficiency | CYP11B1 gene mutations |
| 3β-hydroxysteroid dehydrogenase type 2 deficiency | HSD3B2 gene mutations |
| 17α-Hydroxylase deficiency | CYP17A1 gene mutations |
| P450 Oxidoreductase deficiency | POR gene mutations |
| P450 side-chain cleavage deficiency | CYP11A1 gene mutations |
| Congenital lipoid adrenal hyperplasia | StAR gene mutations |
| Smith-Lemli-Opitz syndrome | DHCR7 gene mutations |
| Adrenal hypoplasia congenita | |
| X-linked | NR0B1 gene mutations |
| Xp21 contiguous gene syndrome | Deletion of the Duchenne muscular dystrophy, glycerol kinase and NR0B1 genes |
| SF-1 linked | NR5A1 gene mutations |
| IMAGe syndrome | CDKN1C gene mutations |
| Kearns-Sayre syndrome | Mitochondrial DNA deletions |
| Wolman’s disease | LIPA gene mutations |
| Sitosterolaimia (also known as phytosterolemia) | ABCG5 and ABCG8 gene mutations |
| Familial glucocorticoid deficiency (FGD, or ACTH insensitivity syndromes) | |
| Type 1 | MC2R gene mutations |
| Type 2 | MRAP gene mutations |
| Variant of FGD | MCM4 gene mutations |
| FGC – Deficiency of mitochondrial ROS detoxification | NNT, TXNRD2, GPX1, PRDX3 gene mutations |
| Primary Generalized Glucocorticoid Resistance or Chrousos syndrome | NR3C1 gene mutations |
| Sphingosine-1-phosphate lyase 1 deficiency | SPGL1 gene mutations |
| Infantile Refsum disease | PHYH, PEX7 gene mutations |
| Zellweger syndrome | PEX1 and other PEX gene mutations |
| Triple A syndrome (Allgrove’s syndrome) | AAAS gene mutations |
Causes of Secondary and Tertiary Adrenal Insufficiency
Secondary adrenal insufficiency may be caused by any disease process that affects the anterior pituitary and interferes with ACTH secretion. The ACTH deficiency may be isolated or occur in association with other pituitary hormone deficits.
Tertiary adrenal insufficiency can be caused by any process that involves the hypothalamus and interferes with CRH secretion. The most common cause of tertiary adrenal insufficiency is chronic administration of synthetic glucocorticoids that suppress the hypothalamic-pituitary-adrenal axis 46.
Other causes of secondary and tertiary adrenal insufficiency are listed in Tables 4 and 5 respectively.
Table 4. Causes of Secondary Adrenal Insufficiency
| Disease | Pathogenetic Mechanism |
| Space occupying lesions or trauma | |
| Pituitary tumors (adenomas, cysts, craniopharyngiomas, ependymomas, meningiomas, rarely carcinomas) or trauma (pituitary stalk lesions) | Decreased ACTH secretion |
| Pituitary surgery or irradiation for pituitary tumors, tumors outside the HPA axis or leukemia | Decreased ACTH secretion |
| Infections or Infiltrative processes (lymphocytic hypophysitis, hemochromatosis, tuberculosis, meningitis, sarcoidosis, actinomycosis, histiocytosis X, Wegener’s granulomatosis) | Decreased ACTH secretion |
| Pituitary apoplexy | Decreased ACTH secretion |
| Sheehan’s syndrome (peripartum pituitary apoplexy and necrosis) | Decreased ACTH secretion |
| Genetic disorders | |
| Transcription factors involved in pituitary development | |
| HESX homeobox 1 | HESX1 gene mutations |
| Orthodentical homeobox 2 | OTX2 gene mutations |
| LIM homeobox 4 | LHX4 gene mutations |
| PROP paired-like homeobox 1 | PROP1 gene mutations |
| SRY (sex-determining region Y) – box 3 | SOX3 gene mutations |
| T-box 19 | TBX19 gene mutations |
| Congenital Proopiomelanocortin (POMC) deficiency | POMC gene mutations |
| Prader-Willi Syndrome (PWS) | Deletion or silencing of genes in the imprinting center for PWS |
Table 5. Causes of Tertiary Adrenal Insufficiency
| Disease | Pathogenetic Mechanism |
| Space occupying lesions or trauma | |
| Hypothalamic tumors (craniopharyngiomas or metastasis from lung, breast cancer) | Decreased CRH secretion |
| Hypothalamic surgery or irradiation for central nervous system or nasopharyngeal tumors | Decreased CRH secretion |
| Infections or Infiltrative processes (lymphocytic hypophysitis, hemochromatosis, tuberculosis, meningitis, sarcoidosis, actinomycosis, histiocytosis X, Wegener’s granulomatosis) | Decreased CRH secretion |
| Trauma, injury (fracture of skull base) | Decreased CRH secretion |
| Drug-induced adrenal insufficiency | |
| Glucocorticoid therapy (systemic or topical) or endogenous glucocorticoid hypersecretion (Cushing’s syndrome) | Decreased CRH and ACTH secretion |
| Mifepristone | Tissue resistance to glucocorticoids through impairment of glucocorticoid signal transduction |
| Antipsychotics (chlorpromazine), antidepressants (imipramine) | Inhibition of glucocorticoid-induced gene transcription |
Adrenal insufficiency in crtically ill patients
Clinical manifestations of adrenal insufficiency are common in critically ill patients, specifically in patients with severe pneumonia, adult respiratory distress syndrome, sepsis, trauma, HIV infection or after treatment with etomidate 47.
The molecular pathogenetic mechanisms underlying adrenal insufficiency in critical illness have not been fully elucidated. However, it seems that both inadequate cortisol secretion and impaired glucocorticoid receptor signaling are convincingly involved. Indeed, proinflammatory cytokines may compete with ACTH on its receptor 48 and/or induce tissue resistance to glucocorticoids 49. Moreover, the widely used medications during the treatment of sepsis may impair both glucocorticoid production and glucocorticoid signaling. Furthermore, other neuropeptides, signaling molecules, components of oxidative stress and the impaired adrenal blood flow contribute to adrenal insufficiency.
To provide recommendations on the diagnosis and management of adrenal insufficiency in critically ill patients, the American College of Critical Care Medicine suggested that the diagnosis is best made by a delta total serum cortisol of < 9 mcg/dL following ACTH (250 microg) administration or a random total cortisol of < 10 mcg/dL 25. Hydrocortisone at a dose of 200 mg/day in four divided doses or as a continuous infusion at a dose of 240 mg/day (10 mg/hr) for at least 7 days is recommended for patients with septic shock 25. Methylprednisolone at a dose of 1 mg/kg/day for at least 14 days is recommended in patients diagnosed with severe early acute respiratory distress syndrome. The role of glucocorticoid therapy in other critically ill patients remains to be further elucidated 50.
Adrenal insufficiency during pregnancy
Although adrenal insufficiency is relatively rare in pregnancy, it may be associated with significant maternal and/or fetal morbidity and mortality if it remains undiagnosed or untreated 51. Symptoms are usually “nonspecific”, such as nausea, vomiting and fatigue, making the diagnosis of adrenal insufficiency challenging. The current diagnostic tests are serum cortisol concentrations and the cosyntropin stimulation test 51. However, it should be emphasized that the peak cortisol response following ACTH stimulation is higher in pregnant than in non-pregnant women during the second and third trimesters, as a result of physiologic pregnancy-associated hypercortisolism and elevations of cortisol-binding globulin 52. Regarding glucocorticoid replacement during pregnancy, hydrocortisone, cortisone acetate, prednisolone or prednisone can be administered; in contrast,, fluorinated glucocorticoids such as dexamethasone should be avoided because they cross the placenta at higher rates 53. Mineralocorticoid replacement is usually more complicated to assess during pregnancy because of the “nonspecific” symptoms often observed in physiologic pregnancy 53. A hydrocortisone stress dose (bolus intravascular injection of 50-100 mg hydrocortisone followed by continuous infusion of 100-200 mg hydrocortisone/24h) should be administered at the beginning of active labor 53.
Adrenal insufficiency in infancy and childhood
Children with primary adrenal insufficiency should be treated with hydrocortisone phosphate at a daily dose of 10-12 mg per meter square body surface area divided into two or three doses 53. Alternatively, cortisone acetate can be administered with safety also as two to three daily doses. Intermediate-acting or long-acting glucocorticoid analogues, such as prednisolone/prednisolone or dexamethasone respectively, are not recommended due to undesirable chronic side effects, such as glucose intolerance or osteopenia/osteoporosis. The hydrocortisone daily dose should be adjusted according to the increasing body surface area of the child. Caution should be paid to decreased growth velocity, excessive weight gain or other clinical manifestations suggestive of iatrogenic Cushing syndrome. Children with primary adrenal insufficiency also require fludrocortisone at a daily dose of 50-300 μg 53. During the first 6 months, infants require supplementation of sodium chloride at a dose of 1-2 g/day administered in multiple feedings, because the infant kidney is physiologically resistant to mineralocorticoids and the infant milk (breast milk or formula) has relatively low sodium content 53.
Glucocorticoid deficiency signs and symptoms
The clinical manifestations of adrenal insufficiency depend upon the extent of loss of adrenal function and whether mineralocorticoid production is preserved. The onset of adrenal insufficiency is often gradual and may go undetected until an illness or other stress precipitates an adrenal crisis 46.
Adrenal Crisis
Adrenal crisis or acute adrenal insufficiency may complicate the course of chronic primary adrenal insufficiency, and may be precipitated by a serious infection, acute stress, bilateral adrenal infarction or hemorrhage. It is rare in patients with secondary or tertiary adrenal insufficiency 25. The main clinical manifestation of adrenal crisis is shock, but patients may also have nonspecific symptoms, such as anorexia, nausea, vomiting, abdominal pain, weakness, fatigue, lethargy, confusion or coma. Hypoglycemia is rare in acute adrenal insufficiency, but more common in secondary adrenal insufficiency.
The major factor precipitating an adrenal crisis is mineralocorticoid deficiency and the main clinical problem is hypotension 25. Adrenal crisis can occur in patients receiving appropriate doses of glucocorticoid if their mineralocorticoid requirements are not met 54, whereas patients with secondary adrenal insufficiency and normal aldosterone secretion rarely present in adrenal crisis. However, glucocorticoid deficiency may also contribute to hypotension by decreasing vascular responsiveness to angiotensin II, norepinephrine and other vasoconstrictive hormones, reducing the synthesis of renin substrate, and increasing the production and effects of prostacyclin and other vasodilatory hormones 55.
Chronic Primary Adrenal Insufficiency
The clinical manifestations of chronic primary adrenal insufficiency are owing to deficient concentrations of all adrenocortical hormones (mineralocorticoids, glucocorticoids, adrenal androgens) and include general malaise, fatigue, weakness, anorexia, weight loss, nausea, vomiting, abdominal pain or diarrhea, which may alternate with constipation, hypotension, electrolyte abnormalities (hyponatremia, hyperkalemia, metabolic acidosis), hyperpigmentation, autoimmune manifestations (vitiligo), decreased axillary and pubic hair, and loss of libido and amenorrhea in women 46. The onset of chronic adrenal insufficiency is often insidious and the diagnosis may be difficult in the early stages of the disease.
Secondary or Tertiary Adrenal Insufficiency
The clinical features of secondary or tertiary adrenal insufficiency are similar to those of primary adrenal insufficiency. However, hyperpigmentation of the skin does not occur, because the secretion of ACTH is not increased. Also, since the production of mineralocorticoids by the zona glomerulosa is mostly preserved, dehydration and hyperkalemia are not present, and hypotension is less prominent. Hyponatremia and increased intravascular volume may be the result of “inappropriate” increase in vasopressin secretion. Hypoglycemia is more common in secondary adrenal insufficiency possibly due to concomitant growth hormone insufficiency and in isolated ACTH deficiency. Clinical manifestations of a pituitary or hypothalamic tumor, such as symptoms and signs of deficiency of other anterior pituitary hormones, headache or visual field defects, may also be present 46.
Glucocorticoid deficiency diagnosis
The clinical diagnosis of adrenal insufficiency can be confirmed by demonstrating inappropriately low cortisol secretion, determining whether the cortisol deficiency is secondary or primary and, hence, dependent or independent of ACTH deficiency, and detecting the cause of the disorder 46.
Basal morning serum cortisol concentrations
The diagnosis of adrenal insufficiency depends upon the demonstration of inappropriately low cortisol secretion. Serum cortisol concentrations are normally highest in the early morning hours (06:00h – 08:00h), ranging between 10 – 20 mcg/dL (275 – 555 nmol/L) than at other times of the day 25. Serum cortisol concentrations determined at 08:00h of less than 3 µg/dL (80 nmol/L) are strongly suggestive of adrenal insufficiency 56, while values below 10 µg/dL (275 nmol/L) make the diagnosis likely. Simultaneous measurements of cortisol and ACTH concentrations confirm in most cases the diagnosis of primary adrenal insufficiency.
Morning salivary cortisol concentrations
Adrenal insufficiency is excluded when salivary cortisol concentration at 08:00h is higher than 5.8 ng/mL (16 nmol/L), whereas the diagnosis is more possible for values lower than 1.8 ng/mL (5 nmol/L) 25.
Urinary free cortisol
Basal urinary cortisol and 17-hydroxycorticosteroid excretion is low in patients with severe adrenal insufficiency, but may be low-normal in patients with partial adrenal insufficiency. Generally, baseline urinary measurements are not recommended for the diagnosis of adrenal insufficiency 25.
Basal plasma ACTH, renin and aldosterone concentrations
Basal plasma ACTH concentration at 08:00h, when determined simultaneously with the measurement of basal serum cortisol concentration, may both confirm the diagnosis of adrenal insufficiency and establish the cause 57. The normal values of basal 08:00h plasma ACTH concentrations range between 20-52 pg/mL (4.5-12 pmol/L). In primary adrenal insufficiency, the 08:00h plasma ACTH concentration is elevated, and is coupled with increased concentration or activity of plasma renin, low aldosterone concentrations, hyperkalemia and hyponatremia. In the cases of secondary or tertiary adrenal insufficiency, plasma ACTH concentrations are low or low normal, associated with normal values of plasma concentrations of renin and aldosterone.
Standard dose ACTH stimulation test
Adrenal insufficiency is usually diagnosed by the standard-dose ACTH test, which determines the ability of the adrenal glands to respond to 250 mcg intravenous or intramuscular administration of ACTH(1-24) by measurement of serum cortisol concentrations at 0, 30 and 60 min following stimulation 25. The test is defined as normal if peak cortisol concentration is higher than 18–20 mcg/dL (500–550 nmol/L), thereby excluding the diagnosis of primary adrenal insufficiency and almost all cases of secondary adrenal insufficiency 25. However, if secondary adrenal insufficiency is of recent onset, the adrenal glands will have not yet atrophied, and will still be capable of responding to ACTH stimulation normally. In these cases, a low-dose ACTH stimulation test or an insulin-induced hypoglycemia test may be required to confirm the diagnosis 58.
Low-Dose ACTH stimulation test
This test theoretically provides a more sensitive index of adrenocortical responsiveness because it results in physiologic plasma ACTH concentrations. This test should be performed at 14:00h, when the endogenous secretion of ACTH is at its lowest 25. The results might not be valid if it is performed at another time. At 14:00h, a blood sample is collected for determination of basal cortisol concentrations. The low dose of ACTH(1-24) (500 nanograms ACTH(1-24)/1.73 m2 ) is then administered as an intravenous bolus. In normal subjects, this dose results in a peak plasma ACTH concentration about twice that of insulin-induced hypoglycemia 58. Subsequently, blood samples are collected at +10 min, +15 min, +20 min, +25 min, +30 min, +35 min, +40 min and +45 min after stimulation for determination of serum cortisol concentrations 59. A value of 18 µg/dL (500 nmol/L) or more at any time during the test is indicative of normal adrenal function. The advantage of this test is that it can detect partial adrenal insufficiency that may be missed by the standard-dose test 60. The low-dose test is also preferred in patients with secondary or tertiary adrenal insufficiency 61.
Prolonged ACTH Stimulation Tests
Prolonged stimulation with exogenous administration of ACTH helps differentiate between primary and secondary or tertiary adrenal insufficiency 25. In secondary or tertiary adrenal insufficiency, the adrenal glands display cortisol secretory capacity following prolonged stimulation with ACTH, whereas in primary adrenal insufficiency, the adrenal glands are partially or completely destroyed and do not respond to ACTH 25. The prolonged ACTH test consists of the intravenous administration of 250 μg of ACTH as an infusion over eight hours (8-hour test) or over 24 hours on two (or three) consecutive days (two-day test), and the measurement of serum cortisol, and 24-hour urinary cortisol and 17-hydroxycorticoid concentrations before and after the infusion 62.
Insulin-induced hypoglycemia test
This test provides an alternative choice for confirmation of the diagnosis when secondary adrenal insufficiency is suspected 25. The insulin tolerance test helps in the investigation of the integrity of the hypothalamic- pituitary-adrenal axis and has the ability to assess growth hormone reserve. Insulin, at a dose of 0.1-0.15 U/kg, is administered to induce hypoglycemia, and measurements of cortisol concentrations are determined at 30 min intervals for at least 120 min 63. This test is contraindicated in patients with cardiovascular disease or a history of seizures, and requires a high degree of supervision.
Corticotropin-releasing hormone (CRH) test
This test is used to differentiate between secondary and tertiary adrenal insufficiency 25. It consists of intravenous administration of CRH (1 mcg/kg up to a maximum of 100 mcg) and determination of serum cortisol and plasma ACTH concentrations at 0, 15, 30, 45, 60, 90 and 120 min following stimulation 25. Patients with secondary adrenal insufficiency demonstrate little or no ACTH response, whereas patients with tertiary adrenal insufficiency show an exaggerated and prolonged response of ACTH to CRH stimulation, which is not followed by an appropriate cortisol response 64.
Autoantibody screen
Adrenocortical antibodies or antibodies against 21-hydroxylase can be detected in more than 90% of patients with recent onset autoimmune adrenalitis 25. Furthermore, antibodies that react against other enzymes involved in the steroidogenesis (P450scc, P450c17) and anti-steroid-producing cell antibodies are present in some patients 25.
Very long chain fatty acids
To exclude adrenoleukodystrophy, plasma very long chain fatty acids should be determined in male patients with isolated Addison’s disease and negative autoantibodies 37.
Imaging
Patients without any associated autoimmune disease and negative autoantibody screen should undergo a computed tomography (CT) scan of the adrenal glands 25. In cases of tuberculous adrenalitis, the CT scan shows initially hyperplasia of the adrenal glands and subsequently spotty calcifications during the late stages of the disease. Bilateral adrenal lymphoma, adrenal metastases or adrenal infiltration (sarcoidosis, amyloidosis, hemochromatosis) may also be detected by CT scan. If central adrenal insufficiency is suspected, a magnetic resonance imaging (MRI) scan of the hypothalamus and pituitary gland should be performed. This may detect any potential disease process, such as craniopharyngiomas, pituitary adenomas, meningiomas, metastases and infiltration by Langerhans cell histiocytosis, sarcoidosis or other granulomatous diseases 65. It should be noted that imaging is not required when adrenal cortex autoantibodies are detected.
Glucocorticoid deficiency treatment
Adrenal insufficiency is one of the most life-threatening disorders. Treatment should be administered to the patients as soon as the diagnosis is established, or even sooner if an adrenal crisis occurs 66.
Treatment of Chronic Adrenal Insufficiency
One of the most important aspects of the management of chronic primary adrenal insufficiency is patient and family education. Patients should understand the reason for life-long replacement therapy, the need to increase the dose of glucocorticoid during minor or major stress and to inject hydrocortisone, methylprednisolone or dexamethasone in emergencies.
Emergency precautions
Patients should wear a medical alert (Medic Alert) bracelet or necklace and carry the Emergency Medical Information Card, which should provide information on the diagnosis, the medications and daily doses, and the physician involved in the patient’s management. Patients should also have supplies of dexamethasone sodium phosphate and should be educated about how and when to administer them.
Glucocorticoid replacement therapy
Patients with adrenal insufficiency should be treated with hydrocortisone, the natural glucocorticoid, or cortisone acetate if hydrocortisone is not available 25. The hydrocortisone daily dose is 10-12 mg per meter square body surface area and can be administered in two to three divided doses with one half to two thirds of the total daily dose being given in the morning 67. Small reductions of bone mineral density (BMD) probably due to higher than recommended doses 68, as well as impaired quality of life 69 were observed in patients treated with hydrocortisone. A longer-acting synthetic glucocorticoid, such as prednisone, prednisolone or dexamethasone, should be avoided because their longer duration of action may produce manifestations of chronic glucocorticoid excess, such as loss of lean body mass and bone density, and gain of visceral fat 70. Recently, preparations of hydrocortisone that lead to both delayed and sustained release of this compound have been developed and are under clinical investigation 71. These formulations maintain stable cortisol concentrations during 24 hours and physiologic circadian rhythmicity with the cortisol peak occurring during the early morning after oral intake of the preparation at bed-time. Furthermore, a novel once-daily dual-release hydrocortisone tablet has been developed to maintain more physiologic circadian-based serum cortisol concentrations. Compared to the conventional treatment, the once-daily dual-release hydrocortisone improved glucose metabolism, cardiovascular risk factors and quality of life 72. Regardless of the type of the formulation used, glucocorticoid replacement should be monitored clinically, evaluating weight gain/loss, arterial blood pressure, annualized growth velocity and presence of Cushing features 53.
Glucocorticoid replacement during minor illness or surgery
During minor illness or surgical procedures, glucocorticoids should be given at a dosage up to three times the usual maintenance dosage for up to three days 25. Depending on the nature and severity of the illness, additional treatment may be required.
Glucocorticoid replacement during major illness or surgery
During major illness or surgery, high doses of glucocorticoid analogues (10 times the daily production rate) are required to avoid an adrenal crisis 25. A continuous infusion of 10 mg of hydrocortisone per hour or the equivalent amount of dexamethasone or prednisolone eliminates the possibility of glucocorticoid deficiency. This dose can be halved the second postoperative day, and the maintenance dose can be resumed at the third postoperative day.
Mineralocorticoid replacement therapy
Mineralocorticoid replacement therapy is required to prevent intravascular volume depletion, hyponatremia and hyperkalemia. For these purposes, fludrocortisone (9-alpha-fluorohydrocortisone) in a dose of 0.05 – 0.2 mg daily should be taken in the morning 25. The dose of fludrocortisone is titrated individually based on the findings of clinical examination (mainly the body weight and arterial blood pressure) and the levels of plasma renin activity. Patients receiving prednisone or dexamethasone may require higher doses of fludrocortisone to lower their plasma renin activity to the upper normal range, while patients receiving hydrocortisone, which has some mineralocorticoid activity, may require lower doses. The mineralocorticoid dose may have to be increased in the summer, particularly if patients are exposed to temperatures above 29ºC (85ºF) 73. If patients receiving mineralocorticoid replacement develop hypertension, the dose of fludrocortisone should be reduced accordingly 53. In case of uncontrolled blood pressure, patients should be encouraged to continue fludrocortisone and initiate antihypertensive therapy, such as angiotensin 2 receptor blockers, angiotensin-converting enzyme inhibitors, or dihydropyridine calcium blockers 53.
Androgen replacement
In women, the adrenal cortex is the primary source of androgen in the form of dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS). Treatment with dehydroepiandrosterone (DHEA) enhances mood and general well being both in adult patients and in children and adolescents with adrenal insufficiency 74. A single oral morning dose of dehydroepiandrosterone (DHEA) of 25-50 mg may be sufficient to maintain normal serum androgen concentrations in premenopausal women with primary adrenal insufficiency, who present with decreased libido, anxiety, depression, and low energy levels 53. If symptoms are still present during a period of 6 months, patients are advised to discontinue DHEA replacement 53. Naturally, women should be encouraged to report any side effects of androgen therapy. Finally, DHEA replacement should be monitored by determining serum DHEA concentrations in the morning before patient receives her daily DHEA dose 53.
Treatment of adrenal crisis
The aim of initial management in adrenal crisis is to treat hypotension, hyponatremia and hyperkalemia, and to reverse glucocorticoid deficiency. Treatment should be started with immediate administration of 100 mg hydrocortisone i.v. and rapid rehydration with normal saline infusion under continuous cardiac monitoring, followed by 100–200 mg hydrocortisone in glucose 5% per 24-hour continuous iv infusion; alternatively, hydrocortisone could be administered iv or im at a dose of 50-100 mg every 6 hours depending on body surface area and age 75. With daily hydrocortisone doses of 50 mg or more, mineralocorticoids in patients with primary adrenal insufficiency can be discontinued or reduced because this dose is equivalent to 0.1 mg fludrocortisone 76. Once the patient’s condition is stable and the diagnosis has been confirmed, parenteral glucocorticoid therapy should be tapered over 3-4 days and converted to an oral maintenance dose 74. Patients with primary adrenal insufficiency require life-long glucocorticoid and mineralocorticoid replacement therapy.
Treatment of chronic secondary and tertiary adrenal insufficiency
In chronic secondary or tertiary adrenal insufficiency, glucocorticoid replacement is similar to that in primary adrenal insufficiency, however, measurement of plasma ACTH concentrations cannot be used to titrate the optimal glucocorticoid dose 25. Mineralocorticoid replacement is rarely required, while replacement of other anterior pituitary deficits might be necessary 25.
Glucocorticoid medications
Glucocorticoid medications are synthetic (manufactured) hormones are analogs of cortisol, also known as steroids. Glucocorticoid medications are used for their anti-inflammatory, immunosuppressive and antiproliferative effects (Tables 7 and 8). Examples of commonly used synthetic compounds include:
- PREDNISONE – perhaps the most widely used synthetic corticosteroid (with a very long list of FDA-approved indications). Prednisone has a high ratio of glucocorticoid vs mineralocorticoid activity, which is desirable for use as an anti-inflammatory and immunosuppressant agent (where effects on sodium and water balance are undesirable). Prednisone is a prodrug that has no biologic activity until it is converted by hepatic metabolism to prednisolone (the active metabolite). Metabolic conversion may be impaired with hepatic dysfunction.
- PREDNISOLONE – The active metabolite of prednisone. It has 4-5 times the anti-inflammatory potency of cortisol, and ~1/4th the mineralocorticoid potency than cortisol. Pregnancy: In pregnancy, the placenta expresses an enzyme (11β-hydroxysteroid dehydrogenase 2) that inactivates either cortisol or prednisolone by converting them to inactive forms (cortisone or prednisone). This protects the fetus from high levels of these glucocorticoids. In contrast, this enzyme does not have the same inactivating effect on some other synthetic glucocorticoids such as dexamethasone.
- METHYLPREDNISOLONE – a methylated analog of prednisolone that has 5-6 times the anti-inflammatory potency of cortisol, and 1/4th cortisol’s potency as a mineralocorticoid.
- DEXAMETHASONE – a potent, long-acting glucocorticoid that has relatively little effect on mineralocorticoid receptors.
- HYDROCORTISONE (Cortisol) – the naturally occurring “stress” hormone that stimulates both glucocorticoid and mineralocorticoid receptors.
- CORTISONE – a naturally occurring inactive form of cortisol. Cortisone is converted to cortisol by 11-β hydroxysteroid dehydrogenase 1 (11β-HSD 1) in the liver.
- FLUDROCORTISONE – has a structure identical to cortisol except for addition of a fluorine atom at the 9α position. This structural change produces a high mineralocorticoid/glucocorticoid potency ratio, and prevents its renal inactivation by 11β-HSD 2. At recommended doses, it is essentially free of glucocorticoid activity. Duration of action is 12-24 hrs. It is used in the treatment of Addison’s disease (in which cells of the zona glomerulosa region of the adrenal cortex that produce aldosterone have been destroyed).
Therapeutically, glucocorticoids are usually given at doses that produce minimal mineralocorticoid stimulation in order to avoid the side effects associated with activation of this pathway (e.g. hypokalemia, volume expansion & hypertension).
Figure 7. Glucocorticoid medications
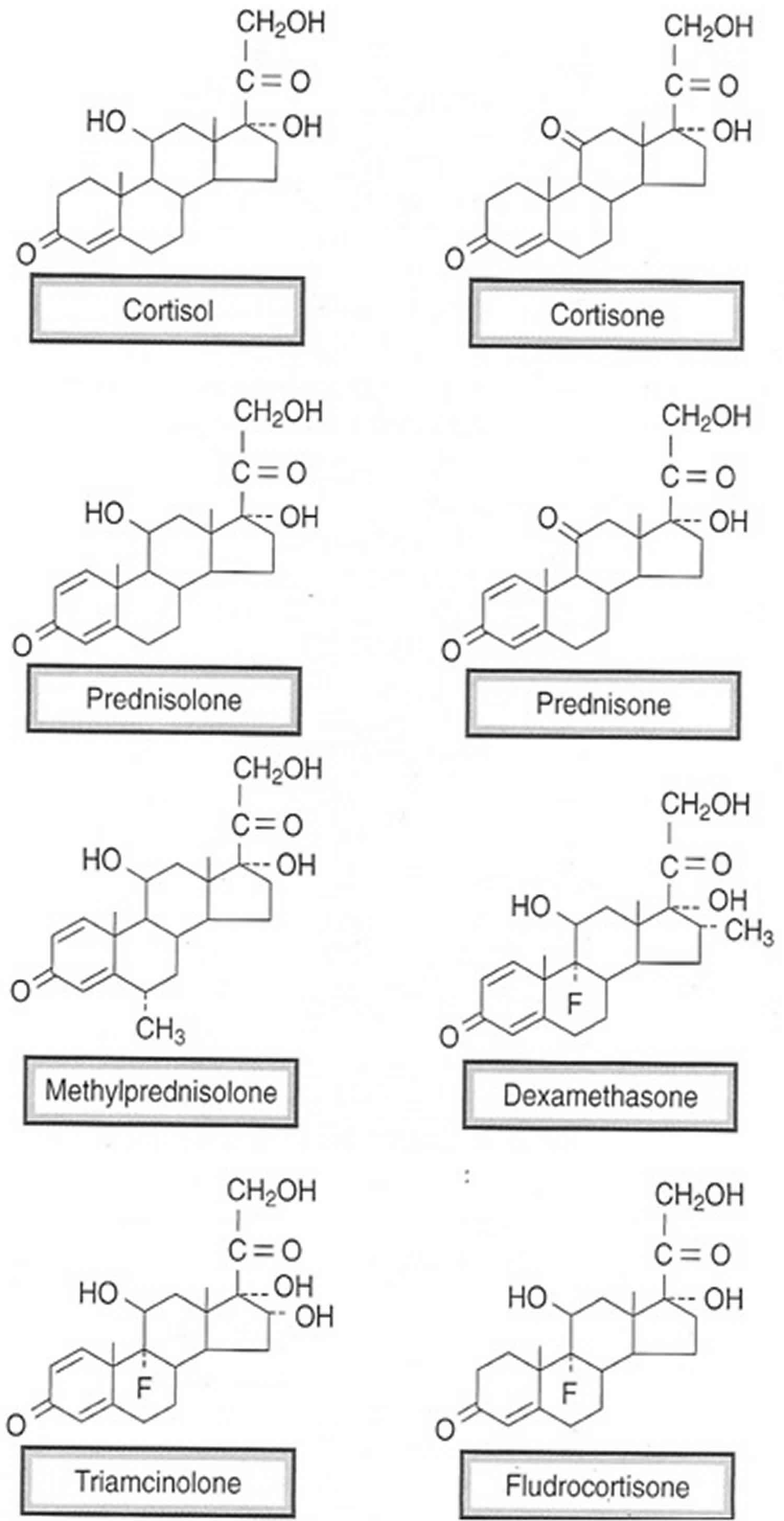
Table 6. Relative Potencies and Duration of Action of Therapeutic Glucocorticoids
| Agent | Relative Glucocorticoid Potency | Relative Mineralocorticoid Potency | Duration of Action |
|---|---|---|---|
| Hydrocortisone (Cortisol) | 1 | 1 | Short |
| Prednisolone | 4-5 | 0.25 | Short |
| Methylprednisolone | 5-6 | 0.25 | Short |
| Dexamethasone | 18 | <0.01 | Long |
| Fludrocortisone | 10 | 125 | Short |
Common conditions treated with glucocorticoid medications include:
- Rheumatoid arthritis, lupus, or other inflammatory joint disease
- Asthma or chronic obstructive pulmonary disease (COPD)
- Inflammatory bowel disease
- Psoriasis or other skin diseases
- Organ transplant (to reduce the risk of rejection)
Glucocorticoid medications can be taken as a pill (by mouth), an injection under the skin or in a vein, a nasal spray or inhaler, or even as a skin ointment or cream. Glucocorticoid medications given by mouth, by vein, or by skin injection are most likely to cause osteoporosis.
Table 7. Therapeutic Effects of Glucocorticoids
| Therapeutic Effect | Mechanism |
|---|---|
| Anti-inflammatory | Inhibit inflammation by blocking the expression & synthesis of inflammatory mediators, and by inducing the expression of anti-inflammatory mediators (e.g. Annexin A1) |
| Immunosuppressive | Directly inhibit T lymphocyte function which suppresses delayed hypersensitivity reactions |
| Antiproliferative | Inhibition of DNA synthesis & epidermal cell turnover |
[Source 4 ]
Table 8. Common Clinical Indications for Systemic Glucocorticoids
| Field of Medicine | Disorders |
|---|---|
| Allergy & Pulmonology | Asthma (moderate/severe), allergic rhinitis, anaphylaxis, urticaria, food/drug allergies |
| Dermatology | Acute severe dermatitis |
| Endocrinology | Adrenal insufficiency |
| GI Diseases | Crohn’s Disease (IBD), ulcerative colitis |
| Hematologic Diseases | Leukemia/lymphoma |
| Ophthalmology | Uveitis (eye inflammation) |
| Rheumatology/Immunology | Rheumatoid arthritis, systemic lupus erythematosus, vasculitis |
| Other | Multiple sclerosis, organ transplant, nephrotic syndrome, cerebral edema |
Figure 8. Glucocorticoid medications effects and side-effects
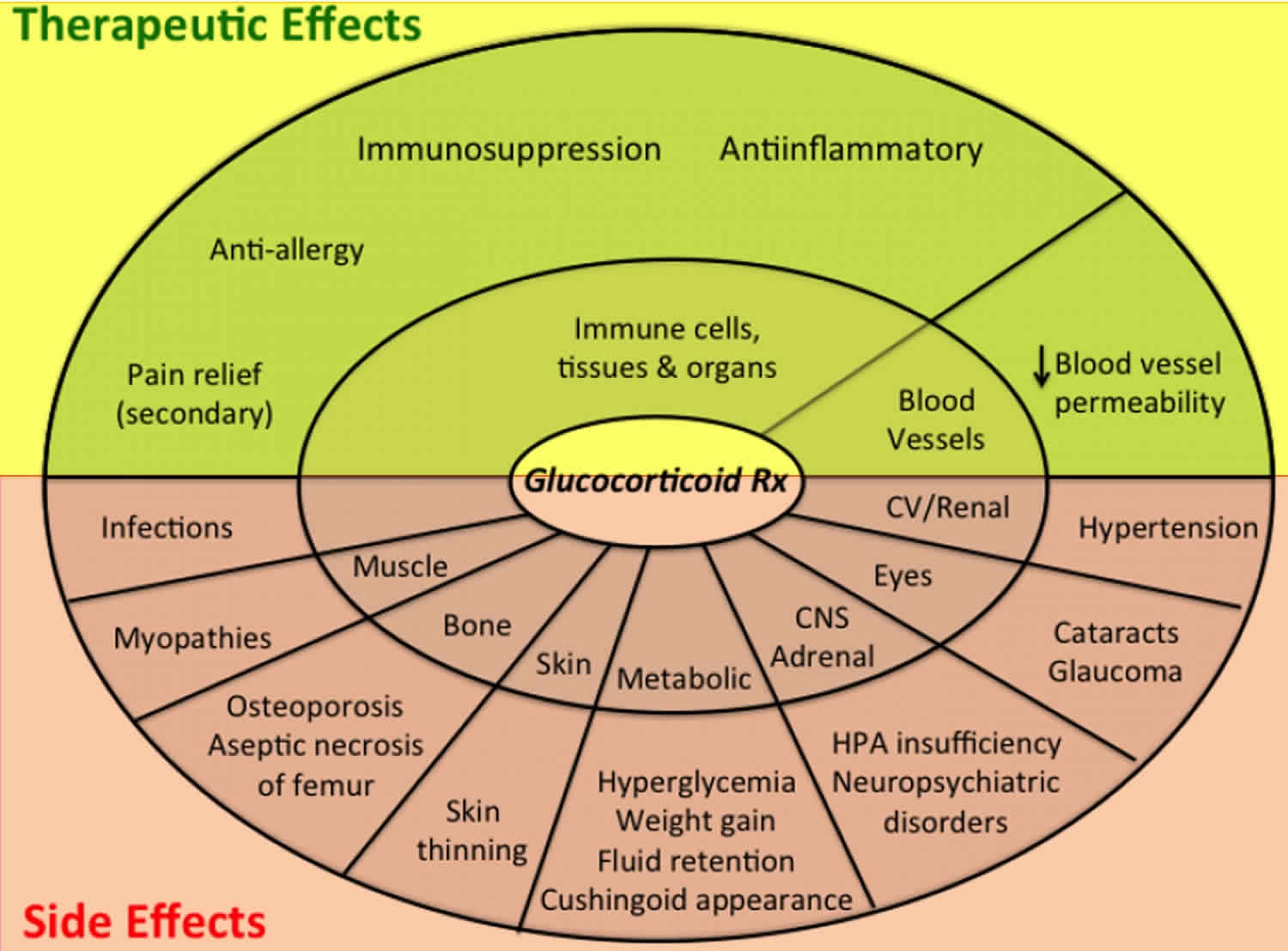
Footnote: The major therapeutic effects (top) and side effects (bottom) produced by glucocorticoids. Many of the side effects are reversible when glucocorticoid treatment is discontinued. Aseptic necrosis of the femur & cataract formation is permanent, and usually requires surgical intervention.
[Source 4 ]Glucocorticoid-induced osteoporosis is a condition in which people who take medicines called glucocorticoids develop osteoporosis—weakening of the bones. Osteoporosis increases the risk of broken bones (fractures).
Glucocorticoid medications start to weaken your bones during the first 3 months of use. The rate of bone loss is greatest within the first 6 months of treatment but continues as long as you take glucocorticoids. The higher your glucocorticoid medications dose, the greater your risk of glucocorticoid-induced osteoporosis. But even low doses can cause glucocorticoid-induced osteoporosis over time. Therefore, experts recommend that doctors prescribe the smallest possible dose for the shortest period of time.
Who is most at risk for glucocorticoid-induced osteoporosis?
Some people who take glucocorticoids are at greater risk for glucocorticoid-induced osteoporosis:
- Women who have gone through menopause
- Men age 50 or older
- Those who have had previous fractures
People who have other risk factors for osteoporosis, including those who:
- Don’t get enough calcium and vitamin D
- Smoke cigarettes
- Drink three or more alcoholic beverages per day
- Have a family history of osteoporosis
Glucocorticoid medication side effects
High-dose, long-term glucocorticoid therapy can produce a constellation of severe toxicities which are summarized in Figures 8 & 9, and Table 9.
Table 9. Glucocorticoids treatment side effects and mechanisms
| Side Effect | Mechanism |
|---|---|
| Hyperglycemia/Diabetes | Increased hepatic glucose output & decreased peripheral glucose utilization (insulin-resistant diabetes mellitus) |
| Central Obesity, Moon Face, Buffalo Hump | Increases effect of lipolytic signals, leading to elevated free fatty acid to fuel gluconeogenesis. Redistribution of fat from extremities to the trunk, back of neck & supraclavicular fossae |
| Muscle Weakness & Wasting | Breakdown of protein & diversion of amino acids to glucose production |
| Weight Gain | Increased appetite (CNS effect) and increased need for insulin over time results in weight gain |
| Osteoporosis | Decreased reabsorption of calcium by the kidney leading to secondary hyperparathyroidism; retardation of bone growth by direct action & decreasing growth hormone. Inhibition of bone deposition. |
| Avascular Necrosis of Femur Head | Animal studies indicate glucocorticoid-induced increased levels of serum lipids result in both increased formation of microemboli in the arteries supplying bone, and fat-related blockage of venous flow from bone. These mechanisms result in increased ischemia in bone, which most commonly affects the femoral head, but can occur in any skeletal site such as the knee, shoulder, ankle or hand |
| Thin Skin that Bruises Easily & Poor Wound Healing | Antiproliferative glucocorticoid effect on fibroblasts & keratinocytes, resulting in dermal atrophy |
| Hirsutism & Acne | due to ACTH-mediated increase of adrenal androgens |
| Hyperpigmentation | direct effect of ACTH on melanocortin 1 receptors |
| Increased Infections | Immunosuppression related to thymic atrophy, decreased production (number) of neutrophils & monocytes, decreased production of cytokines |
| Hypertension | Increased cardiac contractility, increased vascular reactivity to vasoconstrictors (catecholamines, angiotensin 2) |
| Hypomania, Depression, Psychosis | Normal cortisol levels (eucortisolemia) maintains emotional balance |
| Cataract formation & Glaucoma | Increased intraocular pressure & hypoparathyroidism |
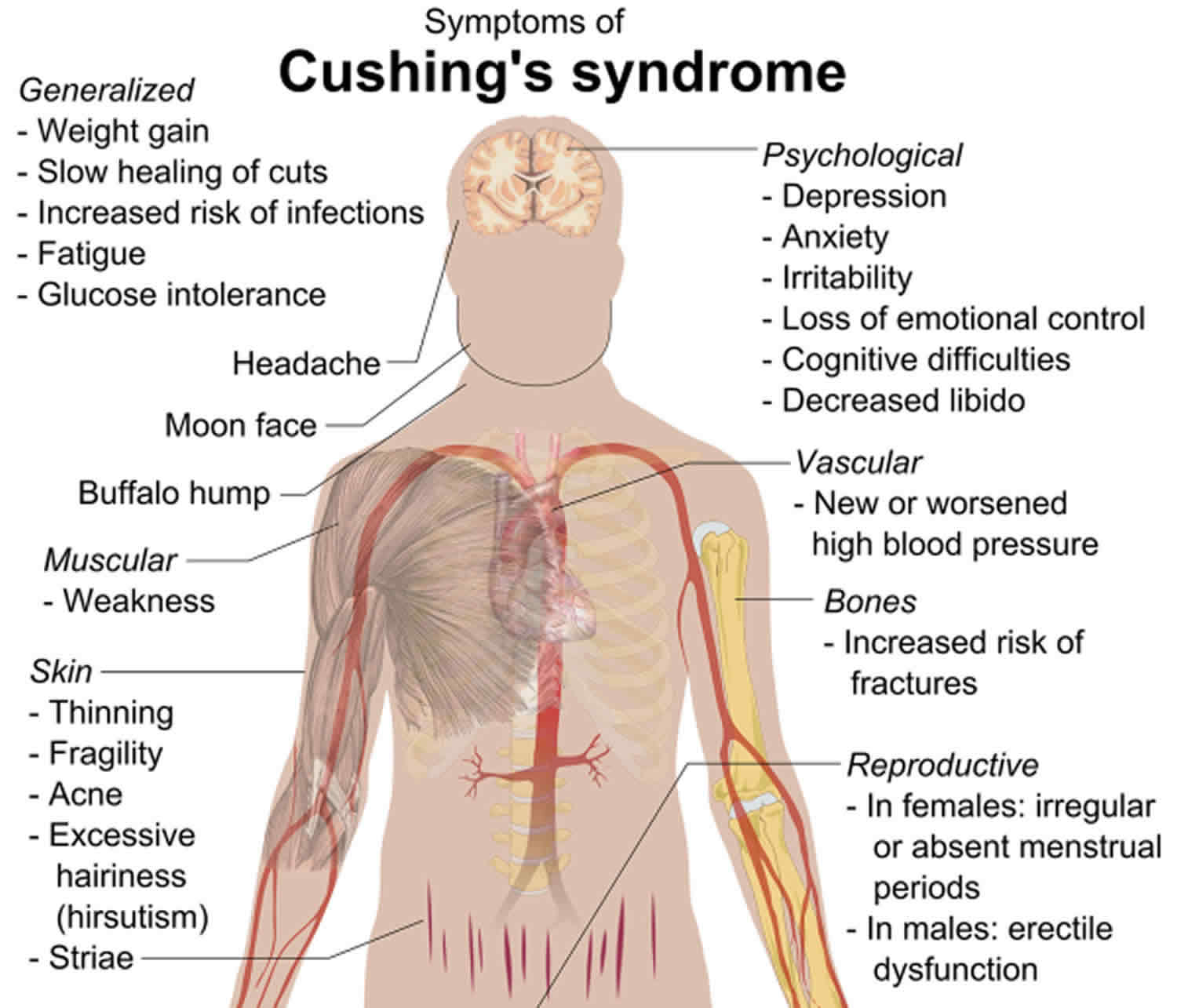
Figure 9. Cushing’s Syndrome
Secondary Adrenal Insufficiency
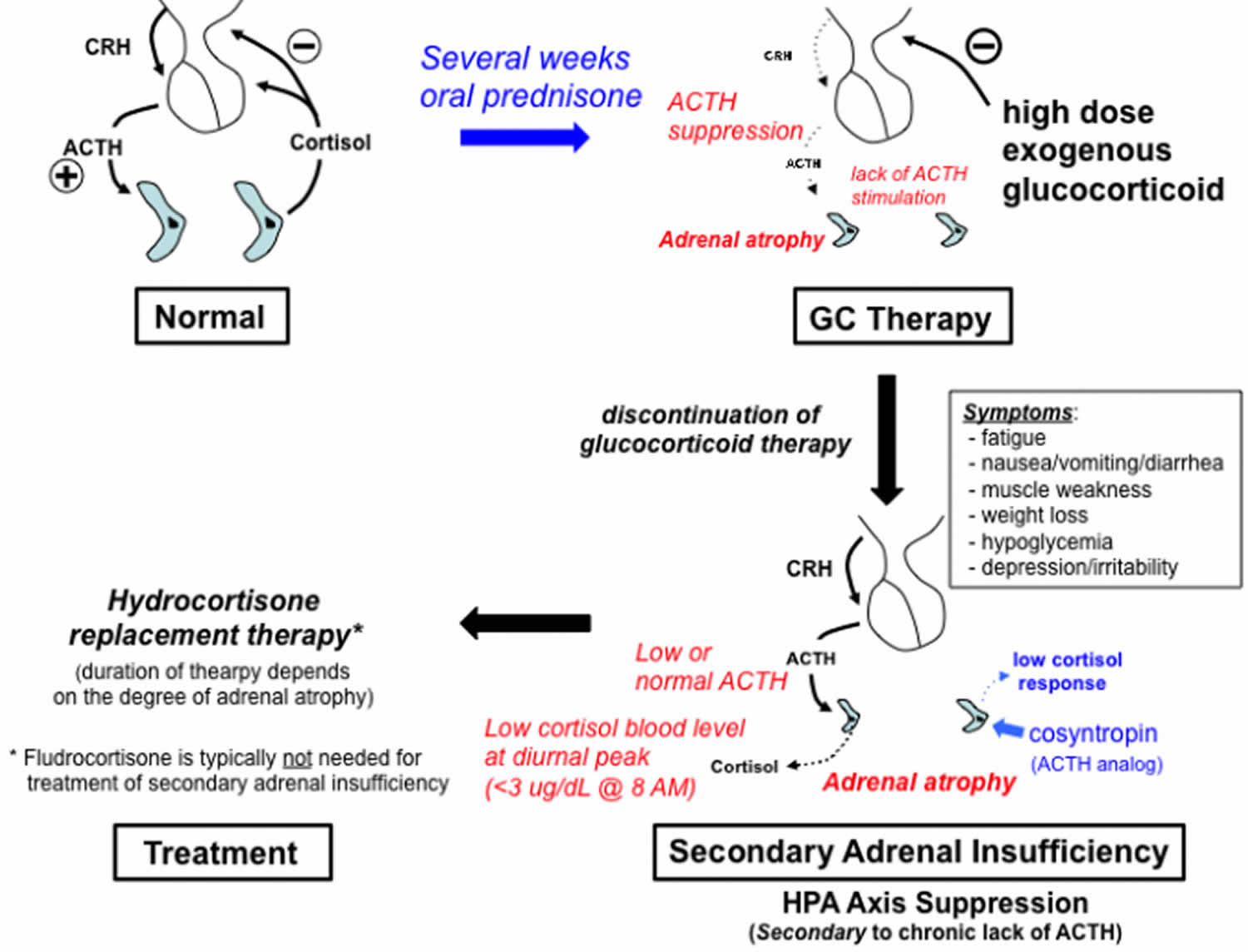
When a patient takes a glucocorticoid for treatment of an illness, the presence of the cortisol-like medication will suppress the release of ACTH by the pituitary (Figure 9). Because the cortisol-producing cells of the adrenal gland (zona fasciculata) depend on ACTH stimulation for normal homeostasis, a reduction in ACTH release for any significant time (e.g. several weeks or more) will result in a progressive atrophy of this zone. Adrenal atrophy reduces the ability of the adrenal gland to produce normal levels of cortisol when therapy with a glucocorticoid (such as prednisone) is discontinued.
Several weeks of exogenous glucocorticoid therapy are typically required for development of adrenal insufficiency (Salvatori, 2005; Chrousos, 2015). Longer-acting glucocorticoids, and higher doses of systemic glucocorticoids (vs topical or inhaled formulations) have been associated with a higher risk of developing adrenal suppression. There are no current evidence-based guidelines regarding the optimal method for discontinuing glucocorticoid therapy, although gradual tapering of dosages over a period of weeks to months, prior to complete discontinuation, is recommended to allow time for recovery of the hypothalamic-pituitary-adrenal axis (Liu et al, 2013).
The signs and symptoms of secondary adrenal insufficiency that result from a sudden withdrawal of glucocorticoids in the presence of HPA axis suppression are typically similar to those seen with Addison’s disease, but don’t include changes in skin pigmentation (because ACTH secretion is not increased), or significant electrolyte disturbances (because aldosterone is regulated by the renin-angiotensin system rather than ACTH)(Nieman 2013).
Hydrocortisone (given twice or three times per day) is commonly preferred for the treatment of adrenal insufficiency because its short half-life allows it to more closely mimic the normal circadian rhythm for cortisol.
Figure 10. Secondary adrenal insufficiency caused by chronic glucocorticoid treatment
Glucocorticoid excess
Cushing’s syndrome, also called hypercortisolism, is a rare endocrine disorder caused by chronic exposure of the body’s tissues to excess levels of cortisol (see Figure 9). The most common cause of Cushing syndrome is taking too much glucocorticoid or corticosteroid medicine to treat inflammatory illnesses. This form of Cushing syndrome is called exogenous Cushing syndrome. Prednisone, dexamethasone, and prednisolone are examples of this type of medicine. Glucocorticoids mimic the action of the body’s natural hormone cortisol. These drugs are used to treat many conditions such as asthma, skin inflammation, cancer, bowel disease, joint pain, and rheumatoid arthritis.
Other people develop Cushing syndrome because their body produces too much cortisol. This hormone is made in the adrenal glands. Causes of too much cortisol are:
- Cushing disease, which occurs when the pituitary gland makes too much of the hormone ACTH (adrenocorticotropic hormone, a substance that controls the release of cortisol). ACTH then signals the adrenal glands to produce too much cortisol. Pituitary adenomas (benign tumors of the pituitary gland) that secrete increased amounts of ACTH can also spur overproduction of cortisol.
- Tumor of the adrenal gland.
- Tumor elsewhere in the body that produces corticotropin-releasing hormone (CRH)
- Tumors elsewhere in the body that produce ACTH (ectopic Cushing syndrome) – a condition in which ACTH is produced by various types of potentially malignant tumors that occur in different parts of the body.
Glucocorticoid excess symptoms
Symptoms vary. Common symptoms of Cushing’s syndrome include upper body obesity, severe fatigue and muscle weakness, high blood pressure, backache, elevated blood sugar, easy bruising, and bluish-red stretch marks on the skin. In women, there may be increased growth of facial and body hair, and menstrual periods may become irregular or stop completely. Neurological symptoms include difficulties with memory and neuromuscular disorders.
Not everyone with Cushing syndrome has the same symptoms. Some people have many symptoms while others have hardly any symptoms.
Most people with glucocorticoid excess have:
- Round, red, full face (moon face)
- Slow growth rate (in children)
- Weight gain with fat accumulation on the trunk, but fat loss from the arms, legs, and buttocks (central obesity)
Skin changes can include:
- Skin infections
- Purple stretch marks (1/2 inch or 1 centimeter or more wide) called striae on the skin of the abdomen, upper arms, thighs, and breasts
- Thin skin with easy bruising (especially on the arms and hands)
Muscle and bone changes include:
- Backache, which occurs with routine activities
- Bone pain or tenderness
- Collection of fat between the shoulders and above collar bones
- Rib and spine fractures caused by thinning of the bones
- Weak muscles, especially of the hips and shoulders
Body-wide (systemic) changes include:
- Type 2 diabetes mellitus
- High blood pressure (hypertension)
- Increased cholesterol and triglycerides (hyperlipidemia)
Women with glucocorticoid excess may have:
- Excess hair growth on the face, neck, chest, abdomen, and thighs
- Periods that become irregular or stop
Men may have:
- Decreased or no desire for sex (low libido)
- Erection problems
Other symptoms that may occur with glucocorticoid excess:
- Mental changes, such as depression, anxiety, or changes in behavior
- Fatigue
- Headache
- Increased thirst and urination
Health problems that may result from Cushing syndrome include any of the following:
- Diabetes
- Enlargement of pituitary tumor
- Fractures due to osteoporosis
- High blood pressure
- Kidney stones
- Serious infections
Glucocorticoid excess diagnosis
Your health care provider will perform a physical exam and ask about your symptoms and the medicines you are taking. Tell the your doctor about all medicines you have been taking for the past several months. Also tell your doctor about shots that you received at a provider’s office.
Laboratory tests that may be done to diagnose glucocorticoid excess and identify the cause are:
- Blood cortisol level
- Blood sugar
- Saliva cortisol level
- Dexamethasone suppression test
- 24-hour urine for cortisol and creatinine
- ACTH level
- ACTH stimulation test (rarely)
Tests to determine the cause or complications may include:
- Abdominal CT
- Pituitary MRI
- Bone mineral density
Glucocorticoid excess treatment
Treatment depends on the cause.
Glucocorticoid excess caused by corticosteroid use:
- Your doctor will instruct you to slowly decrease the medicine dosage. Stopping the medicine suddenly can be dangerous.
- If you cannot stop taking the medicine because of disease, your high blood sugar, high cholesterol levels, and bone thinning or osteoporosis should be closely monitored.
With glucocorticoid excess caused by a pituitary or a tumor that releases ACTH (Cushing disease), you may need:
- Surgery to remove the tumor
- Radiation after removal of a pituitary tumor (in some cases)
- Cortisol replacement therapy after surgery
- Medicines to replace pituitary hormones that become deficient
- Medicines to prevent the body from making too much cortisol
With glucocorticoid excess due to an adrenal tumor or other tumors:
- You may need surgery to remove the tumor.
- If the tumor cannot be removed, you may need medicines to help block the release of cortisol.
Cushing syndrome prognosis
Removing the tumor may lead to full recovery, but there is a chance that the condition will return.
Survival for people with Cushing syndrome caused by tumors depends on the tumor type.
Untreated, Cushing syndrome can be life threatening.
References- Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med 1995; 332:1351-1362
- Hoehn K, Marieb EN (2010). Human Anatomy & Physiology. San Francisco: Benjamin Cummings. ISBN 978-0-321-60261-9.
- YONG S. CHYUN, BARBARA E. KREAM, LAWRENCE G. RAISZ; Cortisol Decreases Bone Formation by Inhibiting Periosteal Cell Proliferation, Endocrinology, Volume 114, Issue 2, 1 February 1984, Pages 477–480, https://doi.org/10.1210/endo-114-2-477
- Glucocorticoids. http://tmedweb.tulane.edu/pharmwiki/doku.php/all_steroid_drugs
- George P. Chrousos, Evangelia Charmandari, Tomoshige Kino; Glucocorticoid Action Networks—An Introduction to Systems Biology, The Journal of Clinical Endocrinology & Metabolism, Volume 89, Issue 2, 1 February 2004, Pages 563–564, https://doi.org/10.1210/jc.2003-032026
- Boumpas DT, Chrousos GP, Wilder RL, Cupps TR, Balow JE. Glucocorticoid therapy for immune-mediated diseases: basic and clinical correlates. Ann Intern Med 1993; 119:1198-1208
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell 1995; 83:835-839
- Androutsellis-Theotokis A, Chrousos GP, McKay RD, DeCherney AH, Kino T. Expression profiles of the nuclear receptors and their transcriptional coregulators during differentiation of neural stem cells. Horm Metab Res 2013; 45:159-168
- Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O’Shea JJ, Chrousos GP, Bornstein SR. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J 2002; 16:61-71
- Parente L, Solito E (2004): Annexin 1. More than an anti-phospholipase protein. Inflamm res 53:125-132. DOI 10.1007/s00011-003-1235-z
- Chatham WW (2014): Glucocorticoid effects on the immune system. In: UpToDate, Basow, DS (Ed), Waltham, MA.
- Girol AP et al (2013): Anti-Inflammatory Mechanisms of the Annexin A1 Protein and Its Mimetic Peptide Ac2-26 in Models of Ocular Inflammation In Vivo and In Vitro. J Immunology 190(11):5689-5701. doi: 10.4049/jimmunol.1202030
- Perretti M, D’Acquisto F (2009): Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nature Reviews Immunology 9:62-70. doi:10.1038/nri2470
- Miller ML (2014): Glucocorticoid-induced myopathy. In: UpToDate, Basow, DS (Ed), Waltham, MA.
- Busti AJ (2015): Average Increases in White Blood Cell (WBC) Counts with Glucocorticoids (e.g., Dexamethasone, Methylprednisolone, and Prednisone). EBM Consult. October 2015.
- Schimmer BP, Funder JW (2011): ACTH, Adrenal Steroids, and Pharmacology of the Adrenal Cortex (Chapter 42). In: Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12th Edition. Brunton LL (Editor). McGraw Hill
- Molnár Á, Patócs A, Likó I, et al. An unexpected, mild phenotype of glucocorticoid resistance associated with glucocorticoid receptor gene mutation case report and review of the literature. BMC Med Genet. 2018;19(1):37. Published 2018 Mar 6. doi:10.1186/s12881-018-0552-6 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5840839/
- Nicolaides NC, Kino T, Chrousos G, et al. Primary Generalized Glucocorticoid Resistance or Chrousos Syndrome. [Updated 2017 Sep 10]. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK278930
- Chrousos GP, Detera-Wadleigh SD, Karl M. Syndromes of glucocorticoid resistance. Ann Intern Med. 1993;119:1113–1124. doi: 10.7326/0003-4819-119-11-199312010-00009
- Ruiz M, Lind U, Gåfvels M, Eggertsen G, Carlstedt-Duke J, Nilsson L, et al. Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance. Clin Endocrinol. 2001;55:363–371. doi: 10.1046/j.1365-2265.2001.01323.x
- Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93:1563–1572. doi: 10.1210/jc.2008-0040
- Kino T, Vottero A, Charmandari E, Chrousos GP. Familial/sporadic glucocorticoid resistance syndrome and hypertension. Ann N Y Acad Sci. 2002;970:101–111. doi: 10.1111/j.1749-6632.2002.tb04416.x
- Charmandari E, Kino T, Chrousos GP. Familial/sporadic glucocorticoid resistance: clinical phenotype and molecular mechanisms. Ann N Y Acad Sci. 2004;1024:168–181. doi: 10.1196/annals.1321.014.
- Charmandari E, Kino T, Chrousos GP. Familial/sporadic glucocorticoid resistance: clinical phenotype and molecular mechanisms. Ann N Y Acad Sci. 2004;1024:168–181. doi: 10.1196/annals.1321.014
- Nicolaides NC, Chrousos GP, Charmandari E. Adrenal Insufficiency. [Updated 2017 Oct 14]. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279083
- Kong MF, Jeffocoate W. Eighty-six cases of Addison’s disease. Clin Endocrinol (Oxf ) 1994; 41(6):757-61.
- Aron DC, Findling JW, Tyrrell JB. Glucocorticoids and adrenal androgens. In: Greenspan’s Basic and Clinical Endocrinology. Gardner DG, Shoback D (eds). Mc Graw Hill 2007; pp 367-378.
- Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003; 361:1881-1893
- Tomlinson JW, Holden N, Hills RK, Wheatley K, Clayton RN, Bates AS, Sheppard MC, Stewart PM. Association between premature mortality and hypopituitarism. West Midlands Prospective Hypopituitary Study Group. Lancet 2001; 357(9254):425-31.
- Regal M, Páramo C, Sierra SM, Garcia-Mayor RV. Prevalence and incidence of hypopituitarism in an adult Caucasian population in northwestern Spain. Clin Endocrinol 2001; 55:735-40.
- Gomez MT, Magiakou MA, Mastorakos G, Chrousos GP. The pituitary corticotroph is not the rate-limiting step in the postoperative recovery of the hypothalamic-pituitary-adrenal axis in patients with Cushing syndrome. J Clin Endocrinol Metab 1993; 77(1):173-7.
- Bornstein SR. Predisposing factors for adrenal insufficiency. N Engl J Med 2009; 360(22):2328-39.
- Erichsen MM, Lovas K, Skinningsrud B, Wolff AB, Undlien DE, Svartberg J, Fougner KJ, Berg TJ, Bollerslev J, Mella B, Carlson JA, Erlich H, Husebye ES. Clinical, immunological, and genetic features of autoimmune primary adrenal insufficiency: observation from a Norwegian registry. J Clin Endocrinol Metab 2009; 94(12):4882-90.
- Husebye ES, Løvås K. Immunology of Addison’s disease and premature ovarian failure. Endocrinol Metab Clin North Am 2009; 38(2):389-405.
- Mitchell AL, Pearce SH. Autoimmune Addison disease: pathophysiology and genetic complexity. Nat Rev Endocrinol 2012; 8(5):306-16.
- Akirav EM, Ruddle NH, Herold KC. The role of AIRE in human autoimmune disease. Nat Rev Endocrinol 2011; 7(1):25-33.
- Kemp S, Berger J, Aubourg P. X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta. 2012; 1822(9): 1465-74.
- Rao RH. Bilateral massive adrenal hemorrhage. Med Clin North Am 1995; 79(1):107-29.
- Caron P, Chabannier MH, Cambus JP, Fortenfant F, Otal P, Suc JM. Definitive adrenal insufficiency due to bilateral adrenal hemorrhage and primary antiphospholipid syndrome. J Clin Endocrinol Metab 1998; 83(5):1437-9.
- Bhatia E, Jain SK, Gupta RK, Pandey R. Tuberculous Addison’s disease: lack of normalization of adrenocortical function after anti-tuberculous chemotherapy. Clin Endocrinol (Oxf) 1998; 48(3):355-9.
- Jabre P, Combes X, Lapostolle F, Dhaouadi M, Ricard-Hibon A, Vivien B, Bertrand L, Beltramini A, Gamand P, Albizzati S, Perdrizet D, Lebail G, Chollet-Xemard C, Maxime V, Brun-Buisson C, Lefrant JY, Bollaert PE, Megarbane B, Ricard JD, Anguel N, Vicaut E, Adnet F; KETASED Collaborative Study Group. Etomidate versus ketamine for rapid sequence intubation in acutely ill patients: a multicentre randomised controlled trial. Lancet 2009; 374(9686):293-300.
- Sonino N. The use of ketoconazole as an inhibitor of steroid production. N Engl J Med 1987; 317:812.
- Schoneshofer M, Claus M. Multiple-sites of inhibition by intravenous metyrapone of human adrenal steroidogenesis. Acta Endocrinol (Copenh) 1985; 109(3):378-85.
- Elias AN, Gwinup G. Effects of some clinically encountered drugs on steroid synthesis and degradation. Metabolism 1980; 29:582.
- Rock EP, Goodman V, Jiang JX, Mahjoob K, Verbois SL, Morse D, Dagher R, Justice R, Pazdur R. Food and Drug Administration drug approval summary: Sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist 2007; 12(1):107-13.
- Stewart PM. The adrenal cortex. In: Williams Textbook of Endocrinology. Larsen PR, Kronenberg HM, Melmed S, Polonsky KS (eds). Saunders, Philadelphia, PA. 2003; pp 525-532.
- Annetta M, Maviglia R, Proietti R, Antonelli M. Use of corticosteroids in critically ill septic patients: a review of mechanisms of adrenal insufficiency in sepsis and treatment. Curr Drug Targets 2009; 10(9):887-94.
- Bornstein SR, Engeland WC, Ehrhart-Bornstein M, Herman JP. Dissociation of ACTH and glucocorticoids. Trends Endocrinol Metab 2008; 19(5):175-80.
- Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci 2012; 1261:55-63.
- Marik PE, Pastores SM, Annane D, Meduri GU, Sprung CL, Arlt W, Keh D, Briegel J, Beishuizen A, Dimopoulou I, Tsagarakis S, Singer M, Chrousos GP, Zaloga G, Bokhari F, Vogeser M; American College of Critical Care Medicine. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 2008; 36(6):1937-49.
- Langlois F, Lim DST, Fleseriu M. Update on adrenal insufficiency: diagnosis and management in pregnancy. Curr Opin Endocrinol Diabetes Obes. 2017; 24(3):184-192.
- Suri D, Moran J, Hibbard JU, Kasza K, Weiss RE. Assessment of adrenal reserve in pregnancy: defining the normal response to the adrenocorticotropin stimulation test. J Clin Endocrinol Metab 2006; 91(10):3866-72.
- Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, Husebye ES, Merke DP, Murad MH, Stratakis CA, Torpy DJ. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016; 101(2):364-89.
- Cronin CC, Callaghan N, Kearney PJ, Murnaghan DJ, Shanahan F. Addison disease in patients treated with glucocorticoid therapy. Arch Intern Med 1997; 157:456.
- Ohtani R, Yayama K, Takano M, Itoh N, Okamoto H. Stimulation of angiotensinogen production in primary cultures of rat hepatocytes by glucocorticoid, cyclic adenosine 3′,5′-monophosphate, and interleukin-6. Endocrinology 1992; 130:1331.
- Hägg E, Asplund K, Lithner F. Value of basal plasma cortisol assays in the assessment of pituitary-adrenal insufficiency. Clin Endocrinol 1987; 26:221.
- Oelkers W, Diederich S, Bähr V. Diagnosis and therapy surveillance in Addison’s disease: rapid adrenocorticotropin (ACTH) test and measurement of plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab 1992; 75(1):259-64.
- Nye EJ, Grice JE, Hockings GI, Strakosch CR, Crosbie GV, Walters MM, Jackson RV. Comparison of adrenocorticotropin (ACTH) stimulation tests and insulin hypoglycemia in normal humans: low dose, standard high dose, and 8-hour ACTH-(1-24) infusion tests. J Clin Endocrinol Metab 1999; 84(10):3648-55.
- Bratland E, Husebye ES. Cellular immunity and immunopathology in autoimmune Addison’s disease. Mol Cell Endocrinol 2011; 336(1-2):180-90.
- Abdu TAM, Elhadd TA, Neary E, Clayton RN. Comparison of the low dose short Synacthen test (1 µg), the conventional dose short synacthen test (250 µg), and the insulin tolerance test for assessment of the hypothalamic-pituitary-adrenal axis in patients with pituitary disease. J Clin Endocrinol Metab 1999; 84:838.
- Wade M, Baid S, Calis K, Raff H, Sinaii N, Nieman L. Technical details influence the diagnostic accuracy of the 1 microg ACTH stimulation test. Eur J Endocrinol 2010; 162(1):109-13.
- Rose LI, Williams GH, Jagger PI, Lauler DP. The 48-hour adrenocorticotropin infusion test for adrenocortical insufficiency. Ann Intern Med 1970; 73:49.
- Grossman AB. Clinical Review#: The diagnosis and management of central hypoadrenalism. J Clin Endocrinol Metab 2010; 95(11):4855-63.
- Gold PW, Kling MA, Khan I, Calabrese JR, Kalogeras K, Post RM, Avgerinos PC, Loriaux DL, Chrousos GP. Corticotropin releasing hormone: relevance to normal physiology and to the pathophysiology and differential diagnosis of hypercortisolism and adrenal insufficiency. Adv Biochem Psychopharmacol 1987; 43:183-200.
- Ouyang T, Rothfus WE, Ng JM, Challinor SM. Imaging of the pituitary. Radiol Clin North Am 2011; 49(3):549-71.
- Bleicken B, Hahner S, Loeffler M, Ventz M, Decker O, Allolio B, Quinkler M. Influence of hydrocortisone dosage scheme on health-related quality of life in patients with adrenal insufficiency. Clin Endocrinol (Oxf) 2010; 72(3):297-304.
- Neary N, Nieman L. Adrenal insufficiency: etiology, diagnosis and treatment. Curr Opin Endocrinol Diabetes Obes 2010; 17(3):217-23.
- Bleicken B, Hahner S, Loeffler M, Ventz M, Allolio B, Quinkler M. Impaired subjective health status in chronic adrenal insufficiency: impact of different glucocorticoid replacement regimens. Eur J Endocrinol 2008; 159(6):811-7.
- Callies F, Fassnacht M, van Vlijmen JC, Koehler I, Huebler D, Seibel MJ, Arlt W, Allolio B. Dehydroepiandrosterone replacement in women with adrenal insufficiency: effects on body composition, serum leptin, bone turnover, and exercise capacity. J Clin Endocrinol Metab 2001; 86(5):1968-72.
- Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, Johnson TA, Voss TC, Lightman SL, Hager GL. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol 2009; 11(9):1093-102.
- Debono M, Ghobadi C, Rostami-Hodjegan A, Huatan H, Campbell MJ, Newell- Price J, Darzy K, Merke DP, Arlt W, Ross RJ. Modified-release hydrocortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab 2009; 94(5):1548-54.
- Johannsson G, Nilsson AG, Bergthorsdottir R, Burman P, Dahlqvist P, Ekman B, Engström BE, Olsson T, Ragnarsson O, Ryberg M, Wahlberg J, Biller BM, Monson JP, Stewart PM, Lennernäs H, Skrtic S. Improved cortisol exposure-time profile and outcome in patients with adrenal insufficiency: a prospective randomized trial of a novel hydrocortisone dual-release formulation. J Clin Endocrinol Metab 2012; 97(2):473-81.
- Løvås K, Gjesdal CG, Christensen M, Wolff AB, Almås B, Svartberg J, Fougner KJ, Syversen U, Bollerslev J, Falch JA, Hunt PJ, Chatterjee VK, Husebye ES. Glucocorticoid replacement therapy and pharmacogenetics in Addison’s disease: effects on bone. Eur J Endocrinol 2009; 160(6):993-1002.
- Falorni A, Minarelli V, Morelli S. Therapy of adrenal insufficiency: an update. Endocrine 2013; 43(3):514-28.
- Arlt W. The approach to the adult with newly diagnosed adrenal insufficiency. J Clin Endocrinol Metab 2009; 94(4):1059-67.
- Charmandari E, Kino T, Chrousos GP. Glucocorticoids. In Neonatal and Pediatric Pharmacology, 4th Edition. Yaffe SJ and Aranda JV (eds), Lippincott Williams & Wilkins, Philadelphia, PA, USA. 2010; Chapter 51; pages: 760-772.





{kind=link}