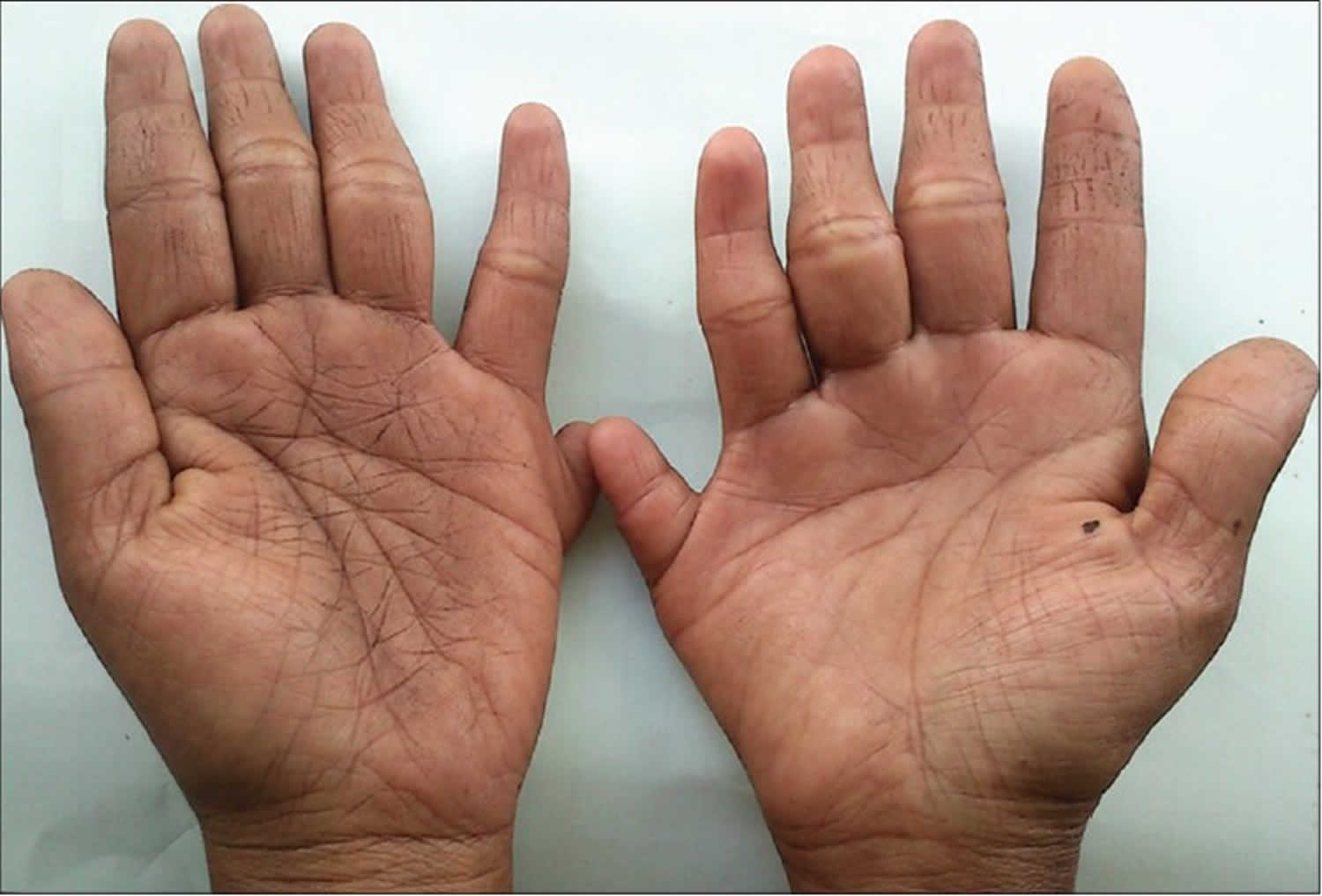
What is Holt Oram syndrome
Holt-Oram syndrome also known as the atriodigital dysplasia syndrome, is a rare autosomal dominant genetic disorder that is characterized by abnormalities in the bones of the upper limb, a family or personal history of a congenital heart malformation, and/or an abnormality in the electrical impulses that coordinate the muscle contractions of the heart (cardiac conduction defect). In some affected individuals, an abnormal wrist (carpal) bone is the only evidence of the disease. Seventy-five percent of those affected have a congenital heart malformation. Holt-Oram syndrome is an autosomal dominant genetic condition that is associated with an abnormality in the TBX5 gene. While this mutation can be inherited, most cases result from a new mutation in patients without a family history of the disorder. Holt-Oram syndrome is estimated to affect 1 in 100,000 individuals 1.
People with Holt-Oram syndrome have abnormally developed bones in their upper limbs. At least one abnormality in the bones of the wrist (carpal bones) is present in affected individuals. Often, these wrist bone abnormalities can be detected only by x-ray. Individuals with Holt-Oram syndrome may have additional bone abnormalities including a missing thumb, a long thumb that looks like a finger, partial or complete absence of bones in the forearm, an underdeveloped bone of the upper arm, and abnormalities of the collar bone or shoulder blades. These skeletal abnormalities may affect one or both of the upper limbs. If both upper limbs are affected, the bone abnormalities can be the same or different on each side. In cases where the skeletal abnormalities are not the same on both sides of the body, the left side is usually more severely affected than the right side.
About 75 percent of individuals with Holt-Oram syndrome have heart (cardiac) problems, which can be life-threatening. The most common problem is a defect in the muscular wall (septum) that separates the right and left sides of the heart. A hole in the septum between the upper chambers of the heart (atria) is called an atrial septal defect (ASD), and a hole in the septum between the lower chambers of the heart (ventricles) is called a ventricular septal defect (VSD). Some people with Holt-Oram syndrome have cardiac conduction disease, which is caused by abnormalities in the electrical system that coordinates contractions of the heart chambers. Cardiac conduction disease can lead to problems such as a slower-than-normal heart rate (bradycardia) or a rapid and uncoordinated contraction of the heart muscle (fibrillation). Cardiac conduction disease can occur along with other heart defects (such as ASD or VSD) or as the only heart problem in people with Holt-Oram syndrome.
The features of Holt-Oram syndrome are similar to those of a condition called Duane-radial ray syndrome; however, these two disorders are caused by mutations in different genes.
What causes Holt-Oram syndrome?
Mutations in the TBX5 gene which has been mapped on the long arm (q) of chromosome 12 (12q24.1) cause Holt-Oram syndrome. TBX5 gene provides instructions for making a protein that plays a role in the development of the heart and upper limbs before birth. In particular, this gene appears to be important for the process that divides the developing heart into four chambers (cardiac septation). The TBX5 gene also appears to play a critical role in regulating the development of bones in the arm and hand. Mutations in this gene probably disrupt the development of the heart and upper limbs, leading to the characteristic features of Holt-Oram syndrome.
A TBX5 gene mutation has been identified in approximately 74% of individuals affected with Holt-Oram syndrome. This is likely to be an underestimate of the true TBX5 mutation frequency, since genetic testing does not always detect all types of mutations. Currently, there are more than 70 known mutations in the TBX5 gene that cause Holt-Oram syndrome. Potential causes for the remainder of affected individuals include incorrect reading and translation of the TBX5 gene during protein production.
Holt-Oram syndrome inheritance pattern
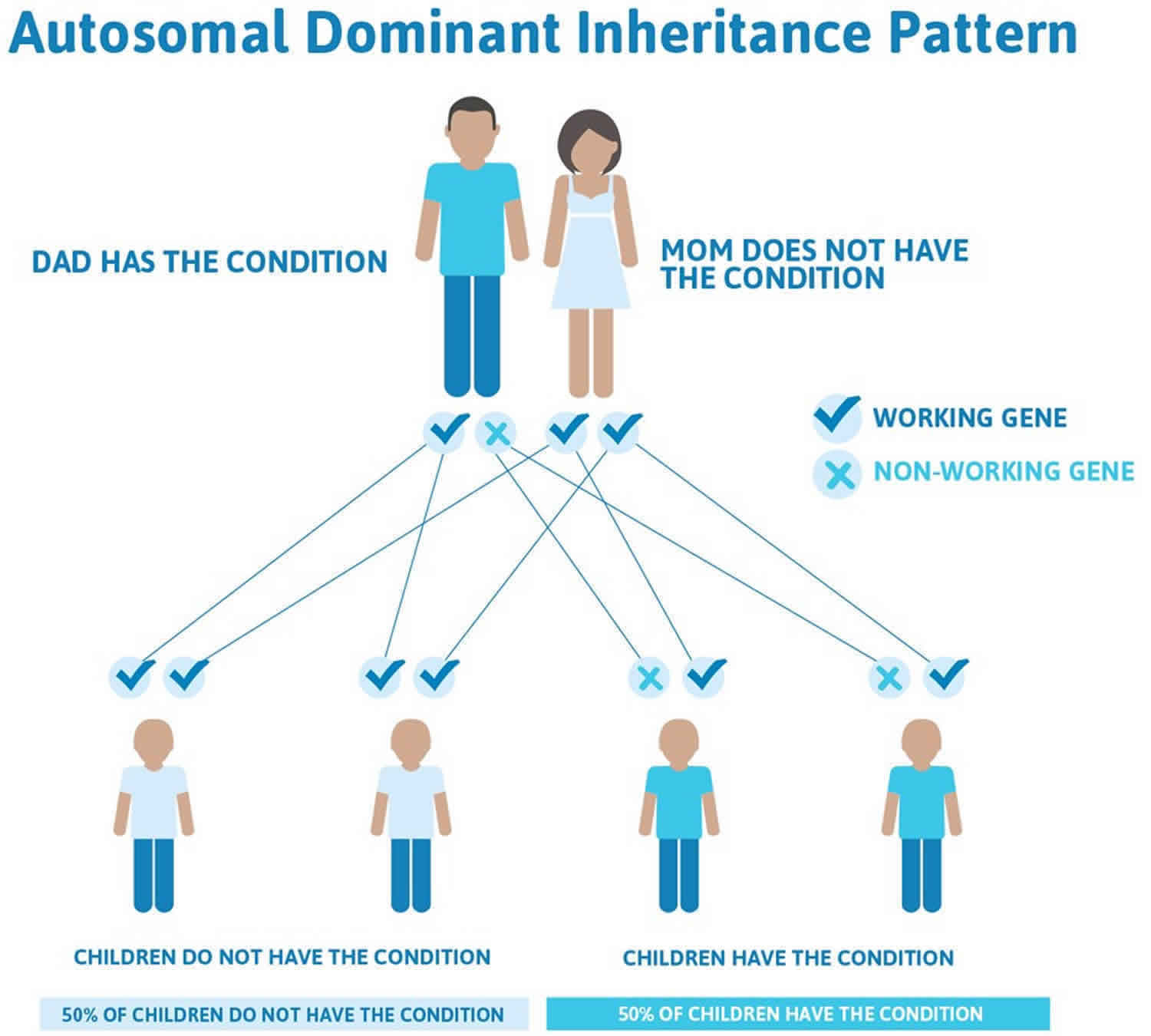
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Approximately 85% of cases of Holt-Oram syndrome are thought to be due to new mutations in the TBX5 gene.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 1 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 1. Holt-Oram syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Holt-Oram syndrome symptoms
The signs and symptoms of Holt-Oram syndrome include birth defects affecting the hands, wrists, arms, and heart. People with Holt-Oram syndrome have at least one of the bones in the wrist, the carpal bones, that is abnormally formed. Other bones of the upper limbs may also have formed abnormally. These may include having a missing thumb, a long thumb that looks like a finger, bones in the forearm or upper arm partially or completely missing, and problems with the shape of the collar bone or shoulder blades. People with Holt-Oram syndrome may be unable to fully extend or rotate their arms 2. In some cases, the bone abnormalities associated with Holt-Oram syndrome may only be visible on x-ray.
About 75% of people who have Holt-Oram syndrome have heart problems. The most common problems are holes in the walls that separate the heart into four areas (chambers). These heart defects are known as atrial septal defects (ASD) or ventricular septal defects (VSD) depending on the exact location of the hole. Other heart defects including patent ductus arteriosus (PDA) have been reported 3. The heart defects associated with Holt-Oram syndrome may not cause any problems or may cause symptoms such as having a hard time breathing, tiring easily (fatigue), having high blood pressure in the arteries of the lungs (pulmonary hypertension), and being smaller than expected (failure to thrive). In some cases, these heart defects may be life-threatening 2.
Some people with Holt-Oram syndrome have cardiac conduction disease, which is when the electrical system that coordinates the heartbeat does not work correctly. Cardiac conduction disease can lead to problems such as a slower than expected heart rate (bradycardia) or a rapid and uncoordinated contraction of the heart muscle (fibrillation). Health problems associated with cardiac conduction disease may be more likely to develop as a person gets older 4.
The symptoms of Holt-Oram syndrome are similar to those of another syndrome called Duane-radial ray syndrome. However, these syndromes are caused by genetic changes (pathogenic variants or mutations) in different genes. Holt-Oram syndrome may be associated with a wide range of signs and symptoms, even among members of the same family. This is called variable expressivity 2.
Holt-Oram syndrome diagnosis
A diagnosis of Holt-Oram syndrome may be suspected when a person is found to have changes in the way the bones of the wrist and other bones of the upper limb are formed. The diagnosis can be confirmed if a person has specific bone changes and a personal or family history of an atrial septal defect, ventricular septal defect, or cardiac conduction disease 4. In order to establish the diagnosis, a doctor may order tests including an x-ray of the hands, wrists, and arms, a test that examines the structure of the heart (echocardiogram), and a test of the electrical rhythm of the heart (electrocardiogram). The diagnosis may also be confirmed with genetic testing of the TBX5 gene 4.
Holt-Oram syndrome treatment
Depending on the severity of the bone and heart problems, treatment for Holt-Oram syndrome may require a team of specialists including pediatricians, surgeons, cardiologists, orthopedists, and geneticists 2. Treatment of wrist bone and other upper limb bone problems may include corrective or reconstructive surgery, the use of limb prosthetics, and physical or occupational therapies. The goal of treatment is to help people with Holt-Oram syndrome have as much use of the upper limbs as possible 4. These therapies may be most effective if they are started as soon as a person is diagnosed with Holt-Oram syndrome. In some cases, the changes in the way the bones formed in the wrist and upper limbs may not cause any problems or need any treatments. In these cases, the bone abnormalities may not even be noticed unless an x-ray is being performed for another reason (incidental finding) 4.
People with mild heart defects or cardiac conduction disease may not require any treatment. In other cases, cardiac conduction disease may be treated with antiarrhythmic medications or a pacemaker to maintain a regular heart rate. Other heart abnormalities may be treated with surgery. The specific surgical procedure will depend on the location and severity of the heart defect 4.
People with heart defects may be at an increased risk for bacterial infection and inflammation of the lining of the heart’s chambers and valves (endocarditis). Antibiotics may be prescribed before surgical procedures to reduce the risk for infection 4.
Holt-Oram syndrome life expectancy
The long-term outlook for people with Holt-Oram syndrome may depend on the severity of heart defects. Some people with Holt-Oram syndrome have no heart problems, or the problems are mild and only require occasional monitoring by a cardiologist. In other cases, heart defects associated with Holt-Oram syndrome may be severe and, in some cases, life-threatening 3.
Differences in the bones of the wrist, hand, arm, and shoulder associated with Holt-Oram syndrome may cause a person to have physical limitations that can affect their everyday lives. These birth defects can also impact social interactions, especially for children whose arms and hands are noticeably different from other children 5.
References- Holt-Oram syndrome. https://ghr.nlm.nih.gov/condition/holt-oram-syndrome
- Holt Oram Syndrome. https://rarediseases.org/rare-diseases/holt-oram-syndrome
- Sinha R and Nema C. Rare cardiac defect in Holt-Oram syndrome. Cardiovascular Journal of Africa. March 12, 2012; 23(2):e3-4. https://www.ncbi.nlm.nih.gov/pubmed/22447508
- McDermott DA, Fong JC, Basson CT. Holt-Oram Syndrome. 2004 Jul 20 [Updated 2019 May 23]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1111
- Holt-Oram Syndrome. https://emedicine.medscape.com/article/159911-overview





{kind=link}