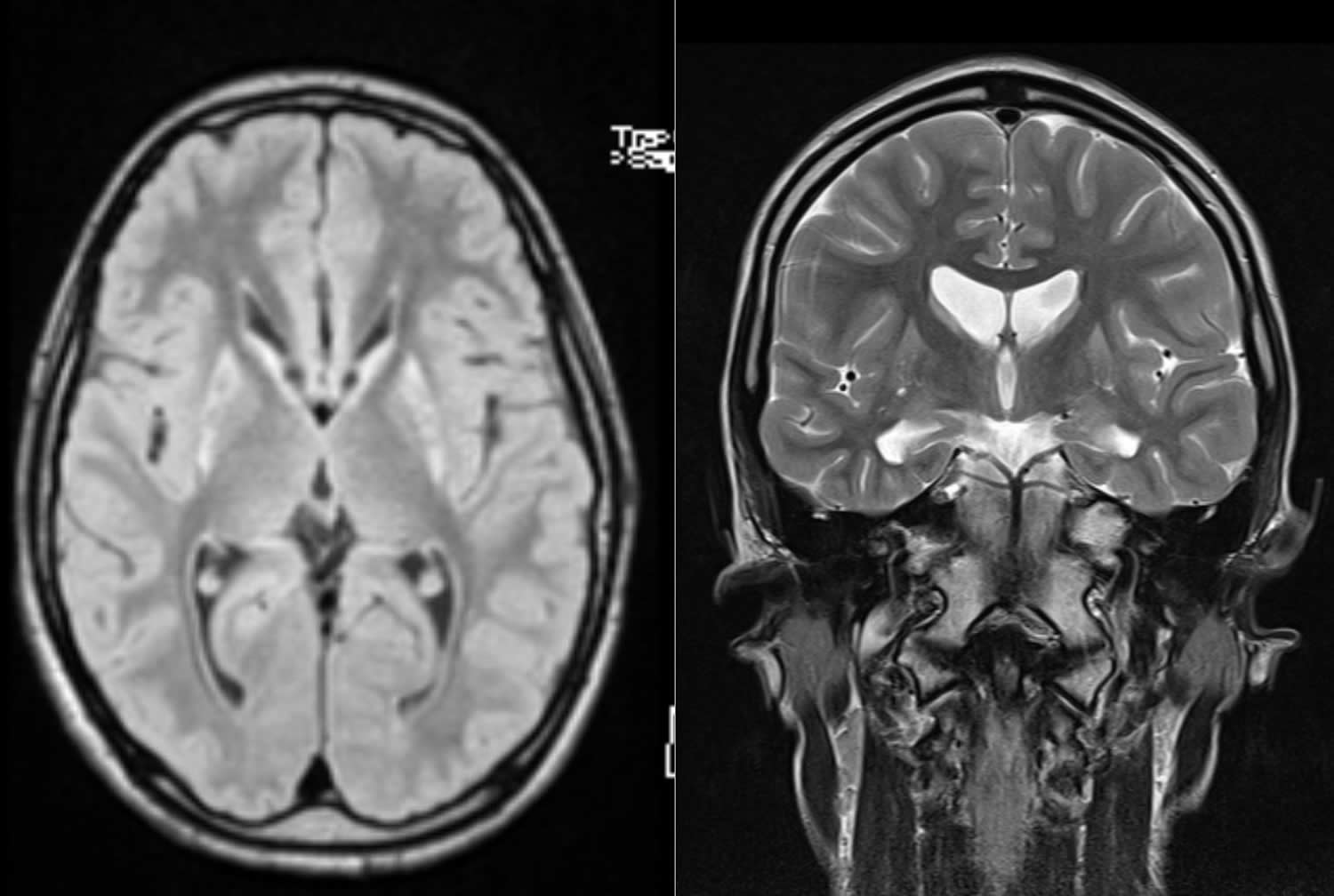
What is Huntington’s disease
Huntington’s disease is a inherited disease that causes certain nerve cells in the brain to progressively waste away 1. Huntington’s Disease causes changes in the central area of the brain, which affect movement, mood, behavior and psychiatric symptoms and thinking skills. Huntington’s Disease affects both sexes and all races and ethnic groups around the world.
The disorder is named for George Huntington, the physician who first described it in the late 1800s 2.
Huntington’s disease is known as the quintessential family disease because every child of a parent with Huntington’s disease has a 50/50 chance of carrying the faulty gene 3. Today, there are approximately 30,000 symptomatic Americans and more than 200,000 at-risk of inheriting the disease, and the devastating effects of the disease touch many more.
Within a family, multiple generations may have inherited the disease. Those at-risk may experience tremendous stress from the uncertainty and sense of responsibility. In the community, lack of knowledge about Huntington’s disease may keep friends and neighbors from offering social and emotional support to the family, fostering unnecessary isolation. The Huntington’s Disease Society of America 4 has a nationwide network that provides support and referrals for individuals with Huntington’s disease and their families.
Symptoms of the disease, which gets progressively worse, include uncontrolled movements (called chorea), abnormal body postures, and changes in behavior, emotion, judgment, and cognition. People with Huntington’s disease also develop impaired coordination, slurred speech, and difficulty feeding and swallowing. Huntington’s disease typically begins between ages 30 and 50. An earlier onset form called juvenile Huntington’s disease, occurs under age 20. Symptoms of juvenile Huntington’s disease differ somewhat from adult onset Huntington’s disease and include unsteadiness, rigidity, difficulty at school, and seizures. More than 30,000 Americans have Huntington’s disease.
What is Juvenile Huntington’s Disease (JHD) ?
In approximately 10% of cases, Huntington’s disease affects children or adolescents 5. The symptoms of Juvenile Huntington’s Disease (JHD) are somewhat different than adult onset Huntington’s disease and may include stiff or awkward walking, increased clumsiness or changes in speech. The ability to learn new information may decline and the child may lose skills they previous had. Juvenile Huntington’s Disease (JHD) typically progresses more rapidly than adult onset Huntington’s disease.
Symptoms of Juvenile Huntington’s Disease
Presentation of Juvenile Huntington’s Disease (JHD) includes changes in personality, coordination, behavior, speech or ability to learn. Physical changes include rigidity, leg stiffness, clumsiness, slowness of movement, tremors or myoclonus. In comparison with adult Huntington’s disease, seizures and rigidity are common, and chorea is uncommon.
Predisposition and typical initial symptoms of Juvenile Onset Huntington’s disease include:
- Positive family history of Huntington’s disease, usually in the father. Huntington’s disease is an inherited disease. One parent usually has the disorder. For genetic reasons, children with a very early onset of Huntington’s disease are far more likely to have an affected father than an affected mother.
- Stiffness of the legs
- Clumsiness of arms and legs
- Decline in cognitive function
- Changes in behavior
- Seizures
- Changes in oral motor function
- Chorea in an adolescent
- Behavioral disturbances
- Swallowing or speech problems.
The dance-like or twisting movements (chorea) often seen in adults with Huntington’s disease is uncommon in children who develop Huntington’s disease before age 10, but may be one of the first symptoms a teenager exhibits. Behavioral disturbances are sometimes the first symptom in an adolescent.
Onset and Progression of Juvenile Huntington’s Disease
Juvenile Huntington’s Disease usually has a more rapid progression rate than adult onset Huntington’s disease; the earlier the onset, the faster Juvenile Huntington’s Disease progresses. Death often occurs within 10 years of Juvenile Huntington’s Disease onset, as opposed to 10-25 years in adult onset Huntington’s disease.
Treatment for Juvenile Huntington’s Disease
There is no cure or treatment to stop, slow or reverse the progression of Juvenile Huntington’s Disease. Medications may be prescribed to manage symptoms. A child psychiatrist or behavior management specialist may address behavior disorders. A speech language pathologist may evaluate communication and swallowing problems. A nutritionist may be consulted to address nutritional needs as the disease progresses. Assistive devices such as wheelchairs, helmets, and communication boards may be used for safety, and to improve quality of life.
Huntington’s disease cause
Huntington’s disease is a progressive brain disorder caused by a single defective gene on chromosome 4 — one of the 23 human chromosomes that carry a person’s entire genetic code. The huntingtin gene called a “CAG repeat expansion” – the mutation results in gradual neuronal degeneration in the basal ganglia of the brain, which is responsible for coordination of movements, thoughts, and emotions (see Figure 3 below). As Huntington’s disease progresses, other regions of the brain undergo neuronal degeneration with diffuse and severe brain atrophy that is comparable to late stage Alzheimer disease. This defect is “dominant,” meaning that anyone who inherits it from a parent with Huntington’s will eventually develop the disease. Each child of a parent with Huntington’s disease has a 50-50 chance of inheriting the Huntington’s disease gene. If a child does not inherit the Huntington’s disease gene, he or she will not develop the disease and generally cannot pass it to subsequent generations. There is a small risk that someone who has a parent with the mutated gene but who did not inherit the Huntington’s disease gene may pass a possibly harmful genetic sequence to her/his children. A person who inherits the Huntington’s disease gene will eventually develop the disease. A genetic test, coupled with a complete medical history and neurological and laboratory tests, helps physicians diagnose Huntington’s disease.
Figure 1. Huntington’s disease inheritance pattern
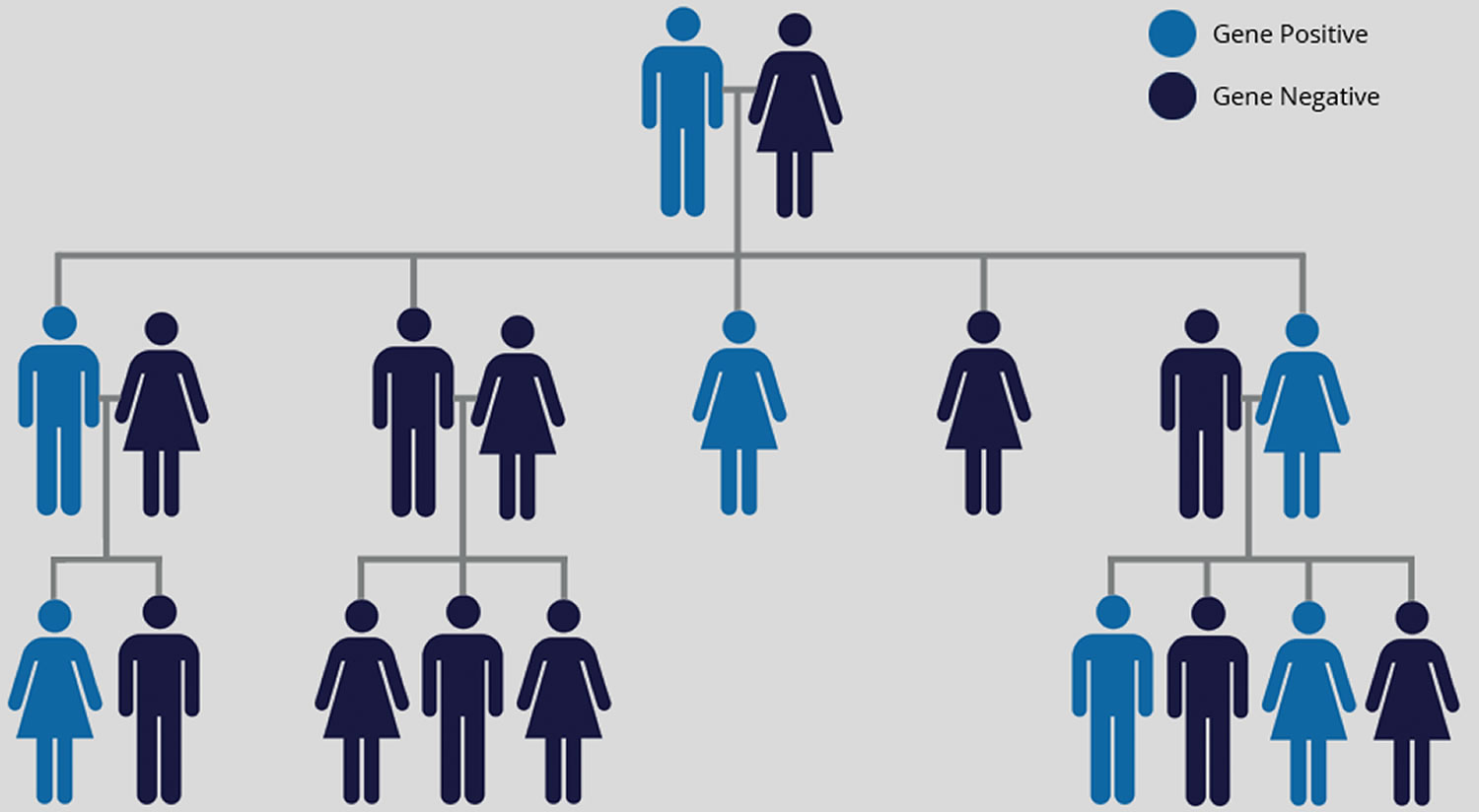
Note: Huntington’s disease is inherited in an autosomal dominant fashion. The probability of each offspring inheriting an affected gene is 50%. Inheritance is independent of gender. If the child has not inherited this expanded gene, he or she will never develop the disease and cannot pass it on to their children.
[Source 3]Figure 2. Huntington’s disease defective gene codes
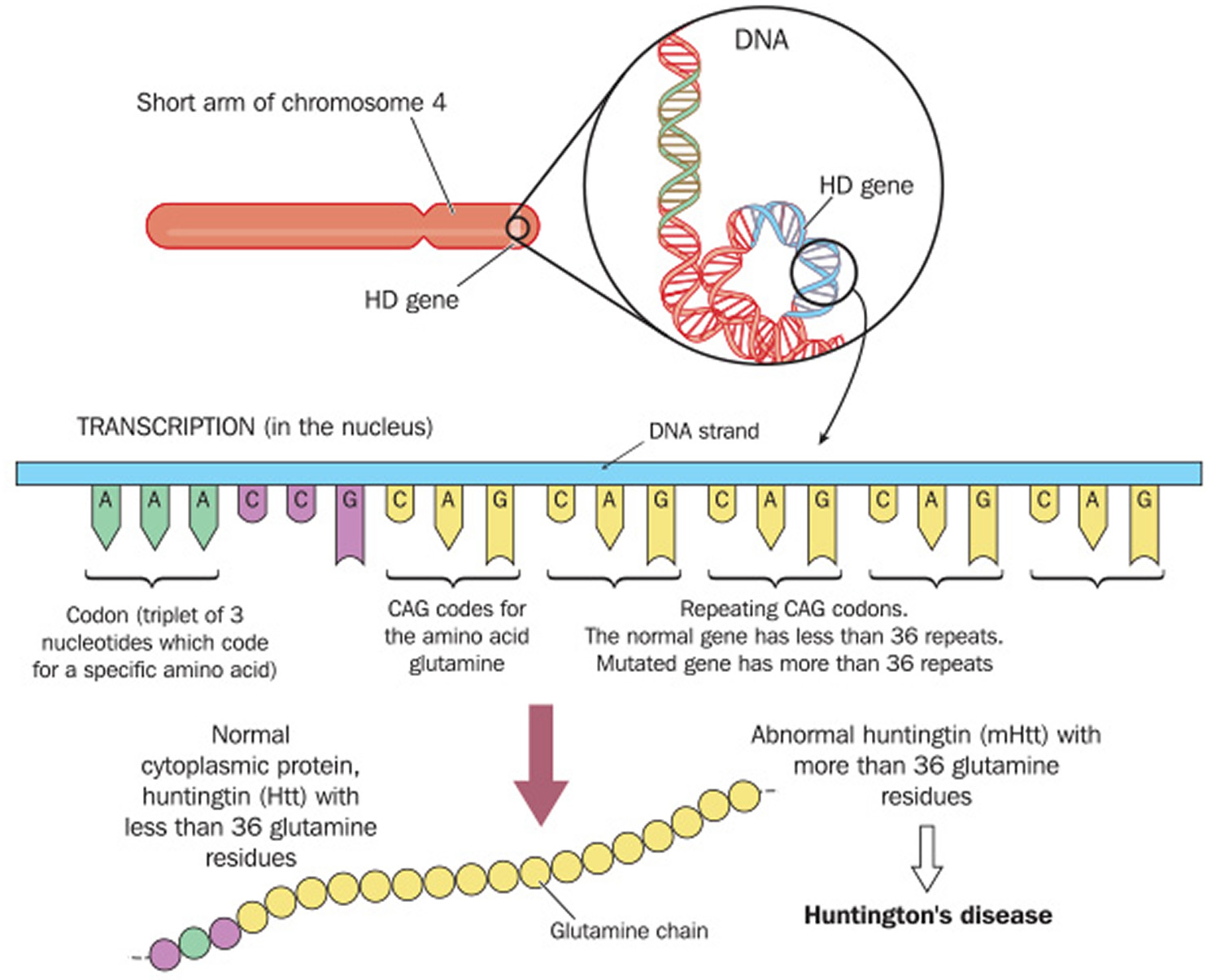
In 1993, researchers identified the gene that causes Huntington’s disease. The defective gene codes the blueprint for a protein called huntingtin. This protein’s normal function isn’t yet known, but it’s called “huntingtin” because scientists identified its defective form as the cause of Huntington’s disease. The defect causes the cytosine, adenine, and guanine (CAG) building blocks of DNA to repeat many more times than is normal. Defective huntingtin protein leads to brain changes that cause abnormal involuntary movements, a severe decline in thinking and reasoning skills, and irritability, depression and other mood changes.
Genetic Testing & Family Planning
People at-risk for the disease face a difficult choice about genetic testing for Huntington’s disease, given the current absence of an effective treatment or cure. Many people see no benefit in knowing that they will someday develop the disease. Others want an end to uncertainty so that they can make informed choices about the future.
- The decision whether to test or not is intensely personal and there is no “right” answer.
The Huntington’s Disease Society of America recommends that at risk individuals who are considering genetic testing do so at a genetic testing center that follows the Huntington’s Disease Society of America guidelines. Testing procedures at these centers involves sessions with professionals who are knowledgeable about Huntington’s disease and the local services available. It may take several weeks to receive the results once the genetic test is complete.
Genetic testing for children is typically prohibited before the age of 18, as the child may not understand the full implications of testing and may be vulnerable to pressure from others. However, a child under the age of 18 may be tested to confirm a diagnosis of juvenile onset Huntington’s disease after a thorough neurological exam.
Prenatal Testing
For families wishing to have a child who does not have the gene that causes Huntington’s disease, there are a few options. Pre-genetic diagnostic (PGD) testing can be used with In Vitro Fertilization (IVF) to make sure that any fertilized egg implanted does not have the abnormal gene. This can be done without informing the at-risk patient whether or not they have the gene that causes Huntington’s disease. If a woman is already pregnant, she can receive testing for the fetus with a chorionic villus biopsy at 10-11 weeks or via amniocentesis at 14-18 weeks.
Symptoms of Huntington’s disease
Symptoms of Huntington’s disease usually develop between ages 30 and 50, but they can appear as early as age 2 or as late as 80. The hallmark symptom of Huntington’s disease is uncontrolled movement of the arms, legs, head, face and upper body. Huntington’s disease also causes a decline in thinking and reasoning skills, including memory, concentration, judgment and ability to plan and organize.
Huntington’s disease brain changes lead to alterations in mood, especially depression, anxiety, and uncharacteristic anger and irritability. Another common symptom is obsessive-compulsive behavior, leading a person to repeat the same question or activity over and over.
Many describe the symptoms of Huntington’s disease as having Amyotrophic lateral sclerosis (ALS), Parkinson’s disease and Alzheimer’s disease– simultaneously.
Symptoms usually appear between the ages of 30 to 50, and worsen over a 10 to 25 year period. Ultimately, the weakened individual succumbs to pneumonia, heart failure or other complications. Everyone has the gene that causes Huntington’s disease, but only those that inherit the expansion of the gene will develop Huntington’s disease and perhaps pass it on to each of their children. Every person who inherits the expanded Huntington’s disease gene will eventually develop the disease. Over time, Huntington’s disease affects the individual’s ability to reason, walk and speak.
Symptoms Include:
- Personality changes, mood swings & depression
- Forgetfulness & impaired judgment
- Unsteady gait & involuntary movements (chorea)
- Slurred speech, difficulty in swallowing & significant weight loss
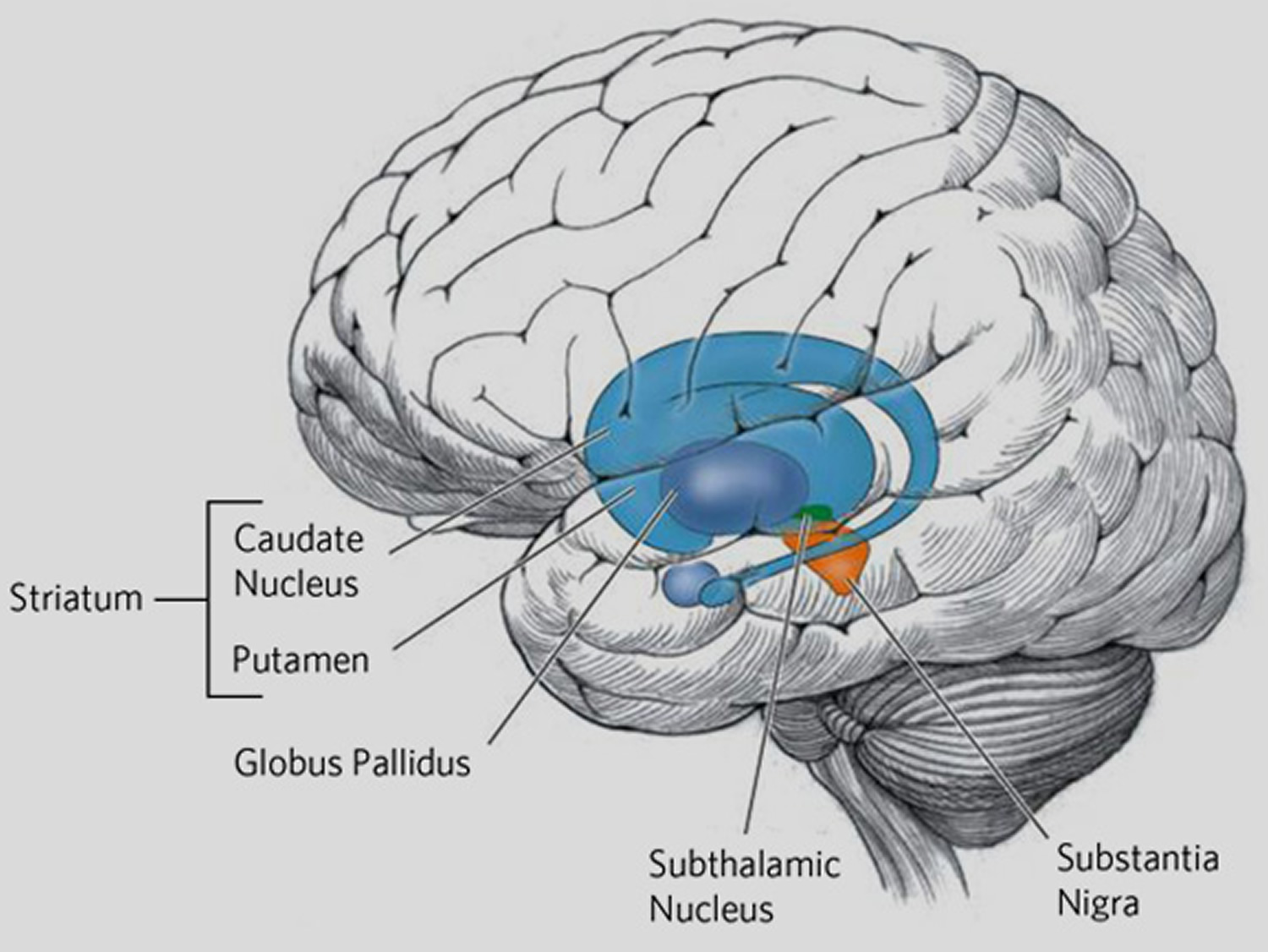
Huntington’s disease affects the whole brain, but certain areas are more vulnerable than others. Figure 3 below are the basal ganglia – a group of nerves cell clusters, called nuclei. These nuclei play a key role in movement and behavior control and are the parts of the brain most prominently affected in early Huntington’s disease.
Figure 3. Basal ganglia most predominantly affected by Huntington’s disease
Stages of Huntington’s disease
Although symptoms of Huntington’s disease vary from person to person, even within the same family, the progression of the disease can be roughly divided into three stages.
Early stage Huntington’s disease
Early stage Huntington’s disease usually includes subtle changes in coordination, perhaps some involuntary movements (chorea), difficulty thinking through problems and often a depressed or irritable mood. Medications are often effective in treating depression or other emotional problems. The effects of the disease may make the person less able to work at their customary level and less functional in their regular activities at home.
Middle stage Huntington’s disease
In the middle stage Huntington’s disease, the movement disorder may become more of a problem. Medication for chorea may be considered to provide relief from involuntary movements. Occupational and physical therapists may be needed to help maintain control of voluntary movements and to deal with changes in thinking and reasoning abilities. Diminished speech and difficulty swallowing may require help from a speech language pathologist. Ordinary activities will become harder to do.
Late stage Huntington’s disease
In the late stage Huntington’s disease, the person with Huntington’s disease is totally dependent on others for their care. Choking becomes a major concern. Chorea may be severe or it may cease. At this stage, the person with Huntington’s disease can no longer walk and will be unable to speak. However, he or she is generally still able to comprehend language and retains an awareness of family and friends. When a person with Huntington’s disease dies, it is typically from complications of the disease, such as choking or infection and not from the disease itself.
In all stages of Huntington’s disease, weight loss can be an important complication that can correspond with worsening symptoms and should be countered by adjusting the diet and maintaining appetite.
References- Huntington’s Disease Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Huntingtons-Disease-Information-Page
- Huntington’s Disease. https://www.alz.org/dementia/huntingtons-disease-symptoms.asp
- What Is Huntington’s Disease? http://hdsa.org/what-is-hd
- Huntington’s Disease Society of America. http://hdsa.org/
- Juvenile Onset HD. http://hdsa.org/living-with-hd/juvenile-onset-hd/




{kind=link}