Immunophenotyping
Immunophenotyping is a process of classifying cells of the immune system based on structural and functional differences. Immunophenotyping uses antibodies to identify white blood cells based on the types of antigens or markers on their surface or their interior. Immunophenotyping detects the presence or absence of white blood cell antigens. These antigens are protein structures found on the surface or interior of white blood cells. Typical groupings of antigens are present on normal white blood cells and are unique to specific cell types. Atypical but characteristic groupings are seen with leukemias and lymphomas. This allows immunophenotyping to be useful in helping to diagnose and classify these blood cell cancers.
The immunophenotyping process is commonly used to analyze and sort T-lymphocytes into subsets based on cell surface antigens called CD antigens (also called cluster of differentiation, cluster of designation or classification determinant) by the technique of flow cytometry, a laser-based instrument capable of analyzing thousands of cells per second. The whole procedure can be performed on cells from the blood, bone marrow or spinal fluid in a matter of a few hours. Immunophenotyping is used in basic research and clinically chiefly to diagnose and categorize lymphomas and leukemias and to help guide their treatment. Immunophenotyping may be ordered as a follow-up test when a complete blood count (CBC) and differential show an increased number of lymphocytes and the presence of immature blood cells or when there is a significant increase or decrease in the number of platelets (thrombocytosis or thrombocytopenia). Immunophenotyping is helpful because in many instances different kinds of benign and malignant lymphoid cells resemble each other in routinely stained tissue sections and smears. For example the small cells of small lymphocytic lymphoma are dead-ringers for benign small lymphocytes. By choosing appropriate antibodies, the origin of leukemic cells can be accurately determined. When lymphoma is suspected, the technique helps to distinguish between malignant and benign lymphoid proliferations, between B- and T-cell processes, and between sub-categories of B- and T-cell lymphomas. Immunophenotyping may also be used to separate cells into different groups based on the markers they have on the surface.
Leukemias and lymphomas are caused by an abnormal cell that begins to clone itself uncontrollably. The abnormal lymphocytes or myeloid (granular) monoclonal cells proliferate, yet do not fight infections or perform other functions like normal white blood cells. Because they do not die at a normal rate, they accumulate in the bone marrow, in a lymph node, or in other tissues, where they grow in numbers. As the number of cells increases in the bone marrow, they may crowd out and inhibit the production of normal red and white blood cells. Eventually, the abnormal cells will also be released into the bloodstream.
Complete blood count (CBC) and differential tests performed on a sample of blood from someone with leukemia or a lymphoma may reveal an increased number of white blood cells with a predominance of one type. These tests may suggest lymphoma or leukemia, but more information is generally needed to confirm a diagnosis. Complete blood count (CBC) and differential testing cannot confirm the presence of monoclonal white blood cells or detect the subtle differences that may exist between different types of blood cell cancers.
With immunophenotyping, a blood, bone marrow, or other tissue sample can be tested to gather this information – information that is then used to identify a specific type of leukemia or lymphoma and, where possible, used to predict its likely aggressiveness and/or responsiveness to certain treatment. The identification of different types of leukemias and lymphomas is based upon the presence or absence of antigens and a typical pattern that has been established with each leukemia/lymphoma.
Immunophenotyping detects the presence or absence of antigens found on the surface or interior of blood cells. Atypical or abnormal cells can demonstrate characteristic antigen groupings that are consistent with specific types of leukemia and lymphoma. The identifications made are based upon a “library” of antigen associations and patterns that have been established over time.
Samples are analyzed using various panels of antibodies that have been established for various types of leukemia or lymphoma. For example, each of these cancers would have a pre-defined panel of antibodies that would be consistent with their diagnosis: acute lymphoblastic leukemia, acute myeloid leukemia, hairy cell leukemia, erythroleukemia, B-cell lymphoma, or T-cell lymphoma.
Typically, a health practitioner will provide information about an individual who they suspect has leukemia or lymphoma. Basic testing of a CBC, differential, and platelet count would be performed in addition to immunophenotyping. The antigen selection, or panel, is made based upon that information.
Testing may sometimes be performed to evaluate the effectiveness of leukemia or lymphoma treatment and to detect residual or recurrent disease by observing the continued presence of abnormal cells.
Most of the antigens that immunophenotyping detects are identified by a CD (clusters of differentiation or cluster designation) number. CD numbers represent a naming convention that is based upon international consensus. While hundreds of antigens have been identified and have received a unique CD number, only a small number of these are routinely used.
The definition of particular subsets of immune cells using cell-surface markers continues to evolve, particularly for cell types that are the subject of intense current research 1. These include regulatory T (TReg) cells, interleukin-17 (IL-17)-secreting T helper (TH17) cells, dendritic cells (DCs) and natural killer (NK) cells. However, even well-characterized subsets, such as naive and memory T cells, are defined differently in various studies. For example, the classical T cell subsets of naive, central memory, effector memory and effector T cells were first defined on the basis of their expression of CC-chemokine receptor 7 (CCR7; also known as CD197) and CD45RA31 (where naive T cells are CCR7+CD45RA+, central memory T cells are CCR7+CD45RA−, effector memory T cells are CCR7−CD45RA− and effector T cells are CCR7−CD45RA+). Other investigators have since used different markers, such as CD62L or CD27 in place of CCR7 2, and CD45RO in place of CD45RA. Although these different combinations of markers generally define similar cell subsets, they introduce an unknown amount of heterogeneity that makes comparisons between studies difficult.
It is reasonable to think that the discovery of new markers and new cell subsets will continue for some time in the future. However, there is sufficient maturity in the field of cellular immunology to reach consensus working definitions for most of the commonly studied subsets of immune cells. At a high level, these definitions are unlikely to change very much as new discoveries are made, although there are likely to be new subsets of these cell types described over time 1. In other words, it should be possible to define a stable set of markers that delineate the major classes of B, T and NK cells, monocytes and dendritic cells (DCs) 1. Towards this end, Maecker et al 1 reviewed the literature that indicates what the key differentiation markers for these cell types might be; these markers could then, in their opinion, form the basis of a standard working definition. They further discuss how consensus is being reached in the immunological community regarding these markers and definitions, leading to a standardized immunophenotyping panel for human immune monitoring. Although there may be disagreements about the choice and utility of certain markers, and the names of the subsets so defined, Maecker et al 1 propose that the markers and subsets shown in Figure 2, and discussed below, can form a working definition of cell types that will help to drive standardization in the future.
What are some methods used for immunophenotyping?
Originally, glass slides with fixed tissue sections were treated with an antibody that was specific for a type of antigen typically found on certain abnormal cells associated with a particular leukemia or lymphoma. These antibodies were often linked with a fluorescence or a peroxidase indicator that would make these abnormal cells visible when observed under a microscope. Immunohistochemistry is based upon immunologic cellular properties and has proven to be particularly valuable in evaluating tissue samples that help in establishing a diagnosis or identifying relapse.
Another technique is flow cytometry and is performed by processing a blood, bone marrow, tissue, or fluid sample by adding specific antibodies that have been tagged with fluorescent markers. These antibodies will bind to corresponding antigens on the white blood cells (WBCs), if present, and are often referred to as cell markers. The WBCs are suspended in a physiologic solution and passed through a flow cytometer. The cell suspension is forced through a fluid stream that passes multiple laser beams causing deflection or absorption of the laser light. These light changes are identified by very sensitive detectors that analyze individual cells based on various physical properties.
The flow cytometer rapidly measures characteristics about each cell, such as its size and granularity (internal cellular structures), and evaluates the type and quantity of fluorescent antigen-antibody complexes that are present. The advantage of flow cytometry over Immunohistochemistry is that thousands of cells are evaluated during the test. Based on the physical characteristics of the abnormal cells and the presence (or absence) of fluorescence, the investigator can quickly determine the type of leukemia or lymphoma that may be present.
Flow cytometry has increasingly become a tool of choice for the analysis of cellular phenotype and function in the immune system 1. Flow cytometry is arguably the most powerful technology available for probing human immune phenotypes, because it measures multiple parameters on many individual cells. Flow cytometry thus allows for the characterization of many subsets of cells, including rare subsets, in a complex mixture such as blood. And because of the wide array of antibody reagents and protocols available, flow cytometry can be used to assess not only the expression of cell-surface proteins, but also that of intracellular phosphoproteins 3 and cytokines 4, as well as other functional readouts 5.
Figure 1. Flow cytometry immunophenotyping
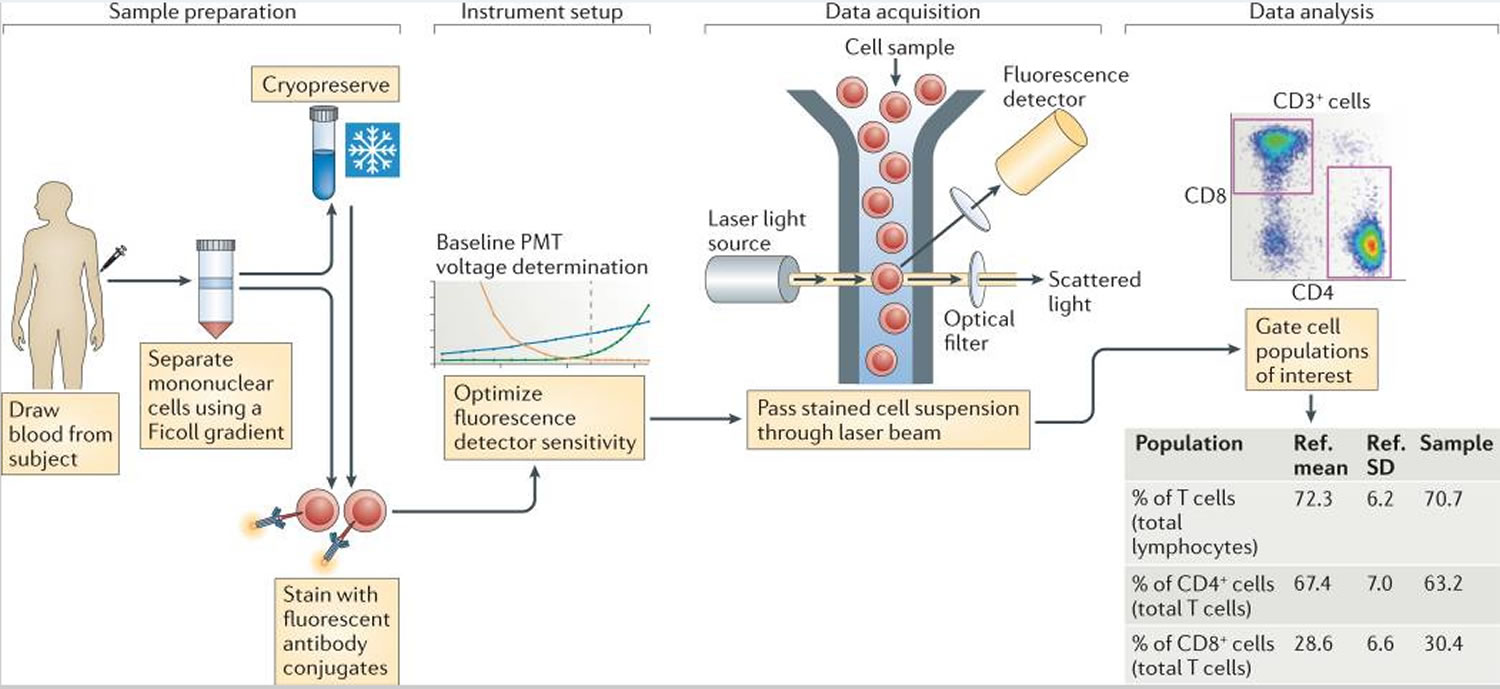
Footnote: A typical flow cytometry experiment. Sample preparation from blood often involves Ficoll gradient separation of mononuclear cells, and sometimes cryopreservation, before staining with fluorescent antibody conjugates. Each of these steps can introduce variability in the assay results. Instrument setup involves setting voltage gains for the photomultiplier tubes (PMTs) so as to achieve optimal sensitivity. To the extent that this is not standardized, it becomes a source of variability as well. Data acquisition involves passing the stained cells through a laser beam and recording the fluorescence emission from all of the bound antibody conjugates. Here, the main variable is the type of instrument, including the lasers and optical filters used. This is followed by data analysis, in which cell populations of interest are defined and reported on, which is another significant source of variation.
Abbreviations: Ref. = reference; SD = standard deviation.
[Source 1 ]Cluster Designation (CD) numbers
Researchers in many scattered laboratories painstakingly identified many lymphoid surface antigens and developed antibodies to them. Every so often, in world congresses, it would become apparent that antibodies originating from different labs and bearing different names were marking the same antigen/molecule. At that point, the antigen would be assigned a cluster designation, or CD number, meaning that a known cluster of antibodies were binding to this known antigen. Any of these antibodies might be referred to by one of several idiosyncratic laboratory names or by the CD number of the antigen. For example, antibodies that recognize CD20, a characteristic B-cell molecule, might be called alternatively L26, B1, or, by association, CD20. This is very confusing and is one reason why hematopathologists keep lots of aspirin in their desks. Nonetheless, if you study lymphomas, you will have to know at least the basics of this scheme. Here are the basics:
- All lymphoid cells (well, almost all): Lymphoid cells are reactive for CD45 (leukocyte common antigen, or LCA).
- B-cells: Almost all of these are reactive for CD19, CD20 and CD22. Certain low-grade B-cell lymphomas are reactive for two markers otherwise usually found on T-cells: CD5 and CD43. Follicular center cell lymphomas (as well as very different fish, lymphoblastic lymphomas) are frequently CD10(+).
- T-cells: Pan T-cell markers (present on almost all T-cells) include CD2, CD3, CD5, and CD7 (the early childhood markers). Most T-cells mark with either CD4 (helper cells) or CD8 (suppressor cells or cytotoxic cells).
- Natural-killer (NK) cells: These tough guys are frequently associated with CD16, CD56, or CD57.
T cells
The major subsets of T cells can be defined by the expression of CD4 and CD8, together with, for example, CCR7 and CD45RA 6. CCR7 has been a difficult marker for use in flow cytometry because of its low expression levels; however, a new CCR7-specific antibody clone (commercially available from multiple vendors) that provides brighter staining has greatly improved this situation. Furthermore, other substitute markers for CCR7 are more problematic. For example, the expression level of CD62L is highly affected by density gradient separation 7 or cryopreservation 8 of peripheral blood mononuclear cells (PBMCs). As peripheral blood mononuclear cell (PBMC) cryopreservation is integral to the workflow of many human studies, owing to the need to batch samples and store cells for future assays, this limits the broad use of CD62L for immunophenotyping. Another possible CCR7 replacement, CD27, is expressed not only by naive and CCR7+ central memory T cells, but also by a population of effector memory T cells that lack expression of CCR7 9, meaning that CD27 is not a full substitute for CCR7. So, a panel containing CD3 (to define T cells), CD4, CD8, CD45RA and CCR7 seems most applicable for distinguishing naive, central memory, effector memory and effector CD4+ and CD8+ T cells. With the addition of activation markers, such as CD38 and HLA-DR, it is possible to define activated subsets of each of these cell types as well (Figure 2; Table 1).
Figure 2. Immunophenotyping test (identification of immune cell subsets by eight-color antibody staining)
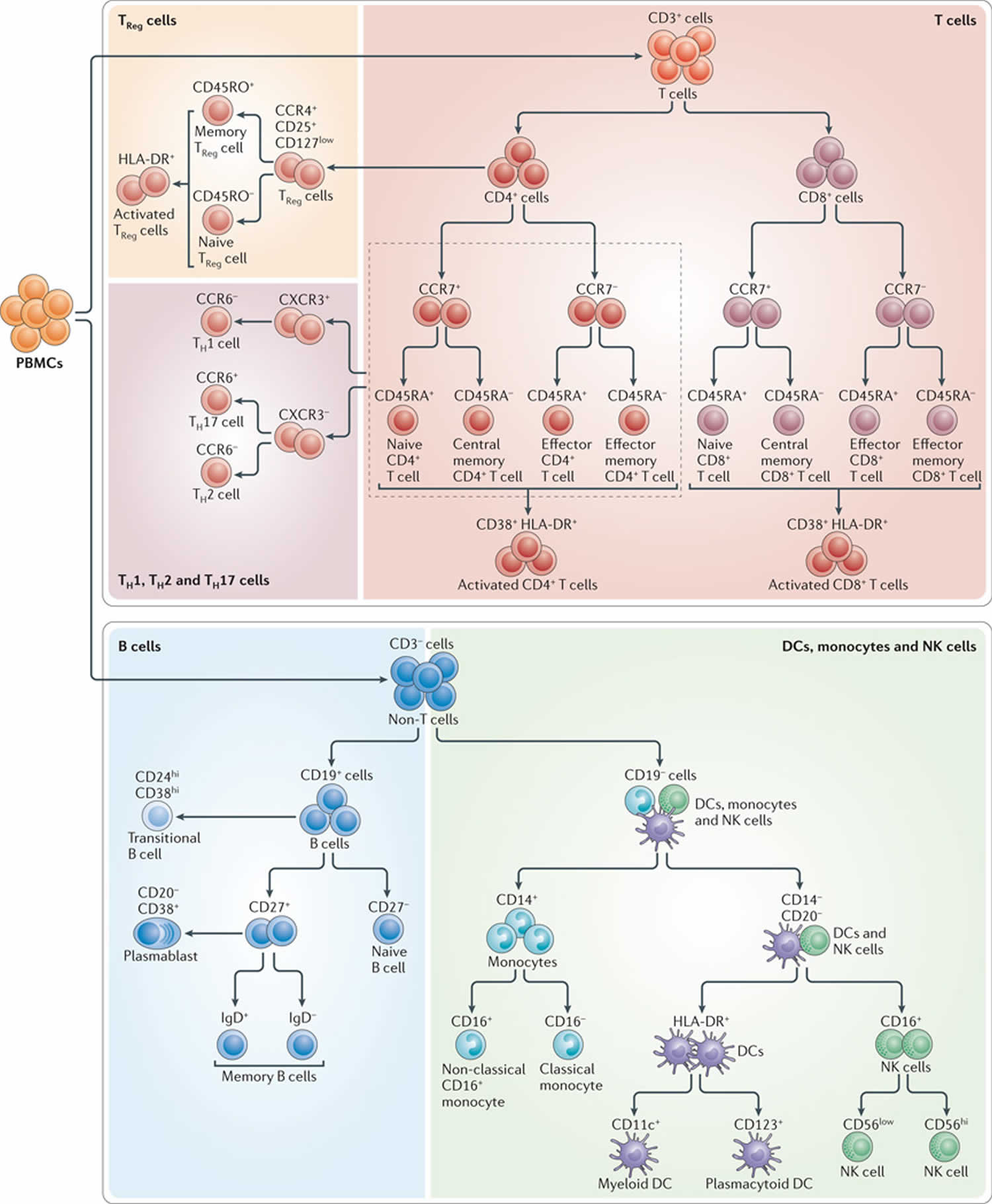
Footnote: The figure shows the cell populations that can be identified using the markers targeted by each of the five antibody cocktails of the Human Immunophenotyping Consortium (HIPC) phenotyping panel shown in TABLE 1.
Abbreviations: CCR = CC-chemokine receptor; CXCR3 = CXC-chemokine receptor 3; DC = dendritic cell; NK = natural killer; PBMC = peripheral blood mononuclear cell; TH = T helper; TReg = regulatory T.
[Source 1 ]Table 1. Eight-color antibody panels proposed by the Human Immunophenotyping Consortium
| Fluorochrome | Marker | |||||
|---|---|---|---|---|---|---|
| T cells | TReg cells | TH1, TH2 and TH17 cells | B cells | DCs, monocytes and NK cells | ||
| FITC | Live or dead | Live or dead | Live or dead | Live or dead | Live or dead | |
| PE | CCR7 | CD25 | CXCR3 | CD24 | CD56 | |
| PerCP-Cy5.5 | CD4 | CD4 | CD4 | CD19 | CD123 | |
| PE-Cy7 | CD45RA | CCR4 | CCR6 | CD27 | CD11c | |
| APC | CD38 | CD127 | CD38 | CD38 | CD16 | |
| APC-H7 | CD8 | CD45RO | CD8 | CD20 | CD3, CD19 and CD20 | |
| V450 | CD3 | CD3 | CD3 | CD3 | CD14 | |
| V500 | HLA-DR | HLA-DR | HLA-DR | IgD | HLA-DR | |
Abbreviations: APC = allophycocyanin; APC-H7 = allophycocyanin–cyanine H7 tandem; CCR = CC-chemokine receptor; CXCR3 = CXC-chemokine receptor 3; DC = dendritic cell; FITC = fluorescein isothiocyanate; NK = natural killer; PE = phycoerythrin; PE-Cy7 = phycoerythrin–cyanine 7 tandem; PerCP-Cy5.5 = peridinin chlorophyll protein–cyanine 5.5 tandem; TH = T helper; TReg = regulatory T; V450 = violet 450; V500 = violet 500.
[Source 1 ]B cells
Tonsillar B cells were first classified into five groups, termed Bm1–Bm5, according to their expression of IgD and CD38 together with that of CD23 and CD77 10. Some versions of all of these cell types can also be found in peripheral blood 11. Bm1 and Bm2 cells are CD38−IgD+, and include mostly resting (CD23−) naive B cells and some activated (CD23+) cells. Bm3 and Bm4 cells are CD38+IgD− germinal centre (or germinal centre precursor) cells, which can be further subdivided by their expression of CD77, a marker of germinal centre location (Bm3 cells are CD77−; Bm4 cells are CD77+). Bm5 cells are CD38−IgD− memory B cells. CD27 can also be used to define naive (CD27−) and memory (CD27+) B cells 12. CD24 has been shown to be useful in defining immature ‘transitional’ B cells (which are CD24hiCD38hi) 13. Recently, circulating plasmablasts were also identified in peripheral blood, using either CD38 and CD138 14 or CD38 and CD27 15. Notably, these plasmablasts are low or negative for CD20 expression and, because of the use of CD20-specific antibody therapy in various diseases, it is prudent to also include CD19 as a marker to define the parental B cell population. Although it is not possible to include all of the potentially relevant B cell markers in a single eight-colour antibody cocktail, a highly useful panel can be constructed using antibodies specific for CD19 and CD20 (to define B cells), CD38 (to identify plasmablasts as well as transitional B cells), CD24 (for transitional B cells), and IgD and CD27 (for naive and memory B cell populations) (see Figure 2).
NK cells
NK cells have been shown in recent years to be much more heterogeneous than previously thought, with a large variety of activating and inhibitory receptors expressed by overlapping subsets of cells that vary widely between individuals 16. However, at a more basic level, NK cells can be subdivided into two major categories based only on the markers CD16 and CD56. The vast majority of peripheral blood NK cells are CD16+CD56low, whereas a smaller subset is CD16+CD56hi. These populations differ in terms of function and tissue localization, and the latter subset has been shown to be an intermediate phenotype that can give rise to the former subset 17.
Dendritic cells
Circulating blood DCs have long been identified as HLA-DRhi cells that are negative for various lineage-specific markers, including CD3 (which defines T cells), CD14 (which is monocyte specific), and CD19 and CD20 (which define B cells) 18. DCs can be divided into cells of the myeloid lineage (CD11c+ cells) and the plasmacytoid lineage (CD123+ cells). Myeloid DCs can be further subdivided by their expression of CD16, CD1c and CD141 19. Although these myeloid DC subsets may have functional significance, their enumeration in cryopreserved peripheral blood mononuclear cell (PBMC) populations is too problematic to warrant the inclusion of these markers in a general phenotyping panel. Thus, one can construct a basic DC panel based on a lineage cocktail, and antibodies specific for HLA-DR, CD11c and CD123.
Monocytes
With further study, monocytes might also reveal substantial heterogeneity, but currently two major categories are widely recognized: classical monocytes (which are CD14hiCD16−) and non-classical monocytes (which are CD14low and CD16hi) 20. Because monocyte, NK cell and DC phenotyping can all include CD16, it is possible to imagine a phenotyping cocktail for all three cell types that includes a lineage cocktail, together with antibodies specific for HLA-DR, CD11c, CD14, CD16, CD56 and CD123.
Immunophenotyping test
Immunophenotyping may be ordered when a person has an increased number of lymphocytes (or sometimes an increase in another type of white blood cell (WBC)), an increased or decreased platelet count, or has immature white blood cells that are not normally seen in blood. These are usually findings from a complete blood count (CBC) and differential and may be the first indication that a person might have a blood cell cancer. Symptoms of early leukemia and lymphoma may be unremarkable, mild, or nonspecific.
Examples of signs and symptoms of a blood cell cancer include:
- Feeling tired or rundown, weakness
- Unexplained loss of weight or appetite
- Shortness of breath during normal physical activity
- Pale skin
- Bleeding or bruising easily
- Fever
- Bone and joint pain
- Enlarged lymph nodes, spleen, liver, kidneys, and/or testicles
- Headaches
- Vomiting
- Night sweats
Testing may also be ordered when a person has been treated for a leukemia or lymphoma to evaluate the effectiveness of treatment and detect residual or recurrent disease.
What does the immunophenotyping test result mean?
The presence of certain antigens that are identified by immunophenotyping require expertise to interpret. A pathologist, often one specializing in the study of blood diseases and/or blood cell cancers (a hematopathologist), will consider the results from the complete blood count (CBC), differential, blood smear, bone marrow findings, and immunophenotyping as well as other tests in order to provide a diagnostic interpretation. A laboratory report will typically include specific results from the tests as well as an analysis of what those results mean.
The markers that are present on the cells as detected by immunophenotyping will help characterize the abnormal cells present (if any). This information is considered together with the affected person’s clinical history, physical examination, signs and symptoms as well as all laboratory tests to help make a diagnosis.
It must be kept in mind that while findings represent comparisons to “normal” results and to known antigen associations with leukemias and lymphomas, each person’s condition will also be unique. A person may have (or lack) certain antigens that are typically seen, yet still be diagnosed with a specific type of leukemia or lymphoma.
Abnormal immunophenotype profiles are usually present in: acute myelogenous leukemia (or acute myeloid leukemia), acute lymphoblastic leukemia, chronic lymphocytic or myelocytic leukemias, B-cell and T-cell non-Hodgkin lymphomas, erythroleukemia (RBC leukemia), megaloblastic leukemia (platelets), and multiple myeloma.
Table 2. Markers that are often expressed in certain type of cells
| Cell | Markers |
| Immature precursor cells | HLA-DR, TdT, CD34, CD38, CD117 |
| B-lymphocytes | CD19, CD20, CD22, CD79a, immunoglobulin heavy (gamma, alpha, mu or delta) and light chains (kappa or lambda)CD10 (pre-B cell) |
| T-lymphocytes | CD2, CD3, CD5, CD7, and either CD4 or CD8 |
| Myeloid cells (granulocytes) | MPO (myeloperoxidase), CD11, CD13, CD15, CD16b, CD33, CD66 |
| Natural killer (NK) cells | CD11b, CD16, CD56 |
Table 3. Markers that suggest certain types of cell differentiation
| Cell | Markers |
| Megakaryocytic differentiation; Platelets | CD31, CD36, CD41, CD42, CD61 |
| Red blood cell (erythroid) differentiation | CD235a |
| Monocytic differentiation | CD14, CD33, CD64, CD68 |
| Hairy cell leukemia | CD11c, CD103 |
Can results of immunophenotyping testing be used to determine the course of my cancer?
Diagnosis of leukemia or lymphoma is based on the visual examination of a blood smear and/or bone marrow biopsy and aspiration for the presence of certain cell types. Depending upon the pattern of antigens present and their established association with specific cancers, a health practitioner may determine how likely a cancer will respond to treatment and how aggressive the treatment might be. The course of treatment for that cancer will be determined by the health practitioner and their team based on immunophenotyping and other tests that might be performed.
Will my antigens change?
The antigens on specific monoclonal cancer cells will generally remain the same; however, treatment with chemotherapy and/or radiation will eliminate the abnormal cells. If treatment is successful, normal white blood cells (WBCs) will replace abnormal cells. Because of this, immunophenotyping results will be different by reflecting the current population of WBCs that would be present in an individual in remission.
Immunophenotyping lymphoma
Immunophenotyping refers to the technique of identifying molecules that are associated with lymphoma cells and that help to characterize them. The molecules are identifiable because, in almost all analyzable cases, they are expressed on the outer cell surface membrane. The molecules are identified by using special antibodies that bind to them specifically . In this context these molecules are called “antigens,” and the specific part of the molecule to which the antibody attaches is called the “epitope”.
The antibodies themselves have been manipulated so that they bear artificial reporter moieties. The technique is called “immunophenotyping” for 2 reasons: 1) It is dependent on the activity of antibodies, which are immunological substances 2) It is used chiefly to identify lymphoid and hematopoietic cells, which are part of the immune system.
Immunophenotyping is especially useful in the case of B-cell lymphomas that express surface immunoglobulin and most of them do. Immunoglobulin molecules contain a light chain and a heavy chain. In a random, benign collection of lymphoid cells, the kappa light chains are present on roughly 2/3rds of the cells and lambda light chains on 1/3rd. If you applied an antibody to kappa light chains, it would mark 2/3rds of the cells; an antibody to lambda would mark 1/3rd. On the other hand, a malignant collection of lymphoid cells is monoclonal, at least at first approximation. By definition, then, all the cells will bear identical surface immunoglobulin molecules with either kappa or lambda light chains, but not a mixture. Thus an antibody to a specific kind of light chain (kappa or lambda) will mark either all or none of the cells.
How immunophenotyping for lymphoma works
- A solution of antibodies (called serum) is poured over the lymphoma cells, which may be in liquid suspension or may be in a very thin section of tissue on a slide.
- If the lymphoma cells bear the molecules (antigens) for which the antibodies are specific, the antibodies bind to the cells.
- The cells are washed to prevent non-specific binding; the specific antibodies remain bound.
- The cells with the bound antibodies are manipulated so that the reporter moiety attached to the antibodies declares its presence. This can be done several ways:
- Immunofluorescence: The reporter molecule may fluoresce when exposed to light of the appropriate frequency. If the specimen is a tissue section, it can be examined under a fluoresent microscope, and cells with bound antibodies will glow in the dark (like Halloween).
- Flow cytometry: If the specimen is cells suspended in liquid, the cells can be run through a flow cytometer. This will expose cells in a stream of liquid one at a time to a beam of laser light. If they bear the right bound antibodies, they will fluoresce and the cytometer will record the signal.
- Immunohistochemistry: Sometimes cells in a tissue section are identified with antibodies that have attached enzymes (usually horse radish peroxidase or alkaline phosphatase). When the tissue section is exposed to an appropriate chromogenic substrate, the enzyme on the bound antibodies will cause the substrate to change color and precipitate on the cells.
How the antibodies are made
- Animals (mice, rabbits, goats) are immunized with the antigen for which a corresponding antibody is desired. Serum from the immunized animals is collected and purified. The antibodies thus procured are called “polyclonal antibodies”. Because they are naturally made, they consist of many different kinds of antibodies with varying degrees of specificity for the antigen.
- Alternatively, through the miracles of modern science (invent anything this insanely great, and your mailbox will contain a Nobel Prize), individual cells that secrete the desired antibody can be isolated from immunized animals. The selected cell is then fused with a neoplastic plasma cell that also has a natural propensity to make antibodies. This fused hybrid, called a “hybridoma” cell, secretes only one kind of antibody, without the variation seen in the first method above. The serum from the hybridoma cells thus is rich in a “monoclonal antibody”.
- Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project [published correction appears in Nat Rev Immunol. 2012 Jun;12(6):471]. Nat Rev Immunol. 2012;12(3):191–200. Published 2012 Feb 17. doi:10.1038/nri3158 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3409649
- Appay V, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nature Med. 2002;8:379–385.
- Perez OD, et al. Multiparameter analysis of intracellular phosphoepitopes in immunophenotyped cell populations by flow cytometry. Curr. Protoc. Cytom. 2005;32:6.20.1–6.20.22
- Maecker HT. In: Flow Cytometry Protocols. Hawley TS, Hawley RG, editors. Totowa, New Jersey: Humana Press; 2004. pp. 95–107.
- Parish CR, Glidden MH, Quah BJ, Warren HS. Use of the intracellular fluorescent dye CFSE to monitor lymphocyte migration and proliferation. Curr. Protoc. Immunol. 2009;84:4.9.1–4.9.13.
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712.
- Lin SJ, Chao HC, Yan DC, Huang YJ. Expression of adhesion molecules on T lymphocytes in young children and infants — a comparative study using whole blood lysis or density gradient separation. Clin. Lab. Haematol. 2002;24:353–359.
- Weinberg A, et al. Optimization and limitations of use of cryopreserved peripheral blood mononuclear cells for functional and phenotypic T-cell characterization. Clin. Vaccine Immunol. 2009;16:1176–1186.
- Fritsch RD, et al. Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J. Immunol. 2005;175:6489–6497.
- Pascual V, et al. Analysis of somatic mutation in five B cell subsets of human tonsil. J. Exp. Med. 1994;180:329–339.
- Bohnhorst JO, Bjorgan MB, Thoen JE, Natvig JB, Thompson KM. Bm1–Bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjogren’s syndrome. J. Immunol. 2001;167:3610–3618.
- Agematsu K, et al. B cell subpopulations separated by CD27 and crucial collaboration of CD27+ B cells and helper T cells in immunoglobulin production. Eur. J. Immunol. 1997;27:2073–2079.
- Cuss AK, et al. Expansion of functionally immature transitional B cells is associated with human-immunodeficient states characterized by impaired humoral immunity. J. Immunol. 2006;176:1506–1516.
- Rawstron AC. Immunophenotyping of plasma cells. Curr. Protoc. Cytom. 2006;36:6.23.1–6.23.14.
- Avery DT, et al. Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. J. Immunol. 2005;174:4034–4042.
- Hamerman JA, Ogasawara K, Lanier LL. NK cells in innate immunity. Curr. Opin. Immunol. 2005;17:29–35.
- Beziat V, et al. CD56brightCD16+ NK cells: a functional intermediate stage of NK cell differentiation. J. Immunol. 2011;186:6753–6761.
- Willmann K, Dunne JF. A flow cytometric immune function assay for human peripheral blood dendritic cells. J. Leukoc. Biol. 2000;67:536–544.
- Ju X, Clark G, Hart DN. Review of human DC subtypes. Methods Mol. Biol. 2010;595:3–20.
- Ziegler-Heitbrock HW, et al. Small (CD14+/CD16+) monocytes and regular monocytes in human blood. Pathobiology. 1991;59:127–130.





{kind=link}