Lafora disease
Lafora disease also known as Lafora body disease or Lafora progressive myoclonus epilepsy, is a rare autosomal recessive inherited brain disorder, characterized by severe form of progressive myoclonus epilepsy and a decline in intellectual function 1. The signs and symptoms of Lafora disease most commonly begins with epileptic seizures in late childhood or adolescence. Other signs and symptoms include difficulty walking, muscle spasms (myoclonus) and dementia. Myoclonus is a term used to describe episodes of sudden, involuntary muscle jerking or twitching that can affect part of the body or the entire body. Myoclonus can occur when an affected person is at rest, and it is made worse by motion, excitement, or flashing light (photic stimulation). In the later stages of Lafora progressive myoclonus epilepsy, myoclonus often occurs continuously and affects the entire body.
Several types of seizures commonly occur in people with Lafora disease. Generalized tonic-clonic seizures also known as grand mal seizures affect the entire body, causing muscle rigidity, convulsions, and loss of consciousness. Affected individuals may also experience occipital seizures, which can cause temporary blindness and visual hallucinations. Over time, the seizures worsen and become more difficult to treat. A life-threatening seizure condition called status epilepticus may also develop. Status epilepticus is a continuous state of seizure activity lasting longer than several minutes.
Affected people also experience rapid intellectual function deterioration that begins around the same time as the seizures. Behavioral changes, depression, confusion, and speech difficulties (dysarthria) are among the early signs and symptoms of Lafora progressive myoclonus epilepsy. As the condition worsens, a continued loss of intellectual function (dementia) impairs memory, judgment, and thought. Affected people lose the ability to perform the activities of daily living by their mid-twenties, and they ultimately require comprehensive care. Lafora disease is often fatal within 10 years after symptoms first appear 2.
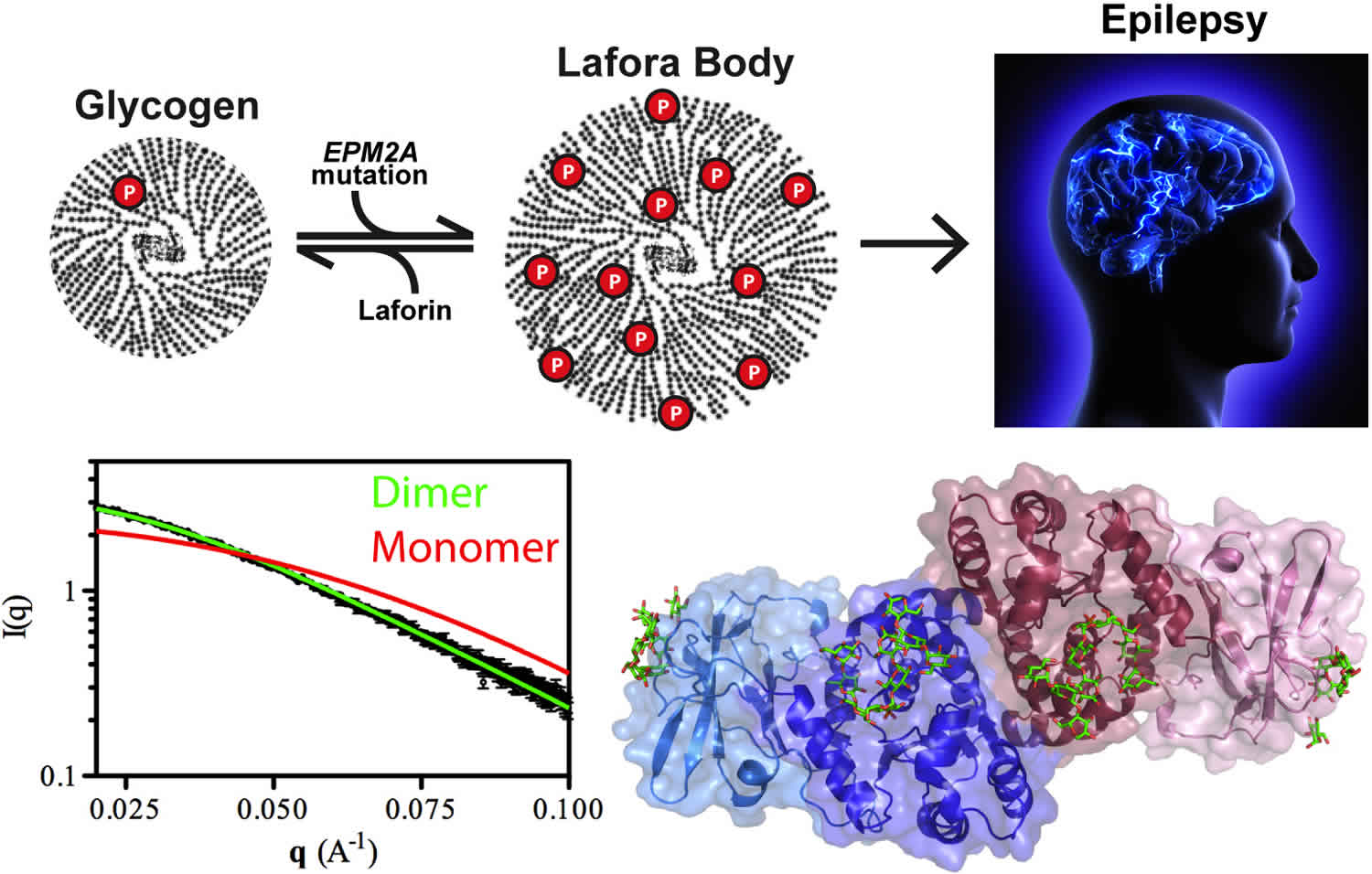
Lafora progressive myoclonus epilepsy was first described by Lafora and Glück over 100 years ago 3. A post-mortem study showed profuse accumulation of small inclusion bodies in many tissues, including the brain. These inclusions, subsequently termed Lafora bodies, became the hallmark of the disease. They were shown to be composed primarily of abnormal glycogen 4, placing Lafora disease in the context of glycogen metabolism disorders.
Most Lafora disease cases are caused by changes (mutations) in either the EPM2A gene or the NHLRC1 gene and are inherited in an autosomal recessive manner. The prevalence of Lafora disease or Lafora progressive myoclonus epilepsy is unknown. Although Lafora disease occurs worldwide, it appears to be most common in Mediterranean countries (Spain, France, and Italy), parts of Central Asia, India, Pakistan, North Africa, and the Middle East 5. Lafora disease has been found in more than 250 families throughout the world, resulting from EPM2A gene (responsible for Laforin) and NHLRC1 gene (EPM2B) (responsible for E3 ubiquitin-protein ligase NHLRC1) mutations, and the prevalence seems to be close to four cases per one million persons 6. However, the number of mis- and undiagnosed patients may be higher, especially in developing countries.
Unfortunately, there is currently no cure or way to slow the progression of Lafora disease. Treatment is based on the signs and symptoms present in each person 7. For example, certain medications may be recommended to manage generalized seizures. In the advanced stages of the condition, a gastrostomy tube may be placed for feeding. Drugs that are known to worsen myoclonus (i.e. phenytoin) should be avoided 7.
Is a ketogenic diet known to help individuals with Lafora disease?
The impact of a long-term ketogenic diet was studied in five people with Lafora disease. Although ketogenic diet was well-tolerated by patients, it did not stop disease progression or improve symptoms. However, given the small study population, the researchers could not exclude the possibility that a ketogenic diet has the potential to slow down the disease progression. Larger studies are needed to evaluate the impact of a ketogenic diet in treating patients with Lafora disease 8.
Can an adult get Lafora disease?
Lafora disease typically begins between ages 12 and 17 years, after a period of apparently normal development. However, there are reports of later onset forms of the condition. In these cases,the affected person often begins showing signs and symptoms of Lafora disease between ages 21 and 28. Some studies suggest that later onset cases of Lafora disease may be associated with a slower disease progression 9.
Have there been any advances that suggest anything can slow the progression of Lafora disease?
There are currently no treatments available to slow the progression of Lafora disease. However, there is ongoing research regarding potential future treatments. For example, there has been research on mice with Lafora disease that were bred with mice that have a knockout of the PTG gene (an inactivated copy of the gene). The PTG gene provides instructions for one of the enzymes that modify glycogen synthase activity. If it is not functional, or “knocked out,” no Lafora disease develops. This research may show that the disease could be prevented by indirectly altering glycogen synthase function.
Why is there little information about Lafora disease available?
Lafora disease affects a small number of people compared to the general population and is considered rare in many parts of the world (a disease is considered to be rare when it affects 1 person per 2000). Unfortunately, the field of rare diseases as a whole suffers from a shortage of medical and scientific knowledge, largely due to lack of awareness and funding sources. Until recently there was no real research or public health policy concerning issues related to the field. Those affected by these diseases face similar difficulties in their quest for relevant information and proper direction towards qualified professionals. On a positive note, there are now several organizations aiming to raise awareness of rare diseases as well as funding programs to support rare disease research and potential therapies.
Lafora disease causes
Lafora progressive myoclonus epilepsy can be caused by mutations in either the EPM2A gene on chromosome 6q24.3 or the NHLRC1 gene (also known as EPM2B) on chromosome 6p22.3 6. EPM2A gene encodes laforin glucan phosphatase (laforin) and NHLRC1 (EPM2B) encodes E3 ubiquitin protein ligase 1 (malin) 1. Laforin and malin play a critical role in the survival of nerve cells (neurons) in the brain.
Studies suggest that laforin and malin work together and may have several functions. One of these is to help regulate the production of a complex sugar called glycogen, which is a major source of stored energy in the body. The body stores this sugar in the liver and muscles, breaking it down when it is needed for fuel. Laforin and malin may prevent a potentially damaging buildup of glycogen in tissues that do not normally store this molecule, such as those of the nervous system.
Researchers have discovered that people with Lafora progressive myoclonus epilepsy have distinctive clumps called Lafora bodies within their cells. Lafora bodies are made up of an abnormal form of glycogen that cannot be broken down and used for fuel. Instead, it builds up to form clumps that can damage cells. Neurons appear to be particularly vulnerable to this type of damage. Although Lafora bodies are found in many of the body’s tissues, the signs and symptoms of Lafora progressive myoclonus epilepsy are limited to the nervous system.
Mutations in the EPM2A gene prevent cells from making functional laforin, while NHLRC1 gene mutations prevent the production of functional malin. It is unclear how a loss of either of these proteins leads to the formation of Lafora bodies. However, a loss of laforin or malin ultimately results in the death of neurons, which interferes with the brain’s normal functions. The condition tends to progress more slowly in some people with NHLRC1 gene mutations than in those with EPM2A gene mutations.
Mutations in the EPM2A and NHLRC1 genes account for 80 percent to 90 percent of all cases of Lafora progressive myoclonus epilepsy. In the remaining cases, the cause of the condition is unknown.
PRMD8 is a recently discovered gene associated with Lafora disease that codes for a protein responsible for laforin and malin translocation to the nucleus and the mutated form cause Laforin and Malin deficiency within the cytoplasm. The PRMD8 gene mutation is associated with early onset Lafora disease 6.
Researchers are still searching for other genetic changes that may underlie Lafora disease.
Lafora bodies
The accumulation of dense cytoplasmic aggregates is a feature of many neurodegenerative diseases, including Parkinson disease and Alzheimer disease 10 and the accumulation of Lafora bodies seems to be the primary cause of Lafora disease progression. This conclusion has been supported by several studies in mouse models of Lafora disease, which lack either laforin or malin and recapitulate the human disease 11. Genetic approaches that reduce or abolish glycogen synthesis prevent Lafora body formation and rescue other features of the Lafora disease phenotype in mice, including autophagy impairment, neurodegeneration and seizure susceptibility 12. In addition, overexpression or increased activation of the glycogen-synthesizing enzyme glycogen synthase in the presence of laforin and malin can result in polyglucosan body formation, as well as down-stream effects such as impaired autophagy and neuro- degeneration 12. Evidence of a link between impaired autophagy and neurodegeneration completes the current view on the pathogenesis of Lafora disease 13. The roles of laforin and malin in glycogen metabolism need to be unravelled to enable us to understand the mechanisms that lead to Lafora body formation and accumulation.
Lafora disease inheritance pattern
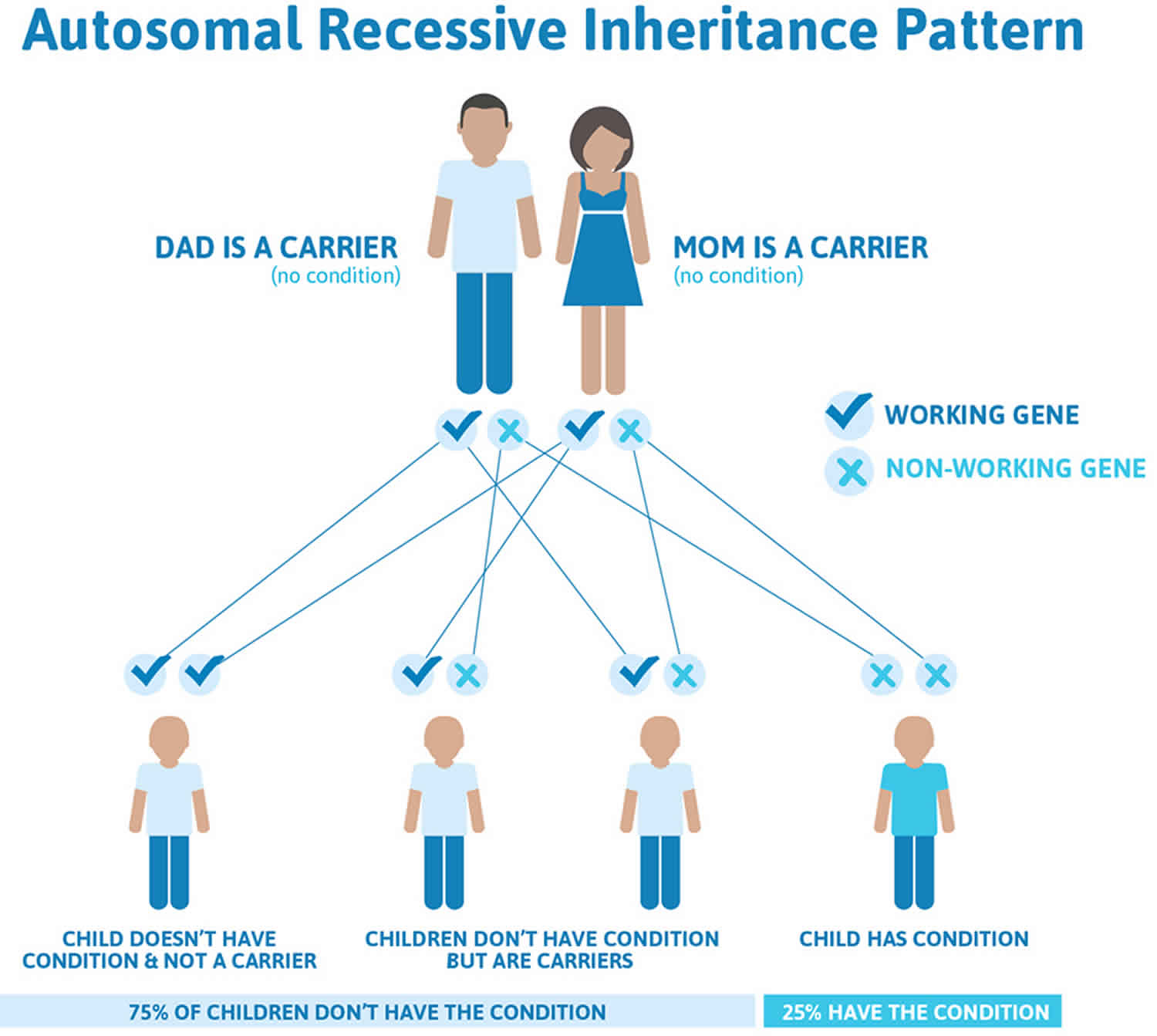
Lafora disease is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Lafora disease autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Lafora disease symptoms
The signs and symptoms of Lafora disease generally appear during late childhood or adolescence. Prior to the onset of symptoms, affected children appear to have normal development although some may have isolated febrile or nonfebrile convulsions in infancy or early childhood 7.
The first 2–3 years of Lafora disease are characterized by the following symptoms 1:
- Ataxia
- Confusion
- Depression
- Grand mal seizures
- Staring spells and/or absence seizures
- Drop in school performance
- Drop attacks
- Myoclonus
- Visual hallucinations
- Headaches
- Dysarthria
The most common feature of Lafora disease is recurrent seizures. At the time of onset, Lafora disease is difficult to distinguish from idiopathic generalized epilepsies 14. Apparently healthy older children or teenagers start having seizures, which can initially be controlled with antiepileptic drugs (AEDs) 15.
Several different types of seizures have been reported including generalized tonic-clonic seizures, occipital seizures (which can cause temporary blindness and visual hallucinations), absence and atonic seizures and myoclonic seizures 16. These seizures are considered “progressive” because they generally become worse and more difficult to treat over time 7. In retrospect, parents often recall that their child had experienced isolated febrile or non-febrile seizures earlier in childhood 16.
With the onset of seizures, people with Lafora disease often begin showing signs of cognitive decline. This may include behavioral changes, depression, confusion, ataxia (difficulty controlling muscles), dysarthria, and eventually, dementia. By the mid-twenties, most affected people lose the ability to perform the activities of daily living; have continuous myoclonus; and require tube feeding and and artificial respiration 7. Death commonly results from status epilepticus or aspiration pneumonia and other complications of chronic neurodegeneration 2.
Lafora disease diagnosis
A diagnosis of Lafora disease is often suspected based on the presence of characteristic signs and symptoms. Additional testing can then be ordered to confirm the diagnosis and rule out other conditions that may cause similar features. “Lafora bodies” (clumps of abnormal glycogen that cannot be broken down and used for fuel) usually accumulate in the skin, muscle liver, and brain tissues. Therefore, the diagnosis can be obtained by performing a biopsy from any of these organs, but the most commonly used and accessible site with high yield is skin biopsy of the axillary skin region 17. Genetic testing for changes (mutations) in either the EPM2A gene or the NHLRC1 gene may be used to confirm the diagnosis in some cases. An EEG and an MRI of the brain are generally recommended in all people with recurrent seizures and are useful in investigating other conditions in the differential diagnosis 7.
EEG in the early stages of the disease may be normal or show generalized slowing and loss of posterior dominant rhythm. With the progression of the disease, asymmetric, irregular, generalized spikes and polyspikes, maximum over the anterior regions associated with photosensitivity on a slowed background can be seen.
In later stages of the disease, myoclonic jerks become almost continuous. EEG usually will show paroxysms of generalized and fast irregular spike-and-wave discharges, exaggerated by photic stimulation at low frequency. These paroxysms are occipital predominantly. Other electrophysiological studies also may be abnormal in Lafora disease, especially visual evoked potentials (VEPs) that may demonstrate increased latencies or absence of response. Somatosensory evoked potentials can reveal aberrant integration of somatosensory stimuli and giant evoked potentials reflecting cortical hyperexcitability.
Neuroimaging including brain MRI is usually normal at the time of diagnosis; however, fluorodeoxyglucose positron emission tomography (FDG-PET) was found positive in two reported Lafora disease cases as it revealed posterior hypometabolism early in the disease.
Lafora disease treatment
Unfortunately, there is no cure available for Lafora disease. Management is only supportive, targeting seizure control and improving the patient’s functional status. Given the diversity of seizure types, including generalized tonic-clonic seizures, wide-spectrum antiepileptic drugs such as levetiracetam, sodium valproate, topiramate, and benzodiazepines are usually considered 18.
Valproate is the treatment of choice as first-line monotherapy, with a dose range from 15 mg/kg to 60 mg/kg, depending on the clinical response. However, it should be avoided in patients with suspected mitochondrial disorders due to inhibition of cytochrome C oxidase (complex 4) leading to decreased respiratory chain activity and carnitine uptake in addition to high ammonia blood levels.
Clonazepam is effective in treating myoclonic seizures; it can be used in Lafora disease, usually as add-on therapy, at doses ranging from 3 to 16 mg/day.
Phenobarbital is another wide spectrum antiepileptic drug that can be used in Lafora disease. The dose ranges from 3 to 8 mg in children and 30 to 200 mg in adults. It should be used with caution when added to valproate to avoid toxicity.
Piracetam, a pyrrolidone derivative, is another effective medication for myoclonus with few adverse effects and a positive tolerability profile. It has long been used for patients with progressive myoclonic epilepsies. One double-blinded, placebo-controlled trial of 20 patients with Unverricht-Lundborg disease showed significant reduction of myoclonic jerks and improvement in gait, particularly with doses close to 24 g/day. A linear dose-effect relationship was also established in this study.
Levetiracetam is a potent wide spectrum antiepileptic drug with few adverse effects. Like piracetam, it is another pyrrolidone derivative. It binds and stimulates synaptic vesicle protein 2A (SV2A), leading to inhibition of neurotransmitter release.
Efficacy of levetiracetam, particularly in the treatment of generalized seizures and myoclonus, has been demonstrated in multiple studies and case series of patients with progressive myoclonic epilepsy. In a study involving 23 patients with Unverricht-Lundborg disease, clinical improvement was seen in over two-thirds of the patients on doses ranging from 1000 to 4000 mg daily.
Brivaracetam is a novel molecule with the same mechanism as levetiracetam but at least a 10-fold higher affinity for the SV2A binding site compared to levetiracetam. It was proposed as a drug with high potential efficacy for myoclonus. In an animal study done on rat models with post-cardiac arrest and anoxic brain injury seizures, low dose brivaracetam of 0.3 mg/kg was superior to 3 mg/kg of levetiracetam in controlling post-anoxic seizures. Anti-seizure activity for both antiepileptic drugs started 30 minutes following intraperitoneal administration and was maintained for 150 minutes
Perampanel is a selective, noncompetitive antagonist of AMPA-type glutamate receptor that is usually used for the treatment of refractory focal onset seizures, but it is also effective for generalized epilepsy.
Two case reports document perampanel effectiveness in Lafora disease when used as first-line monotherapy or add-on. One case was a 15-year-old female with Lafora disease who was treated with 10 mg of perampanel as monotherapy. The treatment resulted in a significant and dramatic decrease in seizure frequency in addition to improvement in neurological and cognitive functioning. The second case was a 21-year-old Turkish female given perampanel at a dose of 8 to 10 mg in addition to a regimen that included clonazepam, levetiracetam, piracetam, valproate, zonisamide, a ketogenic diet, and vagal nerve stimulation (VNS). This was followed by seizure remission for more than 3 months and was associated with decreased epileptiform discharges on EEG.
Topiramate is another wide-spectrum antiepileptic drug, usually used for the treatment of refractory focal seizures as well as generalized seizures. It is a sulfamate-substituted monosaccharide molecule. Beneficial use topiramate for seizure treatment in patients with progressive myoclonic epilepsies and Lafora disease stems mostly from case studies. Effectiveness in treating myoclonus and myoclonic seizure was demonstrated when used as add-on therapy. In one study, five out of eight patients with progressive myoclonic epilepsy improved after adding topiramate to their antiepileptic drug regimen with improvement in myoclonic seizures and functional capacity. However, topiramate efficacy tended to decrease over time, and the drug was discontinued in two out of five patients because of a rapid increase in cognitive impairment and vomiting.
Zonisamide, a sulfonamide derivative, chemically distinct from any of the previously established antiepileptic drugs, is indicated for the treatment of refractory partial epilepsy but also is useful for a variety of generalized epilepsies, including epileptic encephalopathies, such as Lennox-Gastaut (LGS) and West syndromes.
Some case reports and small studies have suggested that zonisamide may be effective in treating patients with progressive myoclonic epilepsy. In long-term observation and clinical follow up of a brother and a sister with Lafora disease who had resistant, repeated attacks of severe myoclonus, tonic, and tonic-clonic seizures, the use of oral zonisamide as add-on therapy resulted in dramatic seizure control for about 12 to 14 years in both patients, not only for myoclonus but generalized tonic-clonic seizures as well.
More specifically, almost all patients with Unverricht-Lundborg disease who were treated with zonisamide as add-on therapy showed a dramatic reduction in myoclonus and a marked improvement in generalized tonic-clonic seizures and functional capacity, although efficacy tended to decrease over time. Doses of up to 6 mg/kg per day were used.
Lamotrigine was noted as an effective treatment for infantile and juvenile neuronal ceroid lipofuscinosis. The use of lamotrigine in addition to some GABA-ergic drugs (vigabatrin and tiagabine) could exacerbate myoclonus in patients with juvenile myoclonic epilepsy. Such exacerbation was also observed in five patients with Unverricht-Lundborg disease, and therefore, the use of lamotrigine in progressive myoclonic epilepsies can be avoided. Clinicians also advised patients to avoid using other sodium channel blockers (phenytoin, carbamazepine, and oxcarbazepine) for the same reason.
Vagal nerve stimulation appears to be effective in reducing the seizure frequency in a few cases of Lafora disease.
A ketogenic diet (low carbohydrate-high cholesterol) that is proven effective in a variety of refractory epilepsies including infantile myoclonic seizures and Lennox-Gastaut syndrome was shown to be ineffective in treating Lafora disease patients in an Italian study of five patients but was unable to stop the disease progression 8.
Lafora disease prognosis
The long-term outlook (prognosis) for people with Lafora disease is unfortunately poor. There is currently no cure for the condition and it is considered progressive (symptoms worsen over time). By the mid-twenties, most affected people lose the ability to perform the activities of daily living; have continuous myoclonus; and require tube feeding and comprehensive care. On average, affected people survive approximately 10 years after the onset of symptoms 7.
References- Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BA. Lafora disease – from pathogenesis to treatment strategies. Nat Rev Neurol. 2018;14(10):606-617. doi:10.1038/s41582-018-0057-0 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6317072
- Girard JM, Turnbull J, Ramachandran N, Minassian BA. Progressive myoclonus epilepsy. Handb Clin Neurol. 2013;113:1731-6. doi: 10.1016/B978-0-444-59565-2.00043-5
- Lafora GR & Glueck B Beitrag zur Histopathologie der myoklonischen Epilepsie [German]. Z. Gesamte Neurol. Psychiatr. 6, 1–14 (1911).
- Sakai M, Austin J, Witmer F, Trueb L. Studies in myoclonus epilepsy (Lafora body form). II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology. 1970 Feb;20(2):160-76. doi: 10.1212/wnl.20.2.160
- Lafora disease. https://ghr.nlm.nih.gov/condition/lafora-progressive-myoclonus-epilepsy
- Ibrahim F, Murr N. Lafora Disease. [Updated 2020 Jul 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482229
- Jansen AC, Andermann E. Progressive Myoclonus Epilepsy, Lafora Type. 2007 Dec 28 [Updated 2019 Feb 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1389
- Cardinali S, Canafoglia L, Bertoli S, Franceschetti S, Lanzi G, Tagliabue A, Veggiotti P. A pilot study of a ketogenic diet in patients with Lafora body disease. Epilepsy Res. 2006 May;69(2):129-34. doi: 10.1016/j.eplepsyres.2006.01.007. Epub 2006 Feb 28. https://doi.org/10.1016/j.eplepsyres.2006.01.007
- Jara-Prado A, Ochoa A, Alonso ME, Lima Villeda GA, Fernández-Valverde F, Ruano-Calderón L, Vargas-Cañas S, Durón RM, Delgado-Escueta AV, Martínez-Juárez IE. Late onset Lafora disease and novel EPM2A mutations: breaking paradigms. Epilepsy Res. 2014 Nov;108(9):1501-10. November 2014; 108(9):1501-1510.
- Nuovo G, Amann V, Williams J, Vandiver P, Quinonez M, Fadda P, Paniccia B, Mezache L, Mikhail A. Increased expression of importin-β, exportin-5 and nuclear transportable proteins in Alzheimer’s disease aids anatomic pathologists in its diagnosis. Ann Diagn Pathol. 2018 Feb;32:10-16. doi: 10.1016/j.anndiagpath.2017.08.003. Epub 2017 Sep 7.
- DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, Roach PJ. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J Biol Chem. 2010 Aug 13;285(33):25372-81. doi: 10.1074/jbc.M110.148668. Epub 2010 Jun 10.
- Duran J, Gruart A, García-Rocha M, Delgado-García JM, Guinovart JJ. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet. 2014 Jun 15;23(12):3147-56. doi: 10.1093/hmg/ddu024. Epub 2014 Jan 22.
- McMahon J, Huang X, Yang J, Komatsu M, Yue Z, Qian J, Zhu X, Huang Y. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci. 2012 Nov 7;32(45):15704-14. doi: 10.1523/JNEUROSCI.2392-12.2012
- Shahwan A, Farrell M, Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol. 2005 Apr;4(4):239-48. doi: 10.1016/S1474-4422(05)70043-0
- Madhavan D, Kuzniecky RI. Lafora disease. Rev Neurol Dis. 2006 Summer;3(3):131-5.
- Minassian BA. Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol. 2001 Jul;25(1):21-9. doi: 10.1016/s0887-8994(00)00276-9
- Parihar R, Rai A, Ganesh S. Lafora disease: from genotype to phenotype. J. Genet. 2018 Jul;97(3):611-624.
- Potes T, Galicchio S, Rosso B, Besocke G, García MDC, Avalos JC. [Progressive myoclonic epilepsy secondary to Lafora’s body disease]. Medicina (B Aires). 2018;78(6):436-439.





{kind=link}