Lower motor neuron lesion
Lower motor neuron lesion is a lesion which affects nerve fibers traveling from the anterior horn of the spinal cord to the relevant muscle(s) the lower motor neuron. Lower motor neuron syndromes are clinically characterized by muscle atrophy, weakness and hyporeflexia without sensory involvement 1. Lower motor neuron syndromes may arise from disease processes affecting the anterior horn cell or the motor axon and/or its surrounding myelin. The lower motor neurons directly innervate skeletal muscle and have cell bodies in the anterior horn of the spinal cord (ventral horn) and at cranial nerve nuclei 2. Lower motor neuron cell bodies are located in specific nuclei in the brainstem as well as in the ventral horn of the spinal cord 3. The remarkable characteristic of lower motor neurons is their axonal extension and connection outside of the central nervous system (CNS). Lower motor neurons are cholinergic and receive inputs from upper motor neurons, sensory neurons as well as from interneurons. Paralysis is a typical clinical symptom of lower motor neuron lesions since once damaged there is no alternative route to convey the information to the muscle targets in the periphery. Lower motor neurons are classified into three groups according to the type of target they innervate: (i) branchial, (ii) visceral, and (iii) somatic motor neurons.
Lower motor neurons transmit impulses via spinal peripheral nerves or cranial nerves to skeletal muscles. Three distinct types of motor neurons are categorized based on the target they innervate: branchial, visceral, and somatic motor neurons.
Branchial motor neurons
These motor neurons are located in the brainstem and are responsible for forming the lower motor neurons of the cranial nerve nuclei. They are called “branchial” because they innervate the muscles formed by the branchial (pharyngeal) arch which includes muscles innervated by cranial nerves V, VII, IX, and X 4. The V3 portion of the trigeminal nerve (CN V) is derived from the 1st branchial arch and innervates the muscles of mastication (temporalis, masseter, medial pterygoid, and lateral pterygoid) as well as smaller muscles supplied by CN V. An abnormality in the development of the 1st branchial arch may result in Pierre Robin sequence or Treacher Collins syndrome. The facial nerve (CN VII) innervates muscles derived from the 2nd branchial arch which includes the muscles of facial expression, allowing one to smile. The glossopharyngeal nerve (CNIX) innervates muscles derived from the 3rd branchial arch which includes the stylopharyngeus muscle which assists in swallowing. Lastly, the vagus nerve (CN X) innervates muscles derived from the 4th and 6th branchial arches which permit swallowing and speaking. Lesions at any region along the nerve from the cranial nerve nuclei in the brainstem to these muscles would result in their respective lower motor neuron deficits.
Visceral motor neurons
These motor neurons are a component of the autonomic nervous system (ANS) and regulate smooth muscles and glands. These nerves can be further broken down into the sympathetic and parasympathetic nervous system. Motor neurons of the sympathetic nervous system are present from T1 to L2 segments of the spinal cord. These segments regulate the “fight or flight” response which can dictate body metabolism, awareness, and energy storage 4. Conversely, the parasympathetic nervous system contributes to CN III, VII, IX, and X as well as the S2 through S4 segments of the sacrum. These segments regulate the “rest and digest” response which has a significant role in GI motility and sexual drive. Because the lower motor neurons don’t directly innervate these visceral targets, visceral motor neurons have been an exception to the role of traditional lower motor neurons.
Somatic motor neurons
These motor neurons are the traditional motor neurons located in the ventral horn of the spinal cord and directly innervate skeletal muscle. They receive stimuli from the upper motor neurons from the primary motor cortex and relay that information from the spinal cord to their target. Somatic motor neurons can be further divided into alpha, beta, and gamma, depending on the type of muscle fiber they innervate 4. Alpha motor neurons innervate extrafusal fibers and are responsible for muscle contraction. Beta motor neurons innervate intrafusal fibers which act like proprioceptors and detects the change in length of a muscle. Gamma muscle fibers also innervate intrafusal muscle fibers. Abnormalities in somatic motor neurons present in classic fashion in spinal muscular atrophy and poliomyelitis.
Upper motor neurons relay information from the brain to the spinal cord and brainstem where they activate lower motor neurons which directly stimulate muscles to contract 2. Upper motor neurons are first-order neurons regulated by the neurotransmitter glutamate, are found in the primary motor cortex (precentral gyrus), and terminate in the spinal cord or brainstem, unable to leave the central nervous system (CNS).
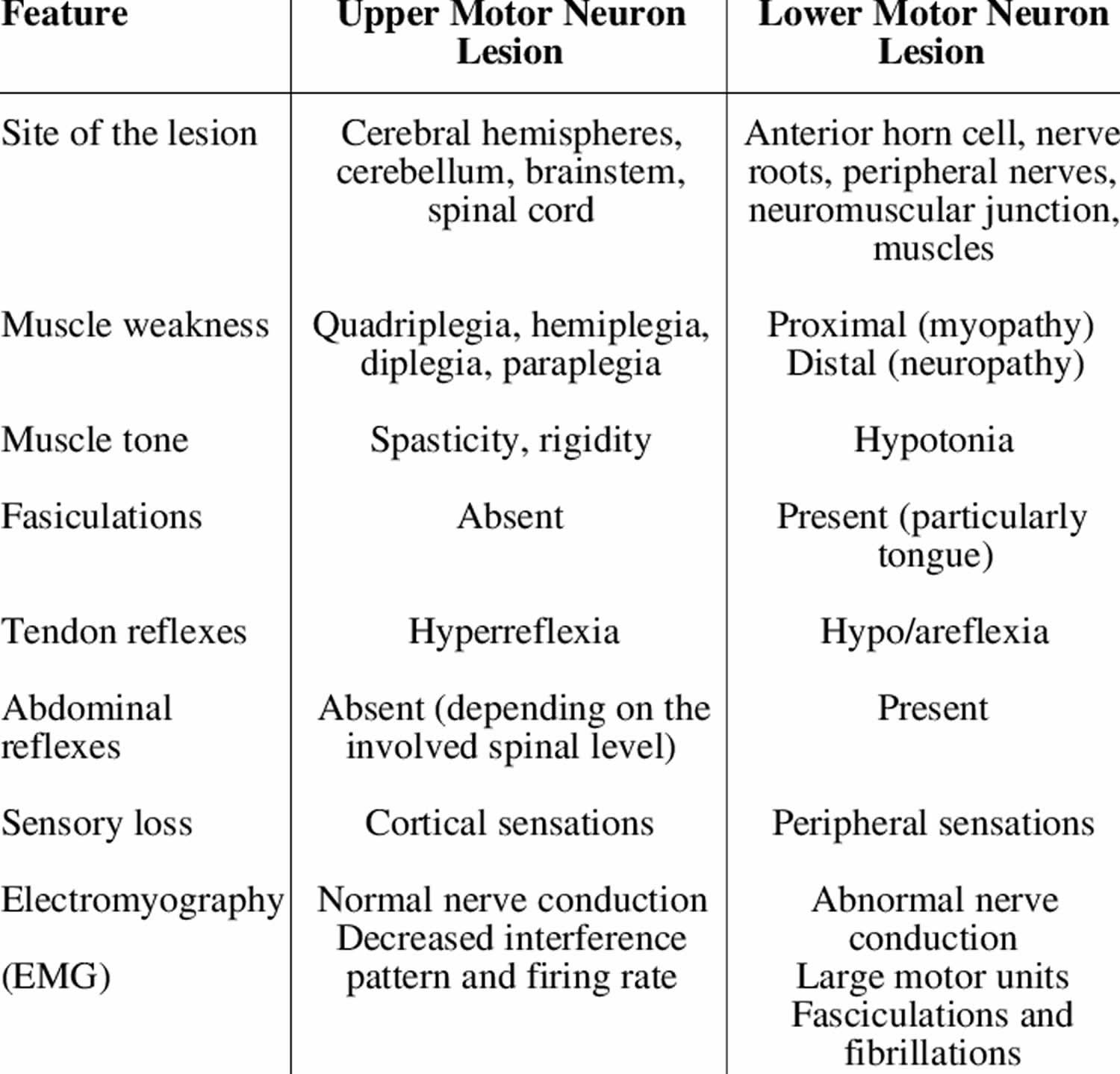
Although both upper and motor neuron lesions result in muscle weakness, they are clinically distinct due to various other manifestations. Unlike upper motor neurons, lower motor neuron lesions present with muscle atrophy, fasciculations (muscle twitching), decreased reflexes, decreased tone, negative Babinsky sign, and flaccid paralysis. These findings are crucial when differentiating upper motor neuron vs. lower motor neuron lesions and must be distinguished from upper motor neuron characteristics to formulate a proper differential diagnosis. Although a variety of diseases involve lower motor neurons, poliomyelitis and spinal muscular atrophy are two classic examples of isolated lower motor neuron disease.
Figure 1. Lower motor neuron
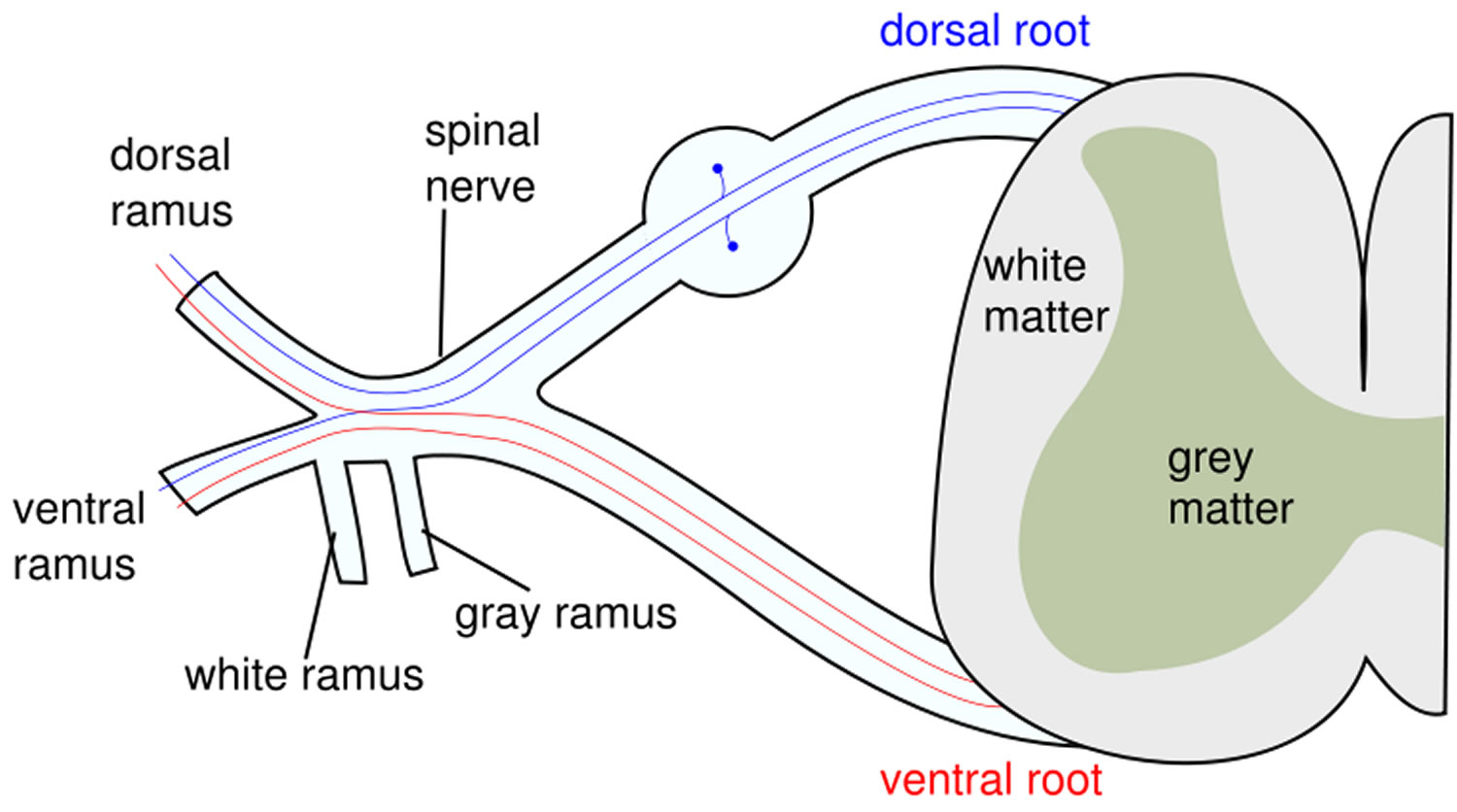
Figure 2. Spinal cord cross section
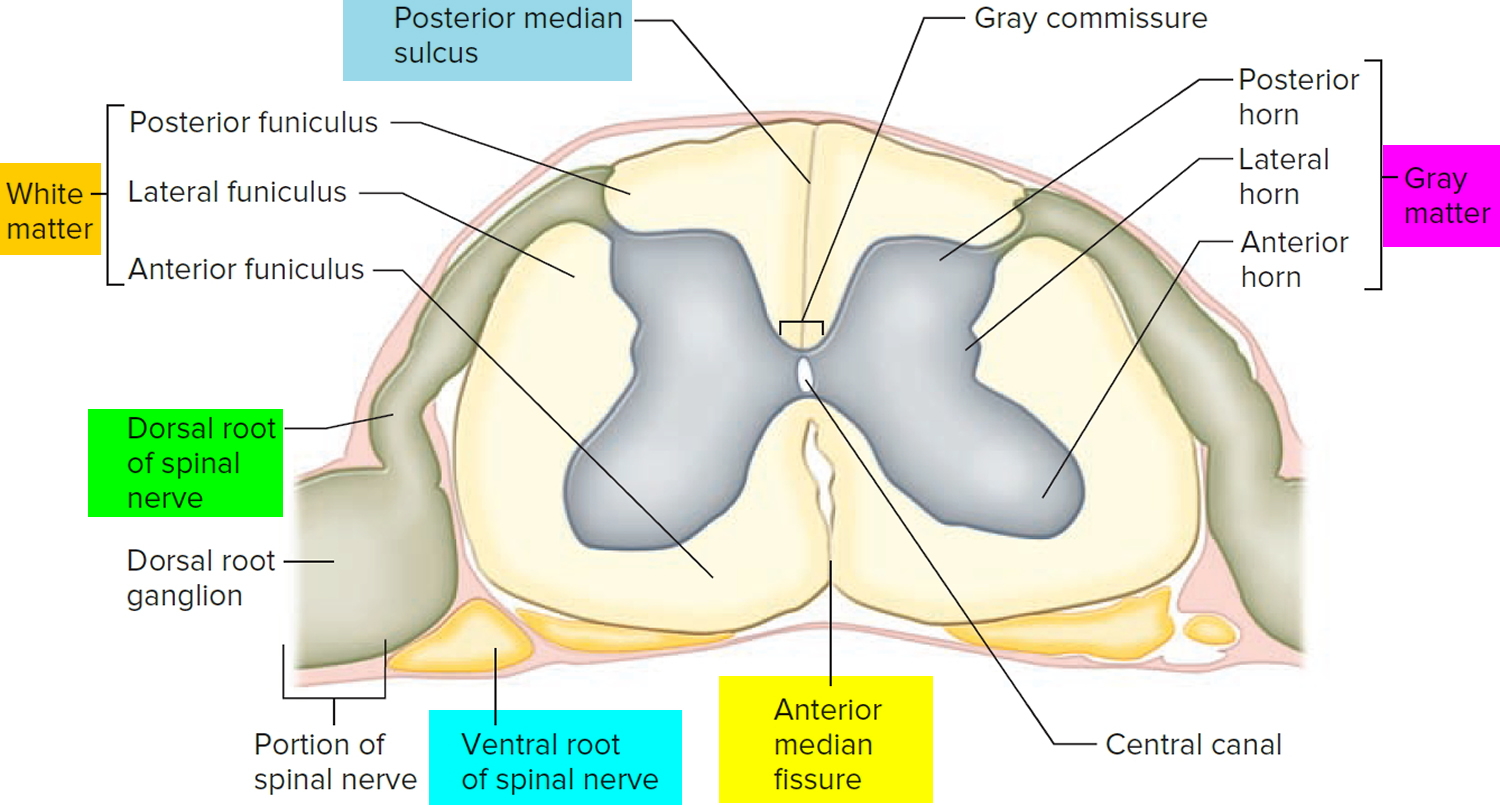
Figure 3. Processing of sensory input and motor output by the spinal cord
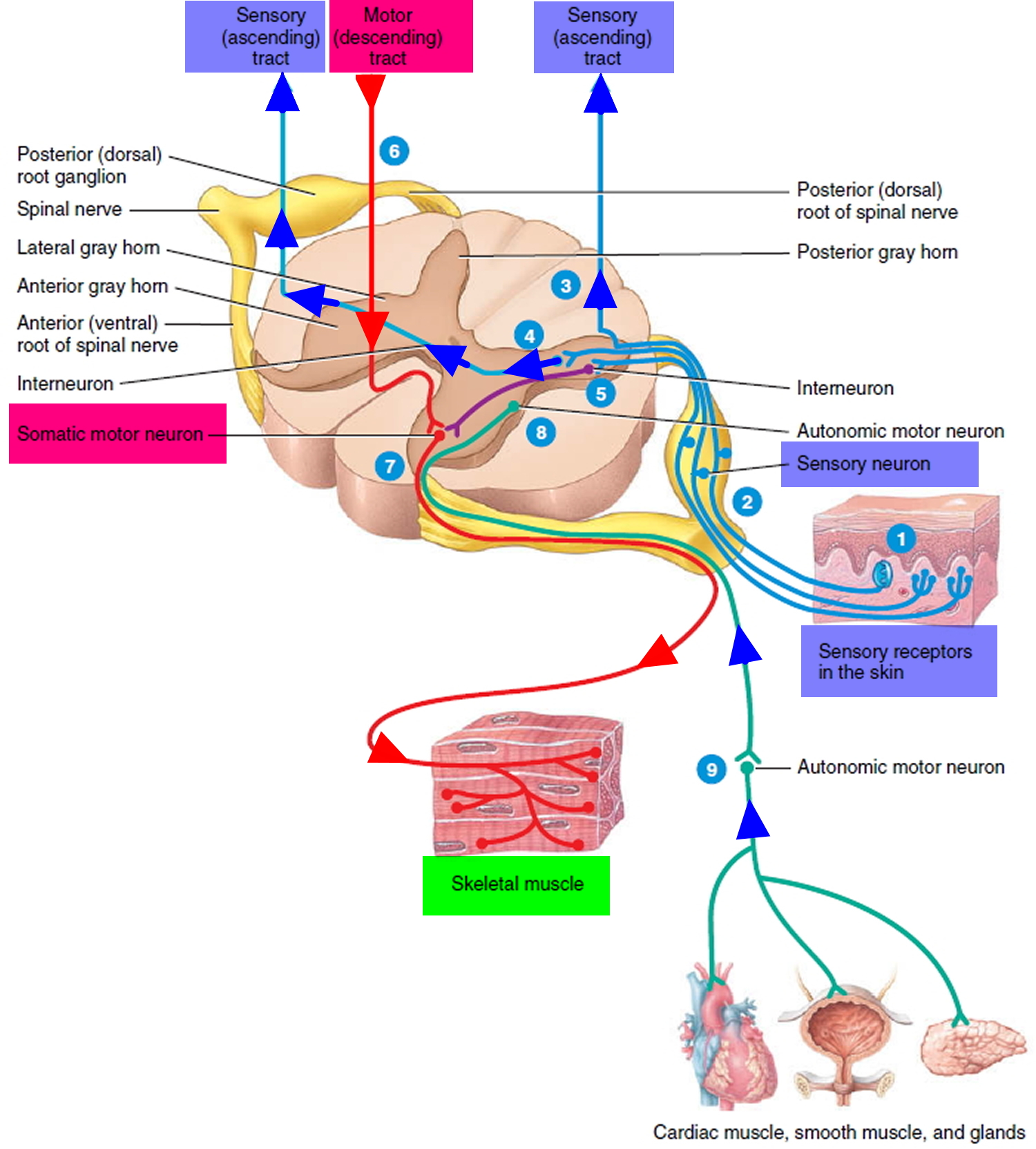
Footnote: Sensory input is conveyed from sensory receptors to the posterior gray horns of the spinal cord, and motor output is conveyed from the anterior and lateral gray horns of the spinal cord to effectors (muscles and glands).
Figure 4. Spinal cord descending tracts from the brain
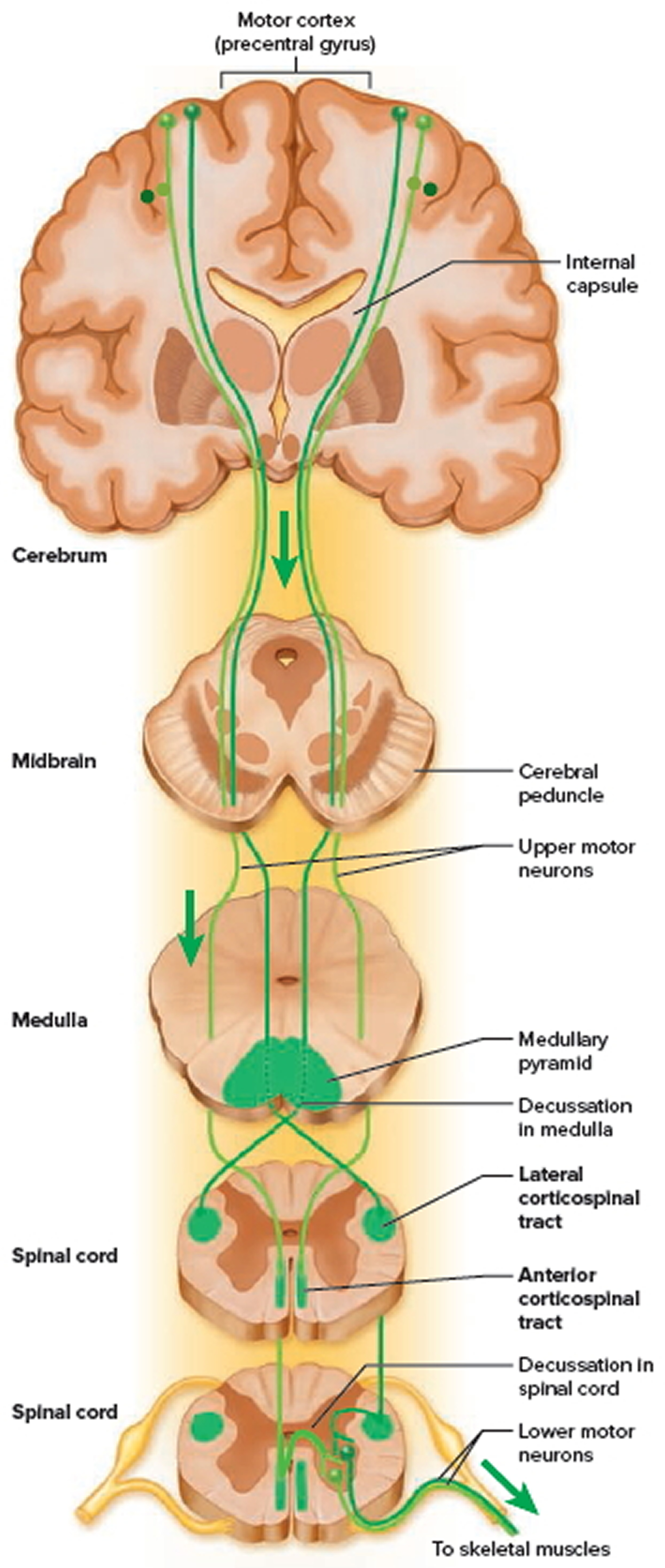
Figure 5. Spinal cord segments
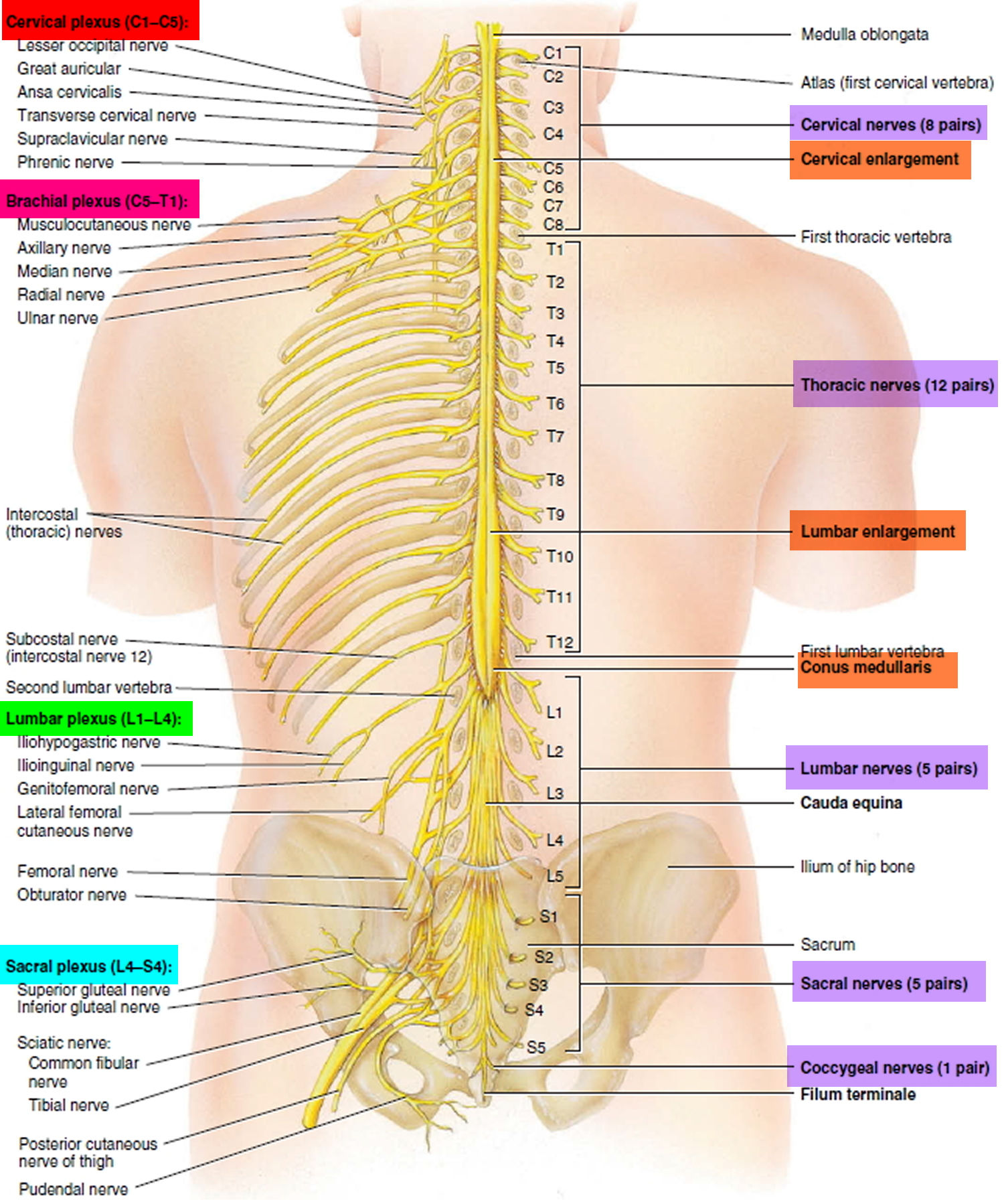
Figure 6. Spinal nerve
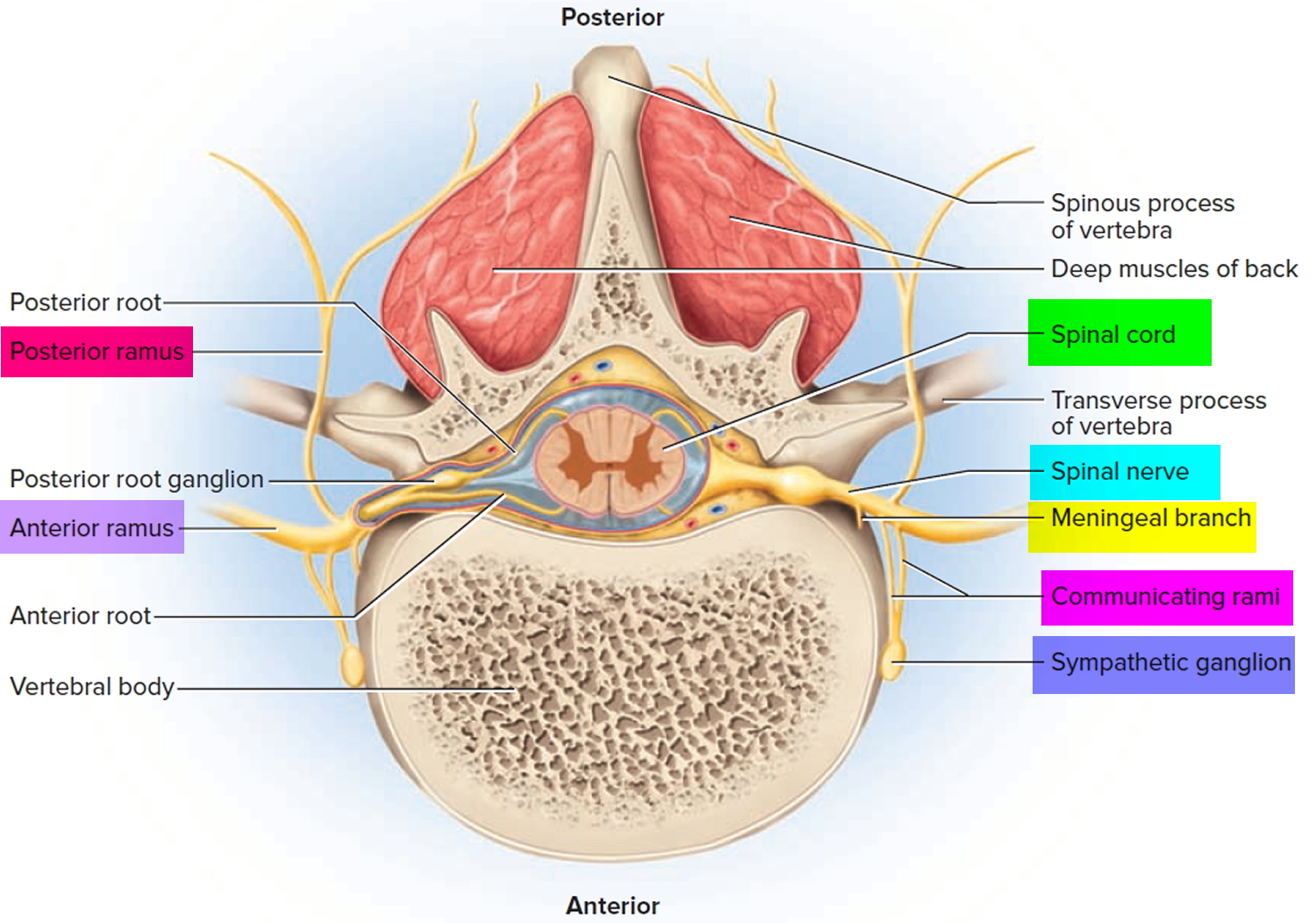
Figure 7. Spinal nerve fiber anatomy
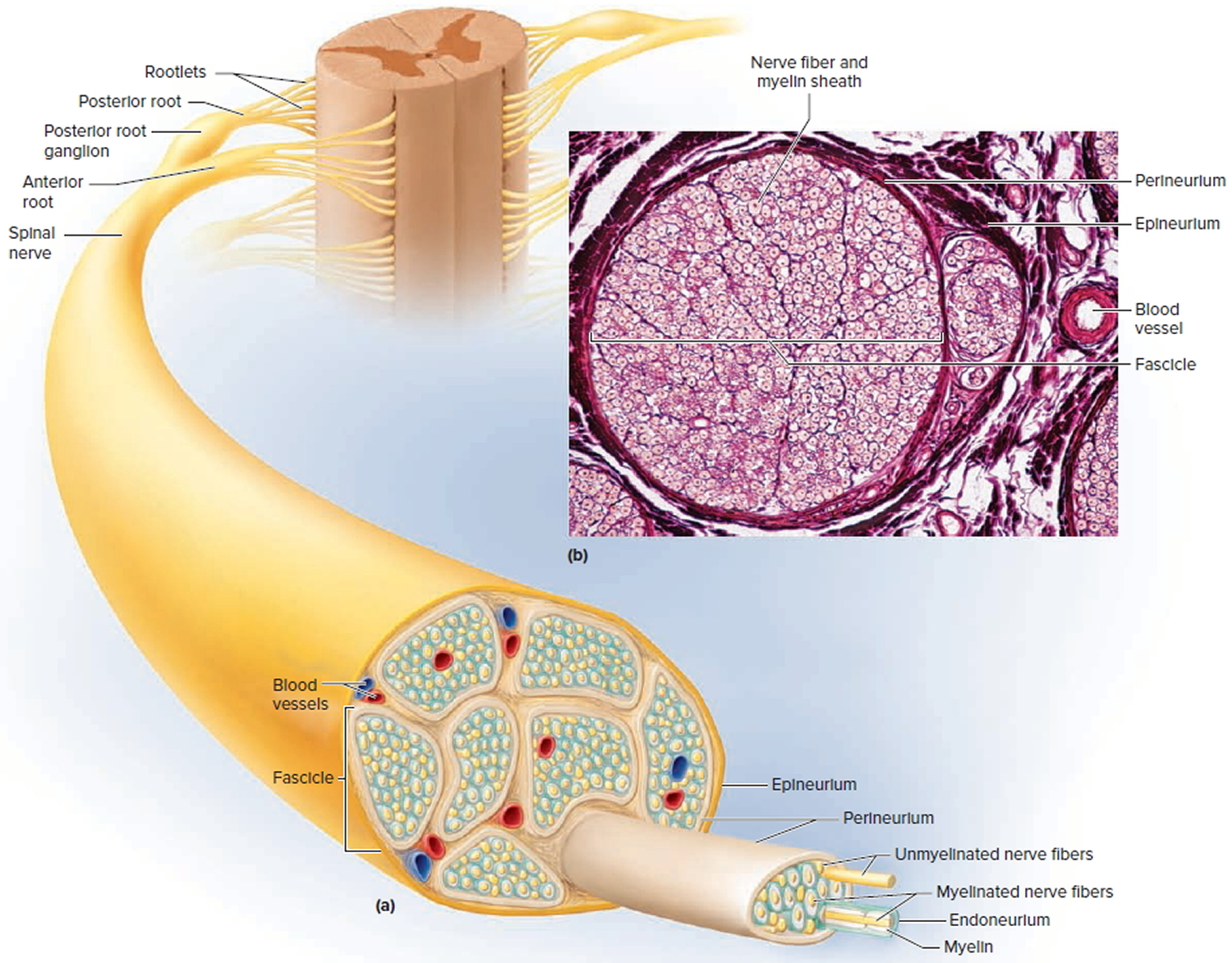
Figure 8. Spinal nerve ganglion
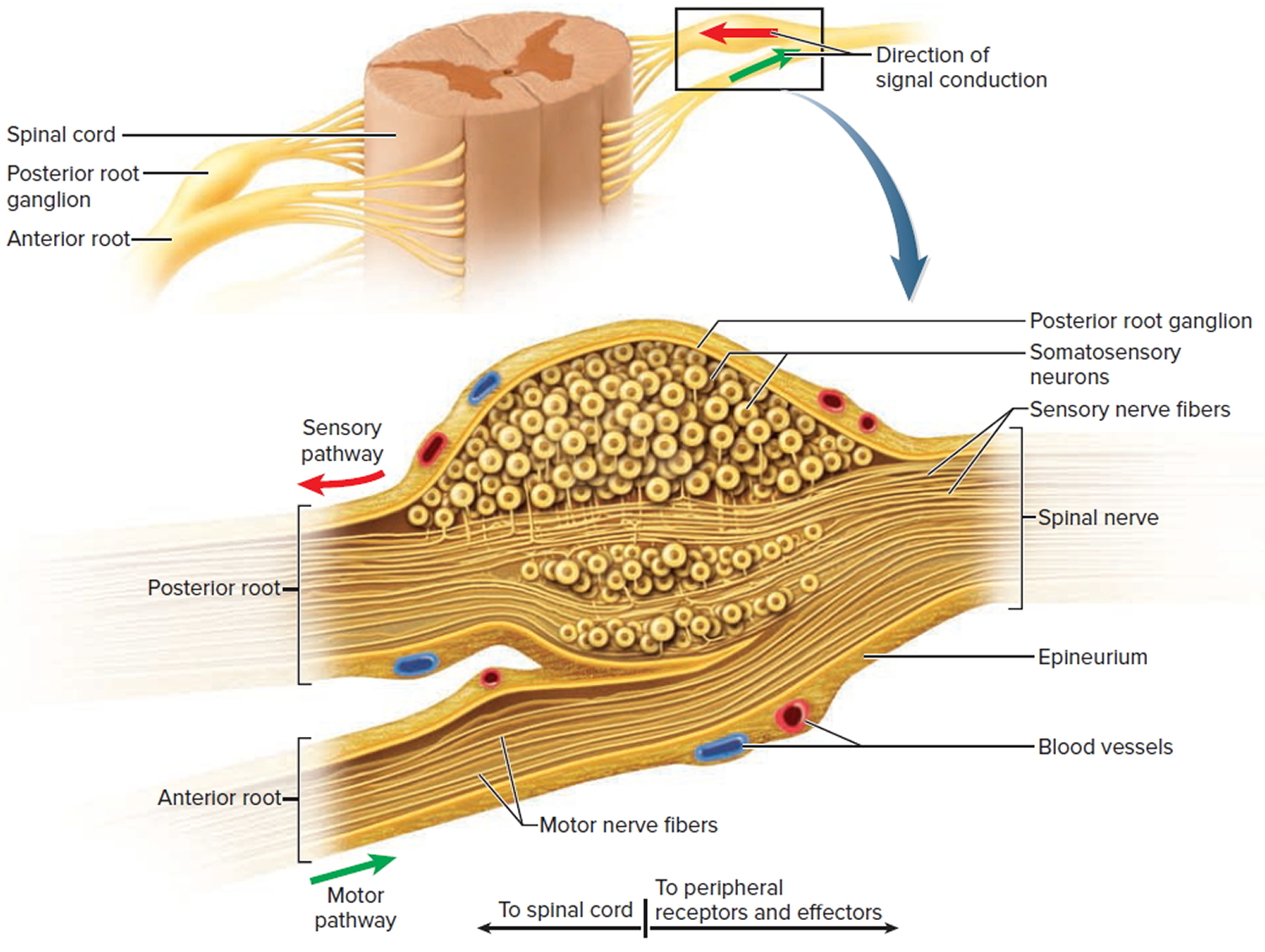
Footnote: The posterior root ganglion contains the somas of unipolar sensory neurons conducting signals from peripheral sense organs toward the spinal cord. Below this is the anterior root of the spinal nerve, which conducts motor signals away from the spinal cord, toward peripheral effectors. Note that the anterior root is not part of the ganglion.
Figure 9. Dermatome (spinal nerves sensory innvervation)
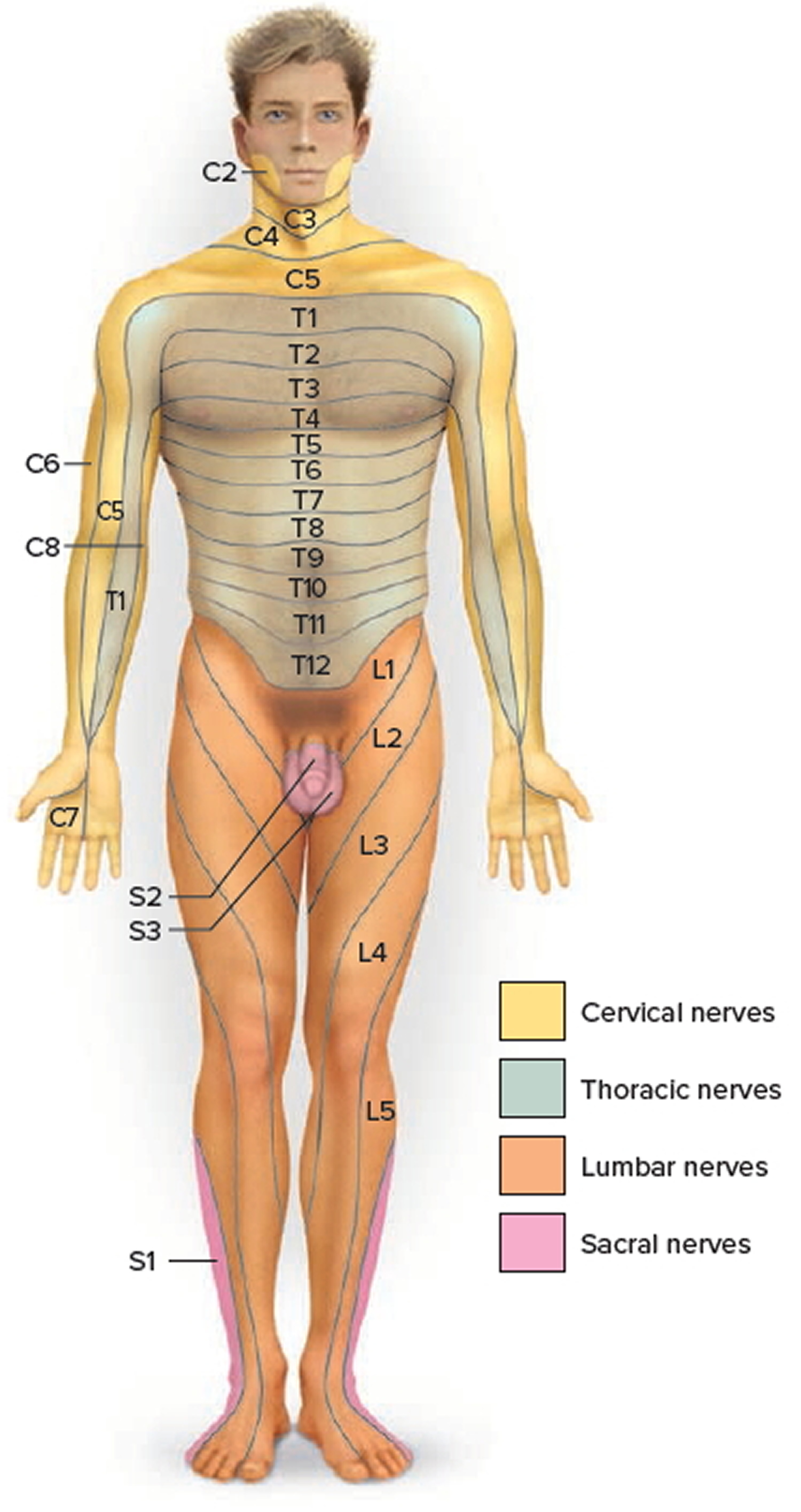
Footnote: Each zone of the skin is innervated by sensory branches of the spinal nerves indicated by the labels. Nerve C1 does not innervate the skin.
Table 1. Differentiating features of upper and lower motor neuron lesions
Branchial motor neurons
Branchial motor neurons are located in the brainstem and form, together with sensory neurons, the cranial nuclei. They innervate branchial arch derived muscles of the face and neck through 5 cranial nuclei: the trigeminal (V), facial (VII), glossopharyngeal (IX), vagus (X) and accessory (XI) nerves. Despite their similar function, muscles of the neck and the face differ from other skeletal muscles in their embryological origin since they do not derive from the somites, but instead from the branchial arches. Such developmental difference is mirrored by specific characteristics reviewed in depth by Chandrasekhar (2004).
Visceral motor neurons
Visceral motor neurons belong to the autonomic nervous system (ANS) responsible for the control of smooth muscles (i.e., heart and arteries) and glands. The autonomic nervous system can be described as the association of two components: (i) preganglionic motor neurons located in the CNS connected to ganglionic neurons belonging to the peripheral nervous system (PNS). In turn, peripheral ganglionic neurons target to the final effector organ. Additionally, the autonomic nervous system is anatomically and functionally divided into two structures: (i) the sympathetic system and (ii) the parasympathetic system.
Motor neurons of the sympathetic system
The sympathetic nervous system is involved in the traditional “fight or flight” responses, recruiting energy storage, increasing awareness, and leading to a global activation of the body metabolism. Central motor neurons of the sympathetic system are located in the spinal cord from the thoracic segment 1 (T1) to the lumbar segment 2 (L2). These motor neurons have an intermedio-lateral position and constitute the preganglionic column that will be described below. They connect to 3 different targets: two chains of ganglia adjacent to the spinal cord named (i) paravertebral and (ii) prevertebral as well as directly to (iii) the chromaffin cells of the adrenal medulla responsible for the release of the catecholamines (i.e., adrenaline and noradrenaline) in the circulation, in response to stress stimuli. On the other hand, paravertebral and prevertebral ganglia connect to a wide variety of targets including the heart, lungs, kidneys, intestines and the colon.
Motor neurons of the parasympathetic system
The parasympathetic system controls glands secretion and activates the gastrointestinal tract as well as sexual behavior, which are summarized as “rest and digest” functions. Central motor neurons of the parasympathetic system are located in the brainstem and contribute to the formation of the cranial nerves (III, VII, IX, and X). Parasympathetic motor neurons are also found in sacral segments 2 to 4 (S2–S4) of the spinal cord. They innervate ganglia located in the proximity of the peripheral targets such as the heart, bladder, lungs, kidneys, and pancreas.
In summary, visceral central motor neurons from the sympathetic and parasympathetic systems relay information from the CNS to ganglionic neurons of the peripheral nervous system (PNS). In turn those ganglia antagonistically control a large number of various visceral targets. In contrast to branchial mentioned previously and somatic motor neurons described below, visceral motor neurons do not directly connect to the final effector. As a result, they constitute an anatomical and functional exception among lower motor neurons.
Somatic motor neurons
Somatic motor neurons are located in the Rexed lamina IX in the brainstem and the spinal cord and innervate skeletal muscles responsible for movements 6. Motor neurons form coherent groups connecting to a unique muscle target defined as motor neuron pools. Somatic motor neurons can be divided into 3 groups: (i) alpha, (ii) beta, and (iii) gamma according to the muscle fiber type they innervate to within a specific muscle target (Figure 10). A motor unit defines a single motor neuron together with all the muscle fibers it innervates. Interestingly, motor units are homogeneous: a motor neuron innervates muscle fibers of a single type. This observation suggests selectivity in the establishment of neuromuscular connectivity and/or a coordinated maturation between a motor neuron and its targeted fibers. Intuitively, the diversity of motor neurons mirrors the diversity of targets they innervate. Therefore, to better describe somatic motor neuron diversity, a brief description of skeletal muscle physiology will be provided.
Three classes of muscles can be anatomically and functionally distinguished: (i) cardiac muscles, (ii) smooth muscles and (iii) skeletal muscles. Cardiac muscles are responsible for the rhythmic contraction of the heart while smooth muscles control the diameter of blood vessels and the internal digestive and secretion organs. Both smooth and cardiac muscles are innervated by the autonomic nervous system (ANS) (described above). In contrast, somatic motor neurons exclusively innervate skeletal muscles that are the most abundant muscle class, with around 639 different muscles in the human body 7. Skeletal muscles are firmly attached to the skeleton by the tendons and are responsible for both posture and movement.
Figure 10. Muscle innervation
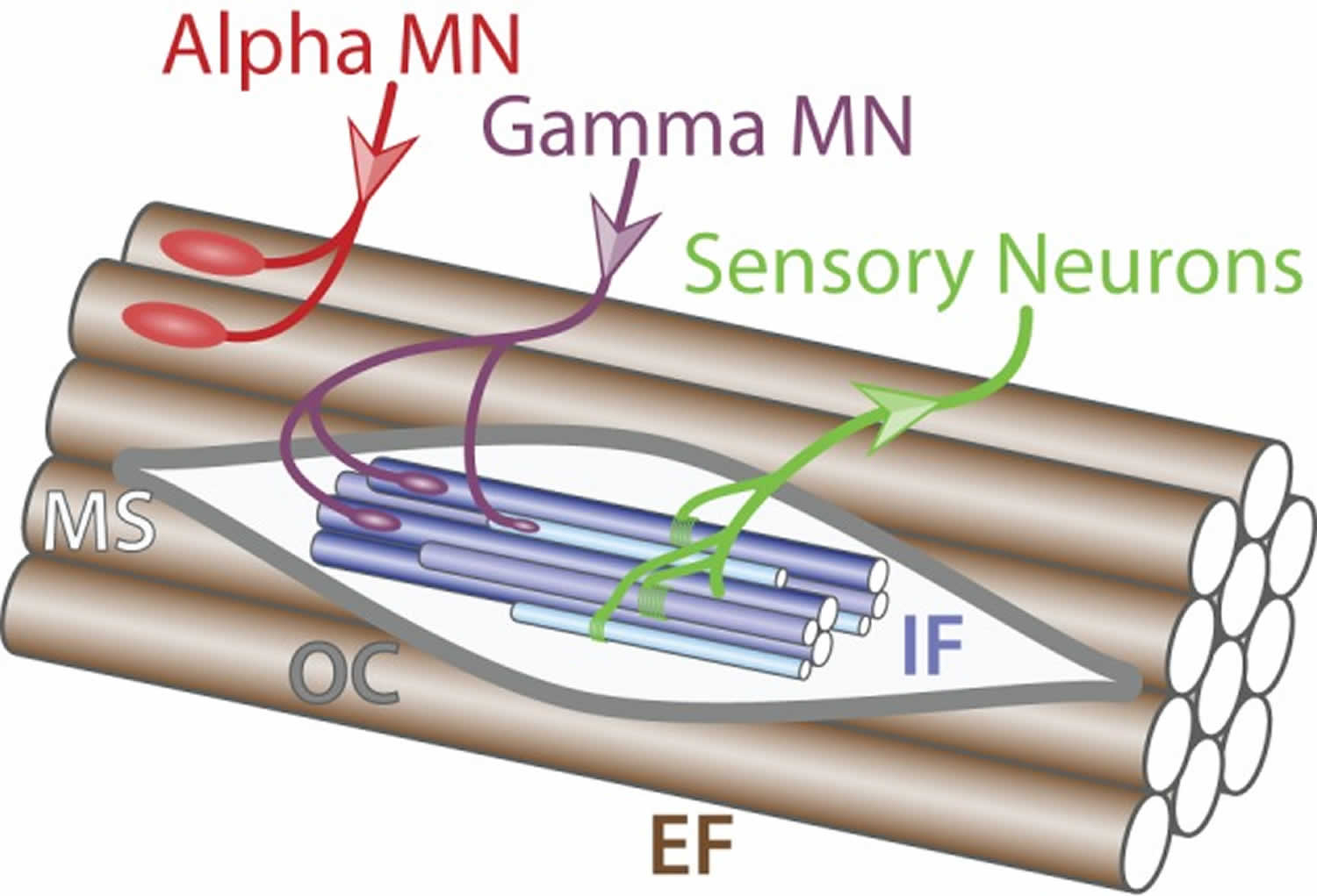
Footnote: Muscle innervation. Schematic of muscle fibers on the longitudinal section. Alpha motor neuron (red) innervates (incoming arrow) extrafusal muscle fibers (EF, brown) whereas gamma motor neuron (purple) connects to intrafusal fibers (IF, blue) within the muscle spindle (MS, light gray) surrounded by the outer capsule (OC, dark gray). Sensory neurons (green) carry information from the intrafusal fibers to the central nervous system (outgoing arrow).
[Source 3 ]Mirroring the diversity of both intra- and extrafusal fiber types in a muscle, somatic motor neurons are further sub-divided into 3 types: (i) alpha, (ii) beta and (iii) gamma that will be further described below.
Alpha motor neurons
Alpha motor neurons exclusively innervate extrafusal muscle fibers and are the key of muscle contraction (Figure 10). Anatomically, alpha motor neurons are characterized by a large cell body and a well-characterized neuromuscular ending. They have an important role in the spinal reflex circuitry by receiving monosynaptic innervation directly from SNs thus minimizing the delay of the response 8. Alpha motor neurons can be further divided into 3 different subtypes depending on the extrafusal fiber type they innervate: (i) SFR, (ii) FFR, and (iii) FF (Figure 3) 9. There is no universal criteria distinguishing alpha motor neurons subtypes; however, some trends are observed in term of size, excitability, and firing pattern. SFR motor neurons tend to have a smaller cell body diameter and thus a higher input resistance making them responsive to a lower stimulation threshold. As a result, SFR motor neurons are recruited first during muscle contraction. They also have the capacity of maintaining a persistent activity even after the stimulation ceased 10. On the other hand, FF motor neurons have often a larger cell body and are firing after the initial recruitment of SFR neurons giving extra strength to the activated muscle. In terms of conduction velocity, motor neurons innervating fast fibers are substantially faster (100 m/s) than SFR motor neurons (85 m/s) 9. Lastly, little is known about FFR motor neurons physiology; yet, they are considered to have intermediate characteristics between FF and SFR motor neurons (Figure 11).
Figure 11. Alpha and gamma motor neurons
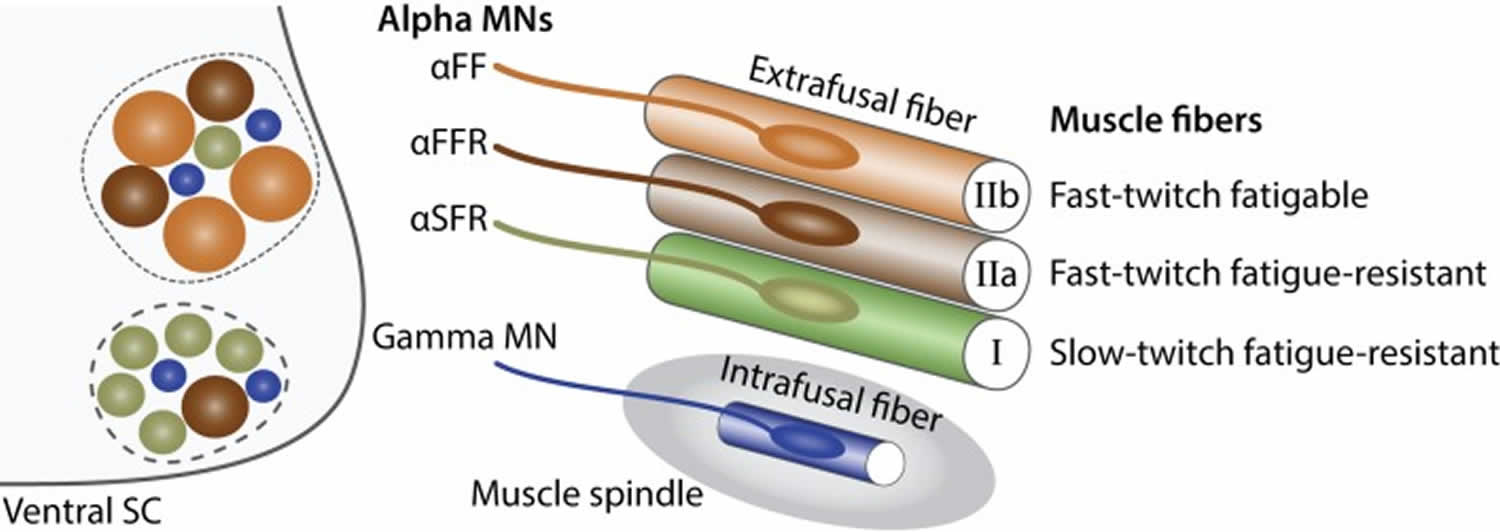
Footnote: Characteristics of alpha and gamma motor neurons. Schematic showing the principal characteristics of alpha and gamma motor neurons. Within the ventral spinal cord (SC light gray), motor neuron pools (dashed lines) are composed of gamma motor neurons (blue) as well as three type of alpha motor neurons: αFF (light brown), αFFR (dark brown), αSFR (green). Alpha motor neurons have a larger diameter than gamma motor neurons. Beta motor neurons are not represented for simplicity. The proportion of alpha motor neuron subtypes varies between motor neuron pools. In the periphery, a muscle is composed of three types of extrafusal fibers: fast-twitch fatigable muscle fibers (light brown, IIb) are innervated by αFF motor neurons, fast-twitch fatigue-resistant muscle fibers (dark brown, IIa) are innervated by alpha αFFR motor neurons and slow-twitch fatigue-resistant muscle fibers (green, I) are innervated by αSFR motor neurons. Intrafusal muscle fibers (blue) reside within a muscle spindle (gray) and are exclusively innervated by gamma motor neurons. A single motor neuron innervate multiple fibers all of the same type; however, for the schematic simplicity only one fiber is represented.
[Source 3 ]Beta motor neurons
Beta motor neurons are smaller and less abundant than other somatic motor neuron subtypes. As a result beta motor neurons are poorly characterized. They innervate both intrafusal and extrafusal muscle fibers (Figure 12) 11. Therefore, beta motor neurons constitute an exception to the homogeneity observed in motor-units and control both muscle contraction and responsiveness of the sensory feedback from muscle spindles. They are further subdivided into two subtypes depending on the type of intrafusal fibers they innervate: (i) static, innervating nuclear chain fibers and (ii) dynamic, innervating the nuclear bag fibers of muscle spindles. Static beta motor neurons increase the firing rate of type Ia and type II sensory fibers at a given muscle length whereas dynamic beta motor neurons increase the stretch-sensitivity of the type Ia sensory fibers by stiffening the nuclear bag fibers. Beta motor neurons are mainly characterized anatomically and functionally, further molecular and electrical properties remain to be identified.
Figure 12. Innervation of a muscle spindle
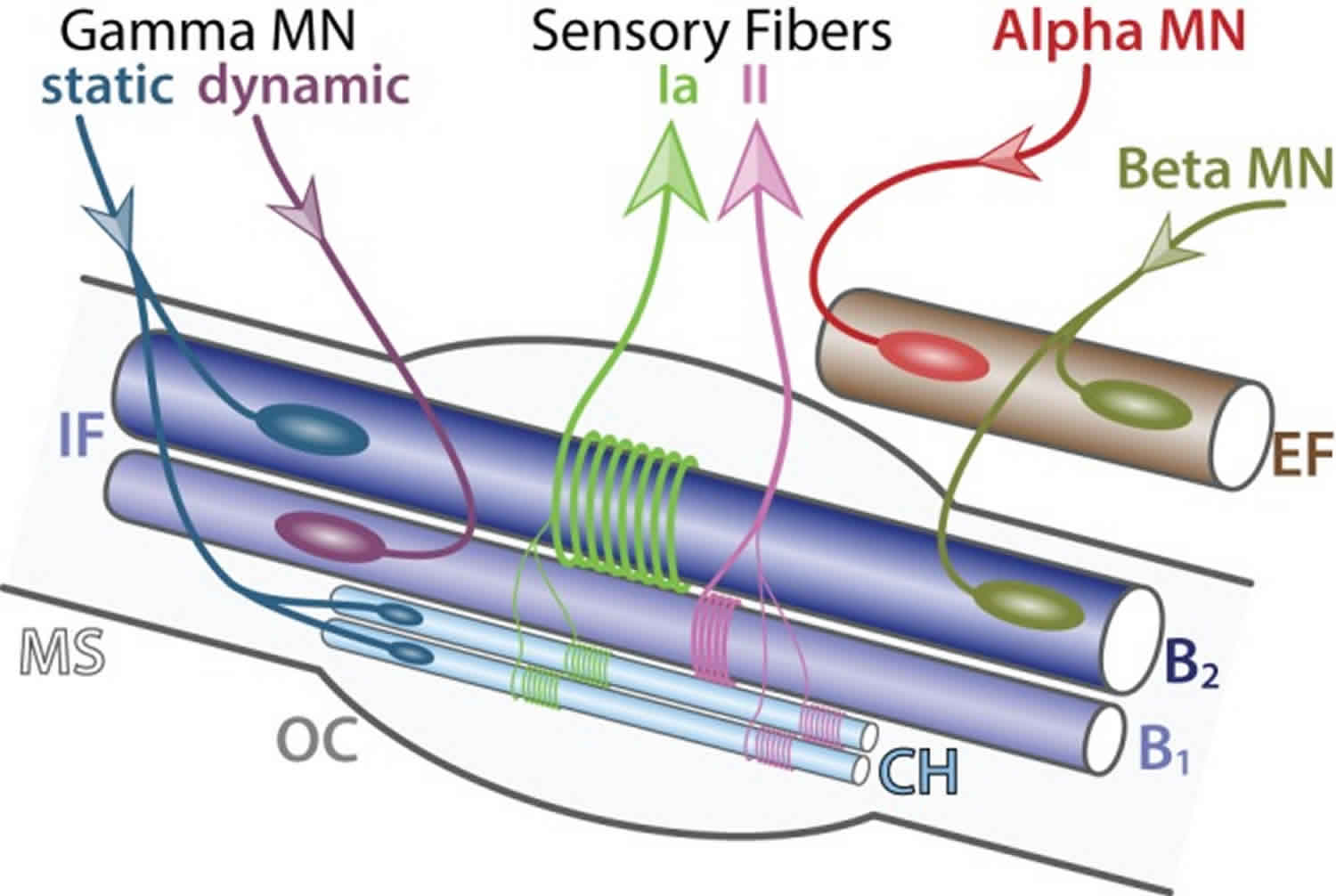
Footnote: Detailed innervation of a muscle spindle. Schematic of an adult muscle spindle (MS, light gray) on the longitudinal section. Alpha motor neuron (red) exclusively innervates (incoming arrow) extrafusal fibers (EF, brown). Beta motor neurons (green-brown) innervate both EF and intrafusal fibers (IF, blue). Gamma motor neurons are divided into two subtypes: static (blue) connecting to nuclear chain (CH, light blue) and nuclear bag 2 (B2, dark blue) fibers and dynamic (purple) connecting to nuclear bag 1 fibers (B1, intermediate blue). Sensory afferent axons Ia (light green) and II (pink) convey information (outgoing arrows) to sensory neurons located in the dorsal root ganglia. The outer capsule (OC) is a dedicated membrane isolating the muscle spindle from the extrafusal fibers. A single motor neuron innervate multiple fibers all of the same type; however, for the schematic simplicity only one fiber is represented.
[Source 3 ]Gamma motor neurons
Gamma motor neurons control exclusively the sensitivity of muscle spindles. Their firing increases the tension of intrafusal muscle fibers and therefore mimics the stretch of the muscle. Like beta motor neurons, gamma motor neurons are functionally divided into two subtypes: (i) static, innervating nuclear chain fibers and static nuclear bag fibers and (ii) dynamic, innervating the dynamic nuclear bag fibers (Figure 4). Gamma motor neurons receive only indirect sensory inputs and do not possess any motor function. Therefore, gamma motor neurons do not directly participate to spinal reflexes 8 but instead contribute to the modulation of muscle contraction.
Causes of lower motor neuron lesion
Lower motor neuron syndromes can be broadly classified as hereditary, sporadic or immune-mediated. Immune-mediated neuropathies, such as multifocal motor neuropathy and chronic inflammatory demyelinating polyneuropathy are important to distinguish from sporadic and hereditary forms, as treatments are available. Lower motor neuron presentations of motor neuron disease are most often sporadic, but several genetic mutations have been described which can be associated with lower motor neuron preponderance. Other hereditary forms of lower motor neuron syndromes include the spinal muscular atrophies (SMAs) and distal hereditary motor neuropathies. The increasing availability of next-generation sequencing, including the ability for multiple genes to be sequenced in parallel, has resulted in an increase in the discovery of novel genetic mutations.
The most common causes of lower motor neuron lesions are trauma to peripheral nerves that serve the axons, and viruses that selectively attack ventral horn cells. Disuse atrophy of the muscle occurs i.e., shrinkage of muscle fiber finally replaced by fibrous tissue (fibrous muscle). Other causes include Guillain–Barré syndrome, Bell’s palsy, poliomyelitis, spinal muscular atrophy, Clostridium botulinum, and cauda equina syndrome; another common cause of lower motor neuron degeneration is amyotrophic lateral sclerosis.
Poliomyelitis
A classic example of solely lower motor neuron paralysis, poliomyelitis has a fecal-oral transmission and is caused by a type of picornavirus: poliovirus. Once infected, the virus replicated in the oropharynx and small intestine before spreading via the bloodstream to the CNS. While replicating in the Peyer’s patches of the small intestine, 95% of patients are asymptomatic and it can only be found in the stool or via an oral swab 12. In the CNS, the virus destroys the anterior (ventral) horn of the spinal cord, resulting in lower motor neuron paralysis. Because lower motor neuron’s originate in the anterior horn of the spinal cord, this results in lower motor neuron signs such as asymmetric weakness, flaccid paralysis, fasciculations, hyporeflexia, and muscle atrophy. Infection could also result in respiratory involvement leading to respiratory paralysis. Other systemic signs of infection include fever, headache, nausea, and malaise. The cerebrospinal fluid would demonstrate increased white blood cell’s and a slight elevation of protein which is consistent with viral infection. Once a prominent cause of paralysis, poliomyelitis has almost been eradicated due to widespread vaccination.
Postpolio syndrome
Postpolio syndrome develops after a period of stability in a proportion of patients who have recovered from acute poliomyelitis. Symptoms may include the development of new weakness and muscle atrophy, fatigue and/or pain 13. The cause of postpolio syndrome remains unclear and may be due to the degeneration of enlarged reinnervated motor units.
Guillain-Barre syndrome
Guillain-Barre syndrome may present as a pure motor disorder. Both the classic acute inflammatory demyelinating polyneuropathy form and the recently described acute motor axonal neuropathy can be pure motor, although the latter is defined by its pure motor phenotype and axonal characteristics. Clinically, acute motor axonal neuropathy is part of the Guillain-Barré syndrome spectrum but distinguished by normal sensation and nerve conduction studies characterised by low distal motor evoked amplitudes, normal sensory conductions and no features of demyelination. Prognosis is similar to that seen in the acute inflammatory demyelinating polyneuropathy form of Guillain-Barré syndrome unless there is secondary axonal degeneration in which case, like in the acute inflammatory demyelinating polyneuropathy form, recovery is delayed. There is a strong association between acute motor axonal neuropathy and IgG antibodies against GM1 or GD1a which may be a result of molecular mimicry resulting from antecedent Campylobacter jejuni infection 14.
Spinal Muscular Atrophy
Spinal Muscular Atrophy represents a group of genetic disorders resulting in progressive degeneration and irreversible loss of the anterior horn cells in the spinal cord (i.e., lower motor neurons) and the brain stem nuclei, predominantly characterized proximal muscle weakness with reduced or absent reflexes. Spinal Muscular Atrophy are classified into four types on the basis of age of onset and clinical course (Spinal Muscular Atrophy I–IV) 15. Spinal Muscular Atrophy 1 and 2 are defined by onset in infancy. Spinal Muscular Atrophy 3 is a milder phenotype with signs of weakness presenting at or after 1 year of age with patients attaining the ability to walk unaided 15. It is associated with significant variability in the age of onset, disease progression and ambulatory period with some patients only developing walking difficulties in adulthood 16. Adult-onset Spinal Muscular Atrophy (Spinal Muscular Atrophy 4) typically presents in the third or fourth decade of life with a slowly progressive and relatively benign course 17. Respiratory insufficiency may occur in Spinal Muscular Atrophy IV, but is usually mild and life expectancy is normal 15.
The vast majority of spinal muscular atrophy is autosomal recessivein inheritance and related to mutations in the SMN1 gene located on chromosome 5q13. Most cases are homozygous for a deletion of exon 7 (94%), but a small percentage are compound heterozygous for a deletion in SMN1 and an intragenic mutation of SMN1 18. Targeted molecular genetic testing is the first-line investigation for spinal muscular atrophy to detect homozygous deletions of SMN1 exon 7 gene. However, if only a single deletion is detected, sequencing the SMN1 gene should be performed to assess for a point mutation. Overall, 4–5% of patients with clinically typical spinal muscular atrophy have no identifiable mutation in SMN1 19. Non-5q spinal muscular atrophy can be inherited in an autosomal dominant, autosomal recessive or X-linked pattern with marked clinical and genetic heterogeneity. Next-generation sequencing technology has facilitated the discovery of a number of non-5q causative genes associated with spinal muscular atrophy 20.
This disease is due to congenital degeneration of the anterior horn of the spinal cord. Unlike polio, this results in symmetric weakness, flaccid paralysis, fasciculations, hyporeflexia, and muscle atrophy. Because it is congenital, it has classically had associations with a “floppy baby” with marked hypotonia and tongue fasciculations. This disease carries an autosomal recessive inheritance and is due to a mutation in the SMN1 gene. Spinal muscular atrophy type I is also known as Werdnig-Hoffmann disease, which is the most severe form of the disease and usually results in childhood death due to respiratory failure 21. Spinal muscular atrophy type II and III are less severe and often result in a lifelong inability to ambulate.
Spinobulbar muscular atrophy (Kennedy’s disease)
Spinobulbar muscular atrophy or Kennedy’s disease is the most common adult-onset spinal muscular atrophy. It is a polyglutamine genetic disorder caused by a CAG trinucleotide repeat expansion in the androgen receptor gene on the X-chromosome 22. Degeneration of motor neurons in the spinal cord and brainstem results in a slowly progressive disorder characterised by weakness and atrophy of facial, bulbar and limb muscles without upper motor neuron signs. Cramps, leg weakness, tremor and orolingual fasciculations (see online supplementary video S1) with bulbar symptoms are the most common presenting symptoms. The syndrome affects only men, although female carriers may experience mild symptoms such as cramps 23. Symptom onset is typically between 30 and 50 years of age, but there is marked variability in age of presentation.9 Weakness is typically noted first in the lower limbs and may be symmetrical or asymmetrical, affecting proximal and/or distal muscles 24. A sensory neuropathy is commonly associated with the syndrome and is usually subclinical. Associated androgen resistance may result in gynaecomastia, testicular atrophy and oligospermia. The diagnosis is confirmed through molecular genetic testing with affected men having >39 CAG repeats 25. Life expectancy may be reduced in selected patients, most commonly due to pneumonia resulting from bulbar dysfunction 24.
Bell’s Palsy
Bell’s palsy is the most common etiology of peripheral facial nerve palsy. Although it is not always a lower motor neuron deficit, it is a perfect example to demonstrate lower motor neuron signs. It usually develops after herpes virus reactivation, but it can also be the result of Lyme disease, herpes zoster (Ramsey-Hunt syndrome), sarcoidosis, tumors of the parotid gland, and diabetes mellitus.
If any part of the corticobulbar tract from the motor cortex to the facial nerve nucleus is damaged, it will result in upper motor neuron deficits; this will result in contralateral facial paralysis involving the lower muscles of facial expression. Because there is bilateral upper motor neuron innervation to the muscles of the forehead, there is sparing of the forehead.
However, if the lesion occurs anywhere from the facial nucleus along CN VII, it will result in lower motor neuron deficits, affecting the ipsilateral side of the face and involve both the upper and lower muscles of facial expressions. This condition presents as incomplete eye closure (orbicularis oculi), dry eyes, corneal ulceration, hyperacusis, and taste sensation loss to the anterior tongue. Because the forehead is involved, the affected individual will be unable to wrinkle their forehead (lift their eyebrows).
Chronic immune-mediated neuropathies
Multifocal motor neuropathy typically presents with asymmetrical distal weakness and wasting, without sensory impairment which is slowly progressive and has an upper limb predilection 26. Weakness may be out of proportion to muscle wasting and involvement of wrist and/or finger extension at onset should prompt consideration of multifocal motor neuropathy as a potential diagnosis. Positive features such as twitching, cramping and spasm are relatively common in multifocal motor neuropathy and may be the presenting symptom 27. Bulbar and respiratory involvement are not typical, although respiratory symptoms may occur due to phrenic nerve involvement.
A definitive diagnosis of multifocal motor neuropathy requires demonstration of focal motor conduction block on neurophysiological studies with normal sensory nerve conduction across the region of block 28. As conduction block may be difficult to demonstrate and may occur in proximal segments, comprehensive neurophysiology should be performed and proximal stimulation may be required. A normal compound muscle action potential amplitude in a weak muscle with neurogenic recruitment on EMG suggests the presence of conduction block. Anti-GM1 IgM is present in ∼50% of cases with a high titre supporting a diagnosis of multifocal motor neuropathy 26. MRI may reveal asymmetrical nerve enlargement and increased signal intensity on T2-weighted images of the brachial plexus.20 Ultrasound imaging may show multiple sites of peripheral nerve enlargement in the arms, including segments without conduction abnormalities 29.
Intravenous immunoglobulin (IVIg) is the accepted treatment for multifocal motor neuropathy 26. Dosing must be individualised and no optimal dosing strategy has been established, although high doses of IVIg are often required 30. Furthermore, despite treatment, multifocal motor neuropathy is often associated with progressive axonal loss and functional decline 26.
A purely ‘axonal’ form of multifocal motor neuropathy has been described which lacks demonstrable partial motor conduction block, demyelinating features and anti-GM1 antibodies, but may respond to IVIg 31. It is important to recognize, however, that at least some of the ‘apparent’ cases of ‘axonal multifocal motor neuropathy’ may represent multifocal motor neuropathy with very proximal conduction blocks which are not detectable with standard neurophysiological techniques 32. A trial of IVIg may be warranted in select cases of asymmetrical adult-onset lower motor neuron syndromes without demonstrable conduction block, particularly those with distal upper limb-onset weakness 33. A pure motor variant of chronic inflammatory demyelinating polyneuropathy with sparing of sensory fibres clinically and neurophysiologically has also been reported. As in multifocal motor neuropathy, the neuropathy appears to be responsive to IVIg, but not corticosteroids, which may cause deterioration 34.
Distal hereditary motor neuropathies
The distal hereditary motor neuropathies share the characteristics of a slowly progressive, length-dependent (ie, distal predominant) pattern of lower motor neuron weakness 35. Distal hereditary motor neuropathies represent a genetically heterogeneous group with significant variability and overlap in clinical phenotypes for many of the known implicated genes. Most are inherited in an autosomal dominant pattern, but autosomal recessive and X-linked inheritance patterns have also been described 36. Onset is often in childhood or teens, but adult onset is not uncommon. Upper limb predominance (distal hereditary motor neuropathy type V), vocal cord paralysis (distal hereditary motor neuropathy type VII), respiratory distress (distal hereditary motor neuropathy type VI) and pyramidal signs may be associated features in some patients. Significant sensory involvement is absent, allowing differentiation from axonal forms of Charcot-Marie-Tooth disease, although some mutations may cause both phenotypes 35.
Despite significant advances in molecular genetics, a disease-causing mutation is only identified in ∼15% of patients with a typical presentation of distal hereditary motor neuropathy 37. Mutations in the HSPB1, HSPB8 and BSCL2 genes are the most frequent causes of autosomal dominant distal hereditary motor neuropathy. Mutations in HSPB1 and HSPB8 are associated with a classical length-dependent motor neuropathy beginning in the lower limbs which may present in childhood (distal hereditary motor neuropathy type I) or adulthood (distal hereditary motor neuropathy type II) 36. Several phenotypes associated with mutations in BSCL2 have been described and include (1) distal hereditary motor neuropathy type II with a length-dependent motor neuropathy, (2) distal hereditary motor neuropathy type V presenting with a predominantly upper limb distal phenotype, (3) distal hereditary motor neuropathy with pyramidal signs and (4) Silver syndrome with atrophy of the intrinsic hand muscles, pyramidal signs and lower limb spasticity 36. The upper limb-onset phenotype ( distal hereditary motor neuropathy type V) may also result from mutations in GARS with most cases presenting in their second decade with progressive weakness and wasting of the thenar eminence and first dorsal interossei muscles.15 Cramping and pain in the hands on exposure to cold may be an early manifestation 38. The GARS mutation may also present with a classical length-dependent neuropathy beginning in the lower limbs, further highlighting the variability in genotype–phenotype correlations. It remains unclear why mutations in ubiquitously expressed proteins may result in such variable and ‘focal’ phenotypes 35. Bulbar involvement is rare in distal hereditary motor neuropathy, but vocal paralysis secondary to recurrent laryngeal nerve involvement is a feature of distal hereditary motor neuropathy type VII which may result from mutations in dynactin (DCTN1) SLC5A7 or TRPV4.11 autosomal recessive forms of distal hereditary motor neuropathy are less common but there are increasing numbers of genes being described for this group 35.
Although targeted molecular genetic testing was the standard approach to genetic testing in the past, next-generation sequencing has become a more efficient and cost-effective means of establishing a diagnosis in many settings. This is particularly true with many lower motor neuron syndromes as there is a large overlap between genotypes and phenotypes. For example, individual genes can cause phenotypes that have been labelled distal hereditary motor neuropathy, amyotrophic lateral sclerosis (ALS) and hereditary spastic paraplegia. Targeted gene panels covering a large number of genes causing these phenotypes are the current preferred next-generation sequencing diagnostic test. Whole exome and whole genome sequencing which sequence genes encoding proteins, or an individual’s entire DNA, respectively, are commonly used in research and increasingly in diagnostic testing and may have particular clinical utility when known suspected genes have been tested by other methods and found to be normal. Such advances in neurogenetics may allow a genetic diagnosis to be established in a greater proportion of patients, but challenges remain including difficulties managing large volumes of data, in the interpretation of sequence variants and determining the pathogenicity of detected mutations. Furthermore, next-generation sequencing may result in failure to detect certain mutations such as chromosomal deletions or insertions and repeat expansions.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that affects an estimated 0.05% of the population. It is also called Lou Gehrig’s disease, after the New York Yankees baseball player who died of the disorder in 1936. ALS is characterized by the slow but inexorable degeneration of α motor neurons in the ventral horn of the spinal cord and brainstem, and eventually of neurons in the motor cortex. Affected individuals show progressive weakness and wasting of skeletal muscles and usually die within 5 years of onset. Sadly, these patients are condemned to watch their own demise, since the intellect remains intact. There is no effective therapy.
Approximately 10% of ALS cases are familial. Familial ALS (FALS) is usually inherited as an autosomal dominant trait, and is largely indistinguishable from other forms of the disease. The familial form, however, has provided an opportunity for geneticists and neurologists to decipher the genetic basis of the disease in this subset of patients: The defect is a mutation in the long arm of chromosome 21. Interestingly, this same chromosomal region contains the gene that encodes the cytosolic antioxidant enzyme copper/zinc superoxide dismutase (SOD). Evidence that superoxide radicals can destroy nerve cells suggested that a mutation of the SOD1 gene may cause FALS. This finding led in turn to the identification of mutations of SOD1 in roughly 20% of families with FALS. Further confirmation of this causal sequence in the familial form of ALS is that transgenic mice that overexpress mutant SOD1 protein develop a neurological disease that mimics ALS both behaviorally and pathologically. Overexpression of normal SOD, however, is not associated with motor neuron disease in mice. Together, these data suggest that the mutant SOD molecule is in some way cytotoxic.
How the mutant SOD1 damages motor neurons is not known. Possibilities include generation of damaging free radicals and death by oxidative stress, enhanced release of copper from the mutant enzyme and neuronal death by copper toxicity, and abnormal folding of mutant SOD1 and neuronal death as a consequence of aberrant protein-protein interactions. It is also unclear what accounts for the curious predilection of the disease for motor neurons. Despite these uncertainties, identification of mutant SOD1 as the cause of familial ALS has given scientists a valuable clue to the molecular pathogenesis of at least some forms of this tragic disorder.
Progressive muscular atrophy
The lower motor neuron phenotype of motor neuron disease (progressive muscular atrophy, PMA) is characterised by progressive lower motor neuropathy signs without clinical evidence of upper motor neuron dysfunction, although a significant proportion develop upper motor neuron signs during the disease course 39. It is estimated that the syndrome represents ∼5% of motor neuron disease cases, and may be characterised by slower progression than other forms of motor neuron disease 39. In the absence of upper motor neuron signs, confident differentiation from other lower motor neuropathy syndromes may be difficult, often requiring a period of observation to assess progression. The novel neurophysiological technique of threshold tracking transcranial magnetic stimulation (TMS) has been a major advance allowing for objective assessment of the functional integrity of the upper motor neuron system 40. Threshold tracking transcranial magnetic stimulation has demonstrated evidence of cortical hyperexcitability in motor neuron disease and may play a role in the differentiation from mimic disorders by providing objective evidence of upper motor neuron dysfunction when it is not evident clinically 41.
Flail arm syndrome
The flail arm syndrome (brachial amyotrophic diplegia or ‘man-in-the-barrel’ syndrome) is a distinct variant of motor neuron disease characterised by a progressive, predominantly lower motor neuropathy pattern of weakness in the upper limbs, typically beginning in proximal muscle groups with progression to distal involvement. Original descriptions were of a symmetrical pattern of weakness, but there may be some asymmetry, particularly early in the disease course. Mild upper motor neuron signs are often present in the lower limbs. There is a striking male predominance with a male-to-female ratio of 4:1 42 and up to 10:1 in some series 43. Prognosis is better than that of classical ALS with a median survival of ∼5 years 42. The flail arm phenotype is associated with cortical hyperexcitability with a similar pattern to that seen in ALS 43. Hence, transcranial magnetic stimulation may be a useful adjunct in differentiating this motor neuron disease variant from other more benign lower motor neuropathy syndromes with upper limb predominance.
Flail leg syndrome
The flail leg variant of motor neuron disease (also known as the pseudopolyneuritic variant) is characterised by a progressive, asymmetrical predominantly lower motor neuropathy pattern of weakness with distal-onset weakness and wasting of the lower limbs. Upper motor neuron signs often emerge over time 42. Progression is slower than classical ALS with a median time of 33 months to involvement of a second region and median survival of almost 6 years. In contrast to the flail arm variant, the flail leg group show an equal male-to-female ratio 42. As with other forms of motor neuron disease, cortical hyperexcitability is a feature of the flail leg syndrome, but only when upper motor neuron signs are present. In contrast, features of cortical hyperexcitability were not demonstrated in patients who lacked upper motor neuron signs on clinical examination 44.
Familial lower motor neuron variants of motor neuron disease
A variety of genetic mutations may be associated with significant lower motor neuron involvement with or without upper motor neuron signs. They include mutations in copper–zinc superoxide dismutase type 1 (SOD1), fused in sarcoma, vesicle-associated membrane protein/synaptobrevin-associated membrane protein B (VAPB) and chromatin-modifying protein 2b (CHMP2B) genes 45. Most of the known forms are inherited in an autosomal dominant pattern.
SOD1 gene mutations account for 20% of autosomal dominant familial motor neuron disease and are the second most common cause of familial motor neuron disease (following the expanded hexanucleotide repeat in the C9ORF72 gene associated with the ALS-frontotemporal dementia spectrum) 46. The A4V missense mutation has been demonstrated to occur in around 40% of patients with SOD1 mutations in North American series and is rare in the European population 47. Lower motor neuron signs predominate with absent or mild upper motor neuron features. Disease progression is particularly rapid with a median survival of 1.2 years from disease onset 47. The A4T mutation is also associated with a similarly rapid disease course and lower motor neuron predominant syndrome 48. In contrast, the G93C mutation has been associated with a pure lower motor neuron clinical phenotype without bulbar involvement and more favourable prognosis with a median survival of 153 months 49. The D101N mutation in exon 4 of the SOD1 gene has been associated with progressive muscular atrophy with limited bulbar involvement and rapid disease course with mean time to death from respiratory failure of 28 months 50.
Mutations in the FUS gene account for ∼5% of familial motor neuron disease and may present with progressive muscular atrophy or lower motor neuron predominant motor neuron disease 51. Mutations in the VAPB gene have been associated with a range of phenotypes including progressive muscular atrophy and late-onset spinal muscular atrophy 52. Mutations in the CHMP2B gene were first linked to frontotemporal dementia, but may be associated with progressive muscular atrophy or amyotrophic lateral sclerosis (ALS). In one series, CHMP2B mutations were found in 10% of patients with lower motor neuron predominant amyotrophic lateral sclerosis (ALS), although most cases exhibited a sporadic phenotype 53.
Monomelic amyotrophy
Monomelic amyotrophy is a lower motor neuron disorder that presents with insidious onset of focal wasting and weakness, most commonly affecting the upper limb unilaterally, although it can rarely affect a lower extremity. Symptoms typically progress over a period of 1–5 years and then plateau.42 Bulbar, sensory and pyramidal signs are absent. The condition has a striking male predominance with a male-to-female ratio of 10:1 54. It is seen more commonly in Asian countries with a median age of onset in the late teens or early 20s 54.
The typical pattern of weakness and wasting in upper extremity monomelic amyotrophy also known as Hirayama disease, is distal predominant affecting the hand and forearm muscles, with C7-T1 innervated muscles classically affected. Preservation of brachioradialis muscle bulk (a C6 innervated muscle) with wasting of C7 innervated forearm muscles may result in the clinical sign described by Hirayama et al 55 as ‘oblique amyotrophy’. Symptoms may be aggravated by cold weather and there may be an associated mild tremor on finger extension. Less severe involvement of the contralateral upper limb may occur in a significant proportion of patients 54. MRI findings in Hirayama disease may reveal lower cervical cord atrophy (C5–C7), asymmetric cord flattening and/or intramedullary hyperintensity. Anterior displacement of the dorsal dura on neck flexion may be seen and venous plexus engorgement may give the appearance of an enhancing epidural crescent along the posterior aspect of the cord on neck flexion views 56.
Lower extremity monomelic amyotrophy presents with weakness and wasting of a unilateral lower limb, although less severe or subclinical involvement of the contralateral limb may occur. It is less common than Hirayama disease but is also characterised by male predominance and benign course. Posterior leg muscles are disproportionately affected with imaging studies demonstrating most severe involvement of gastrocnemius and soleus muscles with marked asymmetry 57. The degree of wasting may be out of proportion to weakness and disability 58.
Segmental lower motor neuron disease
While progression typically arrests within a few years in Hirayama disease, segmental lower motor neuron disease is a localised form of sporadic adult-onset lower motor neuron disease affecting the upper limbs characterised by progression over a longer period extending up to 20 years 59. The clinical presentation is with asymmetrical lower motor neuron signs localised to the upper extremities with unilateral dominance. Both proximal and distal forms are recognised. The clinical course is favourable with progression to generalised motor neuron disease/ALS rare. MRI may reveal a ‘snake eyes’ appearance with T2-signal hyperintensity in the anterior horns of multiple segments of the cervical cord, although this is a non-specific finding and has been associated with a number of other lower motor neuron syndromes including cervical spondylosis and infection 60.
Other lower motor neuron syndromes
Although rare, lead and porphyric neuropathies are briefly discussed here, as they are treatable causes of motor neuropathies. Lead toxicity can lead to a subacute motor neuropathy which classically affects the wrist and finger extensors before spreading to other muscles and hence may be confused with multifocal motor neuropathy 61. Porphyria, an inherited metabolic disorder of heme biosynthesis, may present with an acute or subacute predominantly motor neuropathy also with focal weakness at onset, such as wristdrop or footdrop. The acute onset may lead to confusion with acute motor axonal neuropathy. Both lead and porphyric neuropathies are typically associated with involvement of other organ systems and additional features may include gastrointestinal symptoms, cognitive disturbance and haematological changes 62. A family history of symptoms of porphyria or history of occupational exposure to lead may provide clues to the diagnosis. Lead toxicity is treatable with chelation and porphyria with haematin.
Lower motor neuron lesion signs and symptoms
One major characteristic used to identify a lower motor neuron lesion is flaccid paralysis. This is in contrast to a upper motor neuron lesion, which often presents with spastic paralysis.
Other signs and symptoms of lower motor neuron lesion include:
- Muscle paresis or paralysis
- Fibrillations
- Fasciculations – caused by increased receptor concentration on muscles to compensate for lack of innervation.
- Hypotonia or atonia – Tone is not velocity dependent.
- Hyporeflexia – Along with deep reflexes even cutaneous reflexes are also decreased or absent.
- Strength – weakness is limited to segmental or focal pattern
The extensor plantar reflex is usually absent. Muscle paresis/paralysis, hypotonia/atonia, and hyporeflexia/areflexia are usually seen immediately following an insult. Muscle wasting, fasciculations and fibrillations are typically signs of end-stage muscle denervation and are seen over a longer time period. Another feature is the segmentation of symptoms – only muscles innervated by the damaged nerves will be symptomatic.
Lower motor neuron lesion diagnosis
The clinical evaluation of a patient presenting with a lower motor neuron syndrome includes a thorough assessment of disease onset and progression. This is particularly important to ascertain as a rapid rate of decline may support a diagnosis of MND and remains an important factor in distinguishing motor neuron disease from other relatively indolent conditions, such as spinal muscular atrophy and immune neuropathies. The pattern of weakness should be documented, including (1) symmetry versus asymmetry, (2) proximal versus distal involvement, (3) upper versus lower limb predominance and (4) presence versus absence of bulbar involvement. Nerve conduction studies and electromyography (EMG) are essential to confirm that the disorder is neurogenic and should focus on assessing (1) the pattern of involvement, including symmetry and length dependence, (2) presence of focal motor conduction block or demyelinating features and (3) the presence or absence of subclinical sensory abnormalities. Imaging, genetic testing, antibody markers and advanced neurophysiological techniques are useful adjuncts and form an extension of the clinical assessment.
References- Garg N, Park SB, Vucic S, et al. Differentiating lower motor neuron syndromes. Journal of Neurology, Neurosurgery & Psychiatry 2017;88:474-483. https://jnnp.bmj.com/content/88/6/474.full
- Javed K, Daly DT. Neuroanatomy, Lower Motor Neuron Lesion. [Updated 2019 Mar 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539814
- Stifani N. Motor neurons and the generation of spinal motor neuron diversity. Front Cell Neurosci. 2014;8:293. Published 2014 Oct 9. doi:10.3389/fncel.2014.00293 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4191298
- Stifani N. Motor neurons and the generation of spinal motor neuron diversity. Front Cell Neurosci. 2014;8:293.
- Jan, Mohammed & Al-Buhairi, A & Baeesa, Saleh. (2001). Concise outline of the nervous system examination for the generalist. Neurosciences (Riyadh, Saudi Arabia). 6. 16-22.
- A cytoarchitectonic atlas of the spinal cord in the cat. REXED B. J Comp Neurol. 1954 Apr; 100(2):297-379.
- Stone R. J., Stone J. A. (2009). Atlas of Skeletal Muscles. Boston, MA: McGraw-Hill Higher Education
- Electrophysiological studies on gamma motoneurones. ECCLES JC, ECCLES RM, IGGO A, LUNDBERG A. Acta Physiol Scand. 1960 Sep 30; 50():32-40.
- Physiological types and histochemical profiles in motor units of the cat gastrocnemius. Burke RE, Levine DN, Tsairis P, Zajac FE 3rd. J Physiol. 1973 Nov; 234(3):723-48.
- Bistability in spinal motoneurons in vivo: systematic variations in persistent inward currents. Lee RH, Heckman CJ. J Neurophysiol. 1998 Aug; 80(2):583-93.
- Motor fibres innervating extrafusal and intrafusal muscle fibres in the cat. Bessou P, Emonet-Dénand F, Laporte Y. J Physiol. 1965 Oct; 180(3):649-72.
- Mehndiratta MM, Mehndiratta P, Pande R. Poliomyelitis: historical facts, epidemiology, and current challenges in eradication. Neurohospitalist. 2014 Oct;4(4):223-9.
- Howard RS. Poliomyelitis and the postpolio syndrome. BMJ 2005;330:1314–18. doi:10.1136/bmj.330.7503.1314
- Yuki N, Hartung HP. Guillain-Barre syndrome. N Engl J Med 2012;366:2294–304. doi:10.1056/NEJMra1114525
- Prior TW, Leach ME, Finanger E. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2019 Nov 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352
- Rudnik-Schoneborn S, Hausmanowa-Petrusewicz I, Borkowska J, et al. The predictive value of achieved motor milestones assessed in 441 patients with infantile spinal muscular atrophy types II and III. Eur Neurol 2001;45:174–81. doi:521
- Brahe C, Servidei S, Zappata S, et al. Genetic homogeneity between childhood-onset and adult-onset autosomal recessive spinal muscular atrophy. Lancet 1995;346:741–2. doi:10.1016/S0140-6736(95)91507-9
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 2000;15:228–37. doi:10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2
- Wirth B, Herz M, Wetter A, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet 1999;64:1340–56. doi:10.1086/302369
- Peeters K, Chamova T, Jordanova A. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain 2014;137(Pt 11):2879–96. doi:10.1093/brain/awu169
- Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015 Feb;51(2):157-67.
- Querin G, Bertolin C, Da Re E, et al. Non-neural phenotype of spinal and bulbar muscular atrophy: results from a large cohort of Italian patients. J Neurol Neurosurg Psychiatr 2016;87:810–16. doi:10.1136/jnnp-2015-311305
- Mariotti C, Castellotti B, Pareyson D, et al. Phenotypic manifestations associated with CAG-repeat expansion in the androgen receptor gene in male patients and heterozygous females: a clinical and molecular study of 30 families. Neuromuscul Disord 2000;10:391–7. doi:10.1016/S0960-8966(99)00132-7
- Atsuta N, Watanabe H, Ito M, et al. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain 2006;129(Pt 6):1446–55. doi:10.1093/brain/awl096
- La Spada AR, Wilson EM, Lubahn DB, et al. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991;352:77–9. doi:10.1038/352077a0
- Vlam L, van der Pol WL, Cats EA, et al. Multifocal motor neuropathy: diagnosis, pathogenesis and treatment strategies. Nat Rev Neurol 2012;8:48–58. doi:10.1038/nrneurol.2011.175
- Garg N, Heard RNS, Kiers L, et al. Multifocal motor neuropathy presenting as pseudodystonia. Mov Disord Clin Pract 2016;doi:10.1002/mdc3.12336 doi:10.1002/mdc3.12336
- Joint Task Force of the E, the PNS. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst 2010;15:295–301.
- Gallardo E, Noto Y, Simon NG. Ultrasound in the diagnosis of peripheral neuropathy: structure meets function in the neuromuscular clinic. J Neurol Neurosurg Psychiatr 2015;86:1066–74. doi:10.1136/jnnp-2014-309599
- Vucic S, Black KR, Chong PS, et al. Multifocal motor neuropathy: decrease in conduction blocks and reinnervation with long-term IVIg. Neurology 2004;63:1264–9. doi:10.1212/01.WNL.0000140497.85952.FA
- Katz JS, Barohn RJ, Kojan S, et al. Axonal multifocal motor neuropathy without conduction block or other features of demyelination. Neurology 2002;58:615–20. doi:10.1212/WNL.58.4.615
- Vucic S, Black K, Chong PS, et al. Multifocal motor neuropathy with conduction block: distribution of demyelination and axonal degeneration. Clin Neurophysiol 2007;118:124–30. doi:10.1016/j.clinph.2006.09.020
- Simon NG, Ayer G, Lomen-Hoerth C. Is IVIg therapy warranted in progressive lower motor neuron syndromes without conduction block? Neurology 2013;81:2116–20. doi:10.1212/01.wnl.0000437301.28441.7e
- Donaghy M, Mills KR, Boniface SJ, et al. Pure motor demyelinating neuropathy: deterioration after steroid treatment and improvement with intravenous immunoglobulin. J Neurol Neurosurg Psychiatr 1994;57:778–83. doi:10.1136/jnnp.57.7.77
- Rossor AM, Kalmar B, Greensmith L, et al. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatr 2012;83:6–14. doi:10.1136/jnnp-2011-300952
- Irobi J, Dierick I, Jordanova A, et al. Unraveling the genetics of distal hereditary motor neuronopathies. Neuromolecular Med 2006;8:131–46. doi:10.1385/NMM:8:1-2:131
- Dierick I, Baets J, Irobi J, et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study. Brain 2008;131(Pt 5):1217–27. doi:10.1093/brain/awn029
- Sivakumar K, Kyriakides T, Puls I, et al. Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain 2005;128(Pt 10):2304–14. doi:10.1093/brain/awh590
- Kim WK, Liu X, Sandner J, et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology 2009;73:1686–92. doi:10.1212/WNL.0b013e3181c1dea3
- Vucic S, Howells J, Trevillion L, et al. Assessment of cortical excitability using threshold tracking techniques. Muscle Nerve 2006;33:477–86. doi:10.1002/mus.20481
- Menon P, Geevasinga N, Yiannikas C, et al. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet Neurol 2015;14:478–84. doi:10.1016/S1474-4422(15)00014-9
- Wijesekera LC, Mathers S, Talman P, et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology 2009;72:1087–94. doi:10.1212/01.wnl.0000345041.83406.a2
- Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatr 2007;78:849–52. doi:10.1136/jnnp.2006.105056
- Menon P, Geevasinga N, Yiannikas C, et al. Cortical contributions to the flail leg syndrome: pathophysiological insights. Amyotroph Lateral Scler Frontotemporal Degener 2016;17:389–96. doi:10.3109/21678421.2016.1145232
- Yamashita S, Ando Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl Neurodegener 2015;4:13. doi:10.1186/s40035-015-0036-y
- Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942–55. doi:10.1016/S0140-6736(10)61156-7
- Bali T, Self W, Liu J, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatr 2016 Published Online First: 3 Jun 2016. doi: 10.1136/jnnp-2016-313521 doi:10.1136/jnnp-2016-313521
- Aksoy H, Dean G, Elian M, et al. A4T mutation in the SOD1 gene causing familial amyotrophic lateral sclerosis. Neuroepidemiology 2003;22:235–8. doi:70564
- Regal L, Vanopdenbosch L, Tilkin P, et al. The G93C mutation in superoxide dismutase 1: clinicopathologic phenotype and prognosis. Arch Neurol 2006;63:262–7. doi:10.1001/archneur.63.2.262
- Cervenakova L, Protas II., Hirano A, et al. Progressive muscular atrophy variant of familial amyotrophic lateral sclerosis (PMA/ALS). J Neurol Sci 2000;177:124–30. doi:10.1016/S0022-510X(00)00350-6
- Hewitt C, Kirby J, Highley JR, et al. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2010;67:455–61. doi:10.1001/archneurol.2010.52
- Nishimura AL, Mitne-Neto M, Silva HC, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 2004;75:822–31. doi:10.1086/425287
- Cox LE, Ferraiuolo L, Goodall EF, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010;5:e9872. doi:10.1371/journal.pone.0009872
- Gourie-Devi M, Nalini A. Long-term follow-up of 44 patients with brachial monomelic amyotrophy. Acta Neurol Scand 2003;107:215–20. doi:10.1034/j.1600-0404.2003.02142.x
- Hirayama K, Tomonaga M, Kitano K, et al. Focal cervical poliopathy causing juvenile muscular atrophy of distal upper extremity: a pathological study. J Neurol Neurosurg Psychiatr 1987;50:285–90. doi:10.1136/jnnp.50.3.285
- Raval M, Kumari R, Dung AA, et al. MRI findings in Hirayama disease. Indian J Radiol Imaging 2010;20:245–9. doi:10.4103/0971-3026.73528
- Hamano T, Mutoh T, Hirayama M, et al. MRI findings of benign monomelic amyotrophy of lower limb. J Neurol Sci 1999;165:184–7. doi:10.1016/S0022-510X(99)00086-6
- Di Muzio A, Delli Pizzi C, Lugaresi A, et al. Benign monomelic amyotrophy of lower limb: a rare entity with a characteristic muscular CT. J Neurol Sci 1994;126:153–61. doi:10.1016/0022-510X(94)90266-6
- O’Sullivan DJ, McLeod JG. Distal chronic spinal muscular atrophy involving the hands. J Neurol Neurosurg Psychiatr 1978;41:653–8. doi:10.1136/jnnp.41.7.653
- Lebouteux MV, Franques J, Guillevin R, et al. Revisiting the spectrum of lower motor neuron diseases with Snake eyes appearance on magnetic resonance imaging. Eur J Neurol 2014;21:1233–41. doi:10.1111/ene.12465
- Thomson RM, Parry GJ. Neuropathies associated with excessive exposure to lead. Muscle Nerve 2006;33:732–41. doi:10.1002/mus.20510
- Lin C, Park SB, Krishnan AV. Porphyric neuropathy. Handbook of clinical neurology, Vol 115 (3rd series). Peripheral nerve disorders. 2013;613–627.





{kind=link}