Metabolic acidosis
Metabolic acidosis also known as non-respiratory acidosis, is a condition in which your blood pH falls below 7.35 due to some metabolic process 1. Human life requires a tightly controlled pH level in the serum of about 7.4 (a slightly alkaline range of 7.35 to 7.45) to survive 2. The ‘acidity’ of human blood is highly stable (pH = 7.35–7.45) in healthy individuals and cancer patients 3. The pH is a number that shows how acidic or alkaline a substance is. A pH of less than 7 is acidic, and greater than 7 is alkaline. The pH of blood is about 7.4. Your blood pH 7.4 is tightly regulated by your kidneys and respiratory system. The primary pH buffering system in the human body is the bicarbonate (HCO3–) and carbon dioxide (CO2). Bicarbonate (HCO3–) functions as an alkalotic substance. Carbon dioxide (CO2) functions as an acidic substance. Therefore, a decrease in serum bicarbonate (HCO3–) or an increase in CO2 (carbon dioxide) will make blood more acidic. The opposite is also true where an increase in bicarbonate (HCO3–) or a decrease in carbon dioxide (CO2) will make blood more alkaline. The carbon dioxide (CO2) levels are physiologically regulated by the pulmonary system through respiration, whereas the serum bicarbonate (HCO3–) levels are regulated through your kidneys by two mechanisms: bicarbonate [HCO3–] (a base) reclamation mainly in the proximal tubule and bicarbonate [HCO3–] (a base) generation predominantly in the distal nephron. Any excess acid is excreted in the urine. Your blood pH is not altered by your dietary intake. The only situation in which blood pH is altered is during metabolic acidosis, when an individual is critically ill. Clinically, metabolic acidosis can be defined in the first instance by a reduction in serum bicarbonate (HCO3–) concentration of less than 22 mmol/L and, in the second instance, by a decrease in arterial partial pressure of carbon dioxide (PaCO2) of ∼1 mmHg for every 1 mmol/L fall in serum bicarbonate (HCO3–) concentration, and a reduction in blood pH below 7.35 1. Causes of metabolic acidosis may be due to the loss of bicarbonate (HCO3–) in your body, kidney disease, electrolyte disturbances, severe vomiting or diarrhea (e.g., hyperchloremic acidosis), ingestion of certain drugs and toxins, and diseases that affect normal metabolism (e.g., diabetes ketoacidosis, lactic acidosis).
Anytime a metabolic acidosis is suspected, it is extremely useful to calculate the anion gap. The calculation of the serum anion gap:
- Serum anion gap = (Na+) – [(HCO3– + Cl–)]
Where Na+ is plasma sodium concentration, HCO3– is plasma bicarbonate concentration, and Cl– is plasma chloride concentration. The anions are negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]. The anion gap is the difference between measured cations (positively charged ions like sodium [Na+] and potassium [K+]) and measured anions (negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]) 4. The most common application of the anion gap is classifying cases of metabolic acidosis, states of lower than normal blood pH. Specifically, classifying into either those that do and those that do not have unmeasured anions in the plasma. The human body is electrically neutral; therefore, in reality, does not have a true anion gap 4. A normal serum anion gap is measured to be 5 to 16 mEq/L, with autoanalyzers using an ion-selective electrode. However, the anion gap value is dependent on the type of instrument used to measure its components 5. Therefore, you should know the reference range of the analyzer used and, if known, the patient’s baseline anion gap, too.
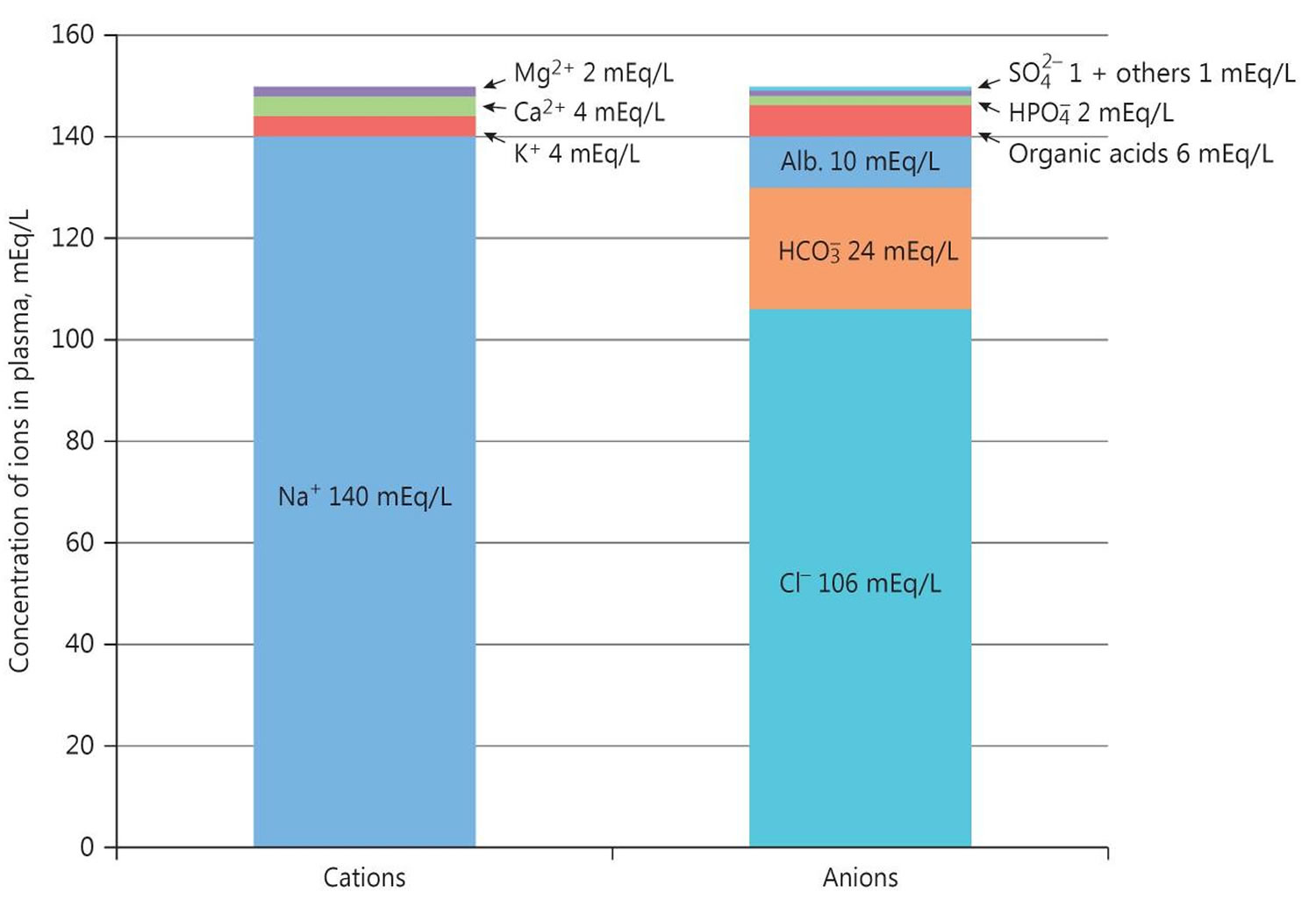
The anion gap is a calculation to determine the quantity of ionically active components within your blood that are not routinely measured. Serum anion gap is affected by the concentrations of all anions and cations which are not included in its calculations: i.e., albumin, globulin, potassium, calcium, magnesium, and organic and inorganic acids (see Figure 1). Because of the narrow extracellular concentration, most ions are omitted from the anion gap calculation. Since there are always components not directly measured, we expect this value to not equal 0. Most of this number is due to albumin (Alb); this anion is not accounted for in the anion gap formula, which is a large reason why the anion gap is not closer to zero. According to James Gamble 6, electrical neutrality in solution demands that the sum of the cations is equal to the sum of the anions (Figure 1). Sodium, chloride, bicarbonate, and albumin are quantitatively the major ions in the extracellular fluid compartment and are therefore used to calculate the anion gap 5. A true “ion gap,” however, does not exist in vivo which makes the anion gap a fundamental tool to evaluate acid-base disorders 7. Albumin is normally 4 mg/dL. Because of the large effect of albumin on anion gap, if a patient’s albumin level is abnormal, their expected anion gap will not be accurate 8. This can be corrected using simple math. The correction factor for albumin is 2.3–2.5 × [albumin], in g/dL 5. Therefore, each g/dL albumin decline will decrease the anion gap with about 2.5 mEq/L. To appreciate these facts, the anion gap formula should be: [Na+] − [Cl−] − [HCO3−] − 2.5 [albumin, in g/dL]. This equation is about zero in health, to stress the balance of ions, and also shows the relevance of albumin as a negative ion 5. As opposed to high anion gap acidosis which involves increased organic acid production, normal anion gap acidosis involves either increased production of chloride (hyperchloremic acidosis) or increased excretion of bicarbonate (HCO3–).
Figure 1. Normal anion gap levels
Metabolic acidosis causes can be divided into the acute forms (lasting minutes to several days) and chronic forms (lasting weeks to years), for which the underlying cause/s and resulting adverse effects may differ. Acute forms of metabolic acidosis most frequently result from the overproduction of organic acids such as ketoacids (e.g., diabetes ketoacidosis) or lactic acid (e.g., lactic acidosis); by contrast, chronic metabolic acidosis often reflects bicarbonate wasting and/or impaired renal acidification (e.g., chronic kidney disease or chronic renal failure). Unmanageable diarrhea and kidney failure are the most common causes of metabolic acidosis. Adverse effects of acute metabolic acidosis primarily include decreased cardiac output, arterial dilatation with hypotension, altered oxygen delivery, decreased ATP production, predisposition to arrhythmias, and impairment of the immune response. The main adverse effects of chronic metabolic acidosis are increased muscle degradation and abnormal bone metabolism. Using base to treat acute metabolic acidosis is controversial because of a lack of definitive benefit and because of potential complications. By contrast, the administration of base for the treatment of chronic metabolic acidosis is associated with improved cellular function and few complications.
There are several types of metabolic acidosis:
- Diabetic acidosis (also called diabetic ketoacidosis or DKA) develops when substances called ketone bodies (which are acidic) build up during uncontrolled diabetes.
- Hyperchloremic acidosis is caused by the loss of too much sodium bicarbonate from the body, which can happen with severe diarrhea.
- Renal tubular acidosis (distal renal tubular acidosis and proximal renal tubular acidosis). Renal tubular acidosis (RTA) occurs when the kidneys do not remove acids from the blood into the urine as they should. The acid level in the blood then becomes too high, a condition called acidosis. Some acid in the blood is normal, but too much acid can disturb many bodily functions. There are three main types of renal tubular acidosis that are characterized by: 1) a normal anion gap metabolic acidosis; 2) abnormalities in renal bicarbonate (HCO3-) absorption or new renal bicarbonate (HCO3-) generation; 3) changes in renal NH4+, Ca2+, K+ and H2O homeostasis; and 4) extrarenal manifestations that provide etiologic diagnostic clues 9, 10.
- Type 1 renal tubular acidosis or distal renal tubular acidosis, occurs when there is a problem at the end or distal part of the tubules.
- Type 2 renal tubular acidosis or proximal renal tubular acidosis, occurs when there is a problem in the beginning or proximal part of the tubules.
- Type 3 renal tubular acidosis is rarely used as a classification now because it is thought to be a combination of type 1 and type 2 renal tubular acidosis.
- Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, occurs when the tubules are unable to remove enough potassium, which also interferes with the kidney’s ability to remove acid from the blood.
- oisoning by aspirin, ethylene glycol (found in antifreeze), or methanol.
- Severe dehydration.
- Drugs induce acidity in the body through a variety of mechanisms. Most important are 11: nonsteroidal anti-inflammatory drugs (NSAIDs); β-blockers; ACE inhibitors and angiotensin 2 type 1 receptor antagonists; K+-sparing diuretics, such as amiloride and triamterene; antibacterials, such as trimethoprim (commonly administered in combination with sulfamethoxazole as cotrimoxazole); and many more.
- Other several diseases disrupt metabolism in ways that cause excessive acidity, most important are pancreatic drainage, biliary fistula, Sjogren’s syndrome, systemic lupus erythematosus, urinary tract obstruction, fever, aldosterone deficiency, and androgen deficiency 12. Interesting, acidosis causes insulin resistance and insulin resistance increases metabolic acidity—another vicious cycle 13.
Metabolic acidosis symptoms depend on the underlying disease or condition. Acute metabolic acidosis itself causes rapid breathing (an increased rate and depth of breathing). Confusion, headaches or lethargy may also occur. Severe metabolic acidosis can lead to seizures, coma and in some cases death.
Blood and urine tests can help diagnose metabolic acidosis.
Metabolic acidosis treatment should address the cause of the underlying acid-base derangement. For example, adequate fluid resuscitation and correction of electrolyte abnormalities are necessary for sepsis and diabetic ketoacidosis. Other therapies to consider include antidotes for poisoning, dialysis, antibiotics, and intravenous sodium bicarbonate (the chemical in baking soda) administration in certain situations.
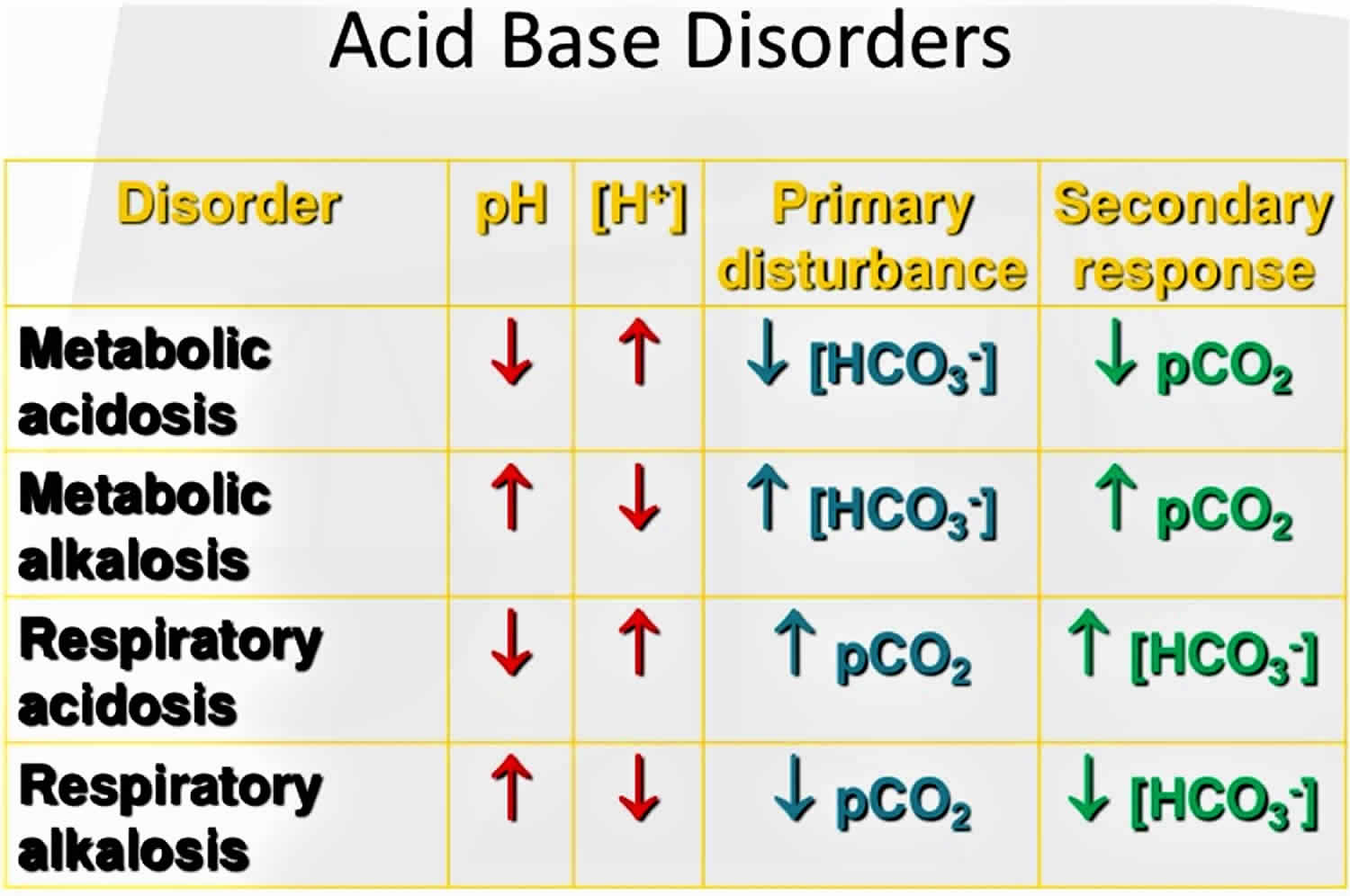
What is the difference between metabolic acidosis and respiratory acidosis?
Metabolic acidosis involves your digestive system and your urinary system. Your kidneys can’t properly filter acids from your bloodstream. Kidney disease, kidney failure, untreated diabetes, loss of bicarbonate and blood poisoning may cause a more acidic pH in your body.
Respiratory acidosis involves your respiratory system. Your lungs can’t remove enough carbon dioxide from your bloodstream. Asthma, brain injuries and excessive or disordered substance use may affect your lungs’ ability to remove carbon dioxide.
Normal anion gap acidosis
The most common cause of normal anion gap acidosis is severe diarrhea with gastrointestinal loss of bicarbonate and a renal tubular acidosis being a distant second 14.
Causes of normal anion gap metabolic acidosis
Causes of normal gap metabolic acidosis include the following:
- Hyperalimentation (e.g. from total parenteral nutrition (TPN) containing ammonium chloride)
- Acetazolamide and other carbonic anhydrase inhibitors
- Renal tubular acidosis 14
- Diarrhea due to a loss of bicarbonate. Because the concentration of bicarbonate in diarrheal fluid is generally greater than that in plasma, large amounts can be lost in severe diarrhea or ileostomy 15. This is compensated by an increase in chloride concentration, thus leading to a normal anion gap metabolic acidosis or hyperchloremic metabolic acidosis. The pathophysiology of increased chloride concentration is the following: fluid secreted into the gut lumen contains higher amounts of sodium (Na+) than chloride (Cl–); large losses of these fluids, particularly if volume is replaced with fluids containing equal amounts of sodium (Na+) and chloride (Cl–), results in a decrease in the plasma sodium (Na+) concentration relative to the chloride (Cl–) concentration. This scenario can be avoided if formulations such as lactated Ringer’s solution are used instead of normal saline to replace gastrointestinal losses 16.
- Ureteral diversion can also lead to a normal anion gap metabolic acidosis. Ureteral implantation into the sigmoid colon or the replacement of the urinary bladder using a short loop of ileum will lead to the exposure of urine to the gastrointestinal mucosa, which will cause gastrointestinal bicarbonate loss and retention of chloride 17.
- Ureteroenteric fistula – an abnormal connection (fistula) between a ureter and the gastrointestinal tract
- Pancreaticoduodenal fistula – an abnormal connection between the pancreas and duodenum
- Normal anion gap metabolic acidosis due to saline infusion. Numerous severely ill patients admitted to the hospital will develop an iatrogenic normal anion gap metabolic acidosis caused by fluid resuscitation with normal saline (NaCL 0.9%). As an example, all patients with severe diabetic ketoacidosis treated with NaCl 0.9% will have a combined high anion gap and normal anion gap metabolic acidosis soon after admission 18. Hyperchloremia develops rapidly, increasing to 50% by 4 h in a previous study 19. Patients treated with therapeutic plasma exchange with a replacement solution of 4% human albumin with a high chloride concentration can also develop a normal anion gap metabolic acidosis 20. Another rare cause in this respect may be the use of NaCl 0.9% for total gut irrigation through the nasogastric route method as a bowel preparation in children undergoing colorectal surgeries 21. Normal saline has a pH of 5.5 and a chloride content of 154 mmol/L and sodium of 154 mmol/L. The low pH has little influence on the development of acidosis after resuscitation. Because plasma has a sodium content of about 140 mmol/L and much lower chloride content of about 106 mmol/L, the chloride increase will be relatively higher than the sodium increase with the infusion of NaCl 0.9%. Because of this increase in chloride, a decrease in bicarbonate follows to maintain electroneutrality. Serum chloride is responsible for about one third of the extracellular fluid tonicity and two thirds of all anionic charges in plasma. Because of its high concentration, chloride is the most important anion to balance extracellular cations 22. All bodily fluids conform to the principle of electrical neutrality, containing an equivalent number of positively and negatively charged ions. Therefore, to maintain electrical neutrality in the face of rising serum chloride anions from normal saline, the serum loses an equal amount of bicarbonate anions resulting in normal anion gap metabolic acidosis 18.
- Cholestyramine is a nonabsorbable anion exchange resin used to bind bile acids in the gut. Cholestyramine has been used in the treatment of hypercholesterolemia, pruritus associated with elevated levels of bile acids, and diarrhea due to bile acid malabsorption in the setting of ileal disease or resection 23. It swaps chloride anions for bile acids in the lumen of the small intestine, resulting in bile acid complexes that are fecal excreted instead of being reabsorbed in the ileum. This exchange causes gastrointestinal secretion of bicarbonate and absorption of chloride. If the kidneys cannot compensate by increasing chloride excretion and bicarbonate retention because of impaired urinary acidification such as renal insufficiency and aldosterone antagonism a normal anion gap metabolic acidosis develops 23.
- Spironolactone
- High ostomy output 14
- Hyperparathyroidism – can cause hyperchloremia and increase renal bicarbonate loss, which may result in a normal anion gap metabolic acidosis. Patients with hyperparathyroidism may have a lower than normal pH, slightly decreased PaCO2 due to respiratory compensation, a decreased bicarbonate level, and a normal anion gap 24.
Increased anion gap metabolic acidosis
The formula for anion gap is:
- Serum anion gap = (Na+) – [(HCO3– + Cl–)]
Where Na+ is plasma sodium concentration, HCO3– is plasma bicarbonate concentration, and Cl– is plasma chloride concentration. The anions are negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]. The anion gap is a calculation to determine the quantity of ionically active components within your blood that are not routinely measured. Since there are always components not directly measured, we expect this value to not equal 0. A normal serum anion gap is measured to be 8 to 16 mEq/L. Most of this number is due to albumin; this anion is not accounted for in the anion gap formula, which is a large reason why the gap is not closer to zero. Albumin is normally 4 mg/dL. Because of the large effect of albumin on anion gap, if a patient’s albumin level is abnormal, their expected anion gap will not be accurate 8. This can be corrected using simple math. The normal anion gap and albumin level differ by a factor of three (normal anion gap of 12, normal albumin of 4 mg/dL). If a patient has an anion gap of 24, that means there are 12 units of the conjugate base present that normally would not be due to the combination of hydrogen ions with bicarbonate. If this same patient has an albumin level of 3 mg/dL, their expected anion gap should actually be about 9. This means that, rather than 12 units of the conjugate base present, there are really 15 units.
If a patient has an anion gap over 12, these mnemonics are helpful to remember the possible causes of the disorder 25, 26. The mnemonic MUDPILES has classically been used by clinicians to summarize the causes of high anion gap metabolic acidosis.
MUDPILES stands for:
- Methanol,
- Uremia,
- Diabetic ketoacidosis,
- Paraldehyde,
- Infection,
- Lactic acidosis,
- Ethylene glycol, and
- Salicylates.
A new mnemonic, GOLDMARK, has been suggested to be an improvement 8.
GOLDMARK is an anagram for:
- Glycols (ethylene and propylene),
- Oxoproline,
- Lactate,
- Methanol,
- Aspirin,
- Renal failure, or chronic kidney disease (CKD) and
- Ketones.
Narrow anion gap metabolic acidosis
If the acidosis involves a normal anion gap, there is a loss of bicarbonate (HCO3–) rather than an increased amount of hydrogen ions (H+), with a concomitant increase in chloride ions. To keep a physiological neutral state, chloride ions migrate out of the cells and into the extracellular space. This causes the patient’s serum chloride to increase and keeps the anion gap at a normal level. This means that a metabolic acidosis without an abnormal anion gap is also a hyperchloremic metabolic acidosis. A metabolic acidosis without an increased anion gap results from many processes including severe diarrhea, type 1 renal tubular acidosis, long-term use of carbonic anhydrase inhibitors, and suctioning of gastric contents. When a patient has a narrow ion gap hyperchloremic acidosis, the physician can calculate the urine anion gap (UAG) to help determine the cause.
The following is the equation for urine anion gap (UAG) where Na is sodium, K is potassium, and Cl is chloride:
- Urine anion gap = Urine Na + Urine K – Urine Cl
The renal system attempts to ameliorate the effects of pathological metabolic acidosis by excreting ammonium (NH4+) into the urine. A urine anion gap between 20 to 90 mEq/L denotes low or normal ammonium (NH4+) secretion. One between minus 20 mEq/L (-20 mEq/L) and minus 50 mEq/L (-50 mEq/L) suggests the main cause of the metabolic acidosis is prolonged severe diarrhea.
Another important formula to use with metabolic acidosis is the Winter formula. This equation provides the clinician with the expected arterial partial pressure of carbon dioxide (PaCO2) value. This is important because there could be another acid-base disorder present.
The Winter formula is:
- Expected arterial partial pressure of carbon dioxide (PaCO2)= (1.5 X HCO3–) + 8 +/- 2
If the arterial partial pressure of carbon dioxide (PaCO2) value is within range of the expected arterial partial pressure of carbon dioxide (PaCO2), there is no mixed disorder, just respiratory compensation. When the value is lower or higher than expected, there is a mixed disorder; lower would mean a respiratory alkalosis and higher a respiratory acidosis. A shortcut for the Winter formula is that the last two digits of the pH +/- 2 is about equal to the expected arterial partial pressure of carbon dioxide (PaCO2) 5, 27.
Hyperchloremic metabolic acidosis
Hyperchloremic metabolic acidosis is a pathological state that results from bicarbonate (HCO3–) loss, rather than acid production or retention 28. Bicarbonate (HCO3–) loss leading to hyperchloremic metabolic acidosis occurs in a variety of ways: gastrointestinal (GI) causes, renal causes, and exogenous causes. Gastrointestinal loss of bicarbonate occurs through severe diarrhea, pancreatic fistula, nasojejunal tube suctioning from the duodenum, and chronic laxative use 29. Kidney sources of hyperchloremic acidosis include proximal renal tubular acidosis, distal renal tubular acidosis, and long-term use of carbonic anhydrase inhibitors 29. Exogenous causes include ingestion of acids such as ammonium chloride and hydrochloric acid and volume resuscitation with 0.9% normal saline 30.
Gastrointestinal causes
Normally, there is a degree of bicarbonate secreted into the intestinal lumen to allow for neutralization of the acidic environment of food from gastric emptying. Over the distance of the small intestines, this bicarbonate is reabsorbed as bile. However, in pathologies with profuse watery diarrhea, bicarbonate within the intestines is lost through the stool due to increased motility of the gut. This leads to further secretion of bicarbonate from the pancreas and intestinal mucosa leading to a net acidification of the blood from bicarbonate loss. Likewise, pancreatic fistula leads to excessive bicarbonate secretion from the pancreas into the intestines. This excess bicarbonate is ultimately lost in stools. Nasojejunal suctioning removes bicarbonate from the duodenal or jejunal space via direct suctioning of the luminal contents.The overarching theme with these pathologies is loss of bicarbonate from the gastrointestinal spaces which leads to an acidotic state in the blood via unopposed hydrogen in the buffering system as above.
Renal causes
Distal renal tubular acidosis (type 1) is a failure of the distal nephron to secrete hydrogen appropriately into urine. This results in alkalotic urine and acidosis of the blood. Failure to secrete hydrogen directly correlates with the ammonium (NH4+) levels in urine and is able to be deduced via a positive urine anion gap as above. Proximal renal tubular acidosis (type 2) is a pathology where bicarbonate is failed to be reabsorbed appropriately. This leads to loss of bicarbonate into the urine. The net result is acidosis of blood and alkalotic urine. Both types of renal tubular acidosis are associated with hypokalemia. Carbonic anhydrase inhibitors such as acetazolamide create a medically induced type 2 proximal renal tubular acidosis scenario by inhibiting bicarbonate reabsorption in the proximal nephron.
Exogenous causes
Many of the exogenous causes of hyperchloremic acidosis as logical evaluations. When substances such as ammonium chloride and hydrochloric acid are supplemented into the body, they react with bicarbonate in an attempt to buffer the pH. However, this will deplete bicarbonate stores leading to an acidotic state. Large volume resuscitation with 0.9% normal saline leads to an overload of chloride ions into the blood. As stated previously, chloride and bicarbonate work together to maintain an ionic balance of the cellular space. Hyperchlorhydria forces bicarbonate to move intracellularly to maintain ionic equilibrium, thus reducing the available bicarbonate for the pH buffering system leading to net acidosis.
Metabolic acidosis causes
The causes of metabolic acidosis are classified into 4 main mechanisms 25, 31, 32, 33:
- Increased production of acid,
- Decreased excretion of acid,
- Acid ingestion, and
- Renal or gastrointestinal bicarbonate losses.
The four leading causes of metabolic acidosis include:
- Diabetes-related acidosis. Diabetes-related acidosis develops when ketone bodies build up in your body from untreated diabetes. Your body produces ketone bodies while it turns (metabolizes) fats into energy. Your body uses ketone bodies for energy when sugars (glucose) aren’t available.
- Hyperchloremic acidosis. Hyperchloremic acidosis develops when your body loses too much sodium bicarbonate. It may occur if you take too many laxatives or have severe diarrhea.
- Lactic acidosis. Lactic acidosis develops when you have too much lactic acid in your body. Lactic acid is an organic acid that your muscle cells and red blood cells produce for energy when you don’t have a lot of oxygen in your body. Causes include liver failure, low blood sugar, alcohol use disorder, cancer and intense exercise.
- Renal tubular acidosis. Renal tubular acidosis (RTA) occurs when the kidneys do not remove acids from the blood into the urine as they should. The acid level in the blood then becomes too high, a condition called acidosis. Some acid in the blood is normal, but too much acid can disturb many bodily functions. There are three main types of renal tubular acidosis that are characterized by: 1) a normal anion gap metabolic acidosis; 2) abnormalities in renal bicarbonate (HCO3-) absorption or new renal bicarbonate (HCO3-) generation; 3) changes in renal NH4+, Ca2+, K+ and H2O homeostasis; and 4) extrarenal manifestations that provide etiologic diagnostic clues 9, 10.
- Type 1 renal tubular acidosis, or distal renal tubular acidosis, occurs when there is a problem at the end or distal part of the tubules.
- Type 2 renal tubular acidosis, or proximal renal tubular acidosis, occurs when there is a problem in the beginning or proximal part of the tubules.
- Type 3 renal tubular acidosis is rarely used as a classification now because it is thought to be a combination of type 1 and type 2 renal tubular acidosis.
- Type 4 renal tubular acidosis, or hyperkalemic renal tubular acidosis, occurs when the tubules are unable to remove enough potassium, which also interferes with the kidney’s ability to remove acid from the blood.
Mnemonic for anion gap metabolic acidosis differential (CAT MUDPILES):
- C: Cyanide and carbon monoxide poisoning
- A: Arsenic
- T: Toluene
- M: Methanol, Metformin
- U: Uremia
- D: Diabetic ketoacidosis (DKA)
- P: Paraldehyde
- I: Iron, Isoniazid
- L: Lactate
- E: Ethylene glycol
- S: Salicylates
Non-gap metabolic acidosis is primarily due to the loss of bicarbonate, and the main causes of this condition are diarrhea and renal tubular acidosis. Additional and rarer causes include Addison’s disease, ureterosigmoid or pancreatic fistulas, acetazolamide use, and hyperalimentation through TPN initiation. Gastrointestinal and renal losses of bicarbonate can be distinguished via urine anion gap analysis:
- Urine anion gap = Urine Na + Urine K – Urine Cl
A positive value is indicative of renal bicarbonate loss, such as renal tubular acidosis. Negative values are found with non-renal bicarbonate losses, such as diarrhea.
Metabolic acidosis pathophysiology
The primary ways the body deals with excessive acidity are through renal adaptations, respiration, and buffering with calcium from bone.
It is vital for life that pH does not waiver too far from normal, and the body will always attempt to return an abnormal pH towards normal when acid-base balance is disturbed. Compensation is the name given to this life-preserving process. To understand compensation, it is important to recall that pH is governed by the ratio bicarbonate [HCO3–] (a base): arterial partial pressure of carbon dioxide (PaCO2) (an acid). So long as the ratio is normal, pH will be normal.
Normal body functions and metabolism generate large quantities of acids that must be neutralized and/or eliminated to maintain blood pH balance. Most of the acid is carbonic acid, which is created from carbon dioxide (CO2) and water (H2O). Carbon dioxide (CO2) is produced as the body uses glucose (sugar) or fat for energy. In its normal state, the body maintains carbon dioxide (arterial partial pressure of carbon dioxide [PaCO2]) in a well-controlled range from 35 to 45 mm Hg by balancing its production and elimination. Lesser quantities of lactic acid, ketoacids, and other organic acids are also produced.
According to the Henderson-Hasselbalch equation (Figure 2), maintaining physiological pH depends on arterial partial pressure of carbon dioxide (PaCO2), which in turn depends on alveolar ventilation (hypoventilation causes acidosis and hyperventilation causes alkalosis). The kidneys participate in maintaining the stable pH by reabsorption of bicarbonate (3,600 mmol of bicarbonate is filtrated in glomeruli during 24 hour) and excretion of hydrogen ions from nonvolatile acids (including sulfur and phosphate) as titratable acidity (0.3 mmol hydrogen ions/kg/day) and in the form of ammonium ion (0.7 mmol hydrogen ions/kg/day) 34, 35.
The lungs and kidneys are the major organs involved in regulating blood pH. And to compensate for the metabolic acidosis, you increase your breathing rate (hyperventilation) to increase carbon dioxide (CO2) elimination 36, 37.
- The lungs flush acid out of your body by exhaling carbon dioxide (CO2). Raising and lowering the respiratory rate alters the amount of carbon dioxide (CO2) that is breathed out, and this can affect blood pH within minutes 38.
- The kidneys excrete acids in the urine, and they regulate the concentration of bicarbonate (HCO3–, a base) in blood. Acid-base changes due to increases or decreases in bicarbonate [HCO3–] concentration occur more slowly than changes in carbon dioxide (CO2), taking hours or days. Bicarbonate (HCO3–) reabsorption occurs in the kidneys in every part of the tubules. About 85–90% of the filtered bicarbonate is reabsorbed in the proximal tubules, 10% in the ascending arms of the Henle loop, 6% in the distal tubules, and 4% in the collecting tubules 34, 35.
Both of these processes are always at work, and they keep the blood pH in healthy people tightly controlled. The absolute quantities of acids or bases are less important than the balance between the two and its effect on blood pH.
Buffering systems that resist changes in pH also contribute to the regulation of acid and base concentrations. The main buffers in blood are hemoglobin (in red blood cells), plasma proteins, carbon dioxide (CO2), bicarbonate (HCO3–) and phosphates.
Carbon dioxide (CO2) plays a remarkable role in the human body mainly through pH regulation of the blood. The pH is the primary stimulus to initiate ventilation. In its normal state, the body maintains carbon dioxide (CO2) in a well-controlled range from 35 to 45 mm Hg by balancing its production and elimination. In a state of hypoventilation (breathing that is too shallow or too slow to meet the needs of the body), the body produces more carbon dioxide (CO2) than it can eliminate, causing a net retention of carbon dioxide (CO2). The increased carbon dioxide (CO2) is what leads to an increase in hydrogen ions (H+) and a slight increase in bicarbonate (HCO3–), as seen by a right shift in the following equilibrium reaction of carbon dioxide:
- Carbon dioxide (CO2) + water (H2O) -> H2CO3 (carbonic acid) -> HCO3– + H+
The buffer system created by carbon dioxide consists of the following three molecules in equilibrium: carbon dioxide (CO2), H2CO3 (carbonic acid), and bicarbonate (HCO3–). When hydrogen ions (H+) is high, bicarbonate (HCO3–) buffers the low pH. When hydroxide (OH–) is high, H2CO3 (carbonic acid) buffers the high pH. In respiratory acidosis, the slight increase in bicarbonate (HCO3–) serves as a buffer for the increase in hydrogen ions (H+), which helps minimize the drop in pH. The increase in hydrogen ions inevitably causes the decrease in pH, which is the mechanism behind metabolic acidosis.
Figure 2. Henderson-Hasselbalch equation
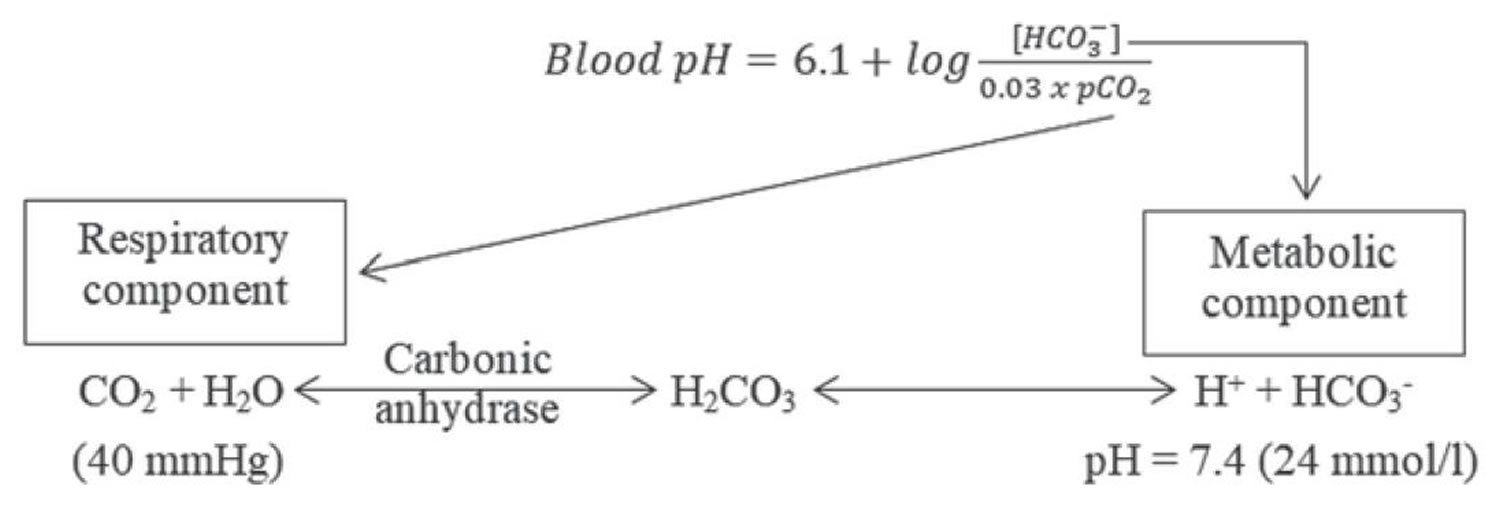
Figure 3. Abnormal acid-base compensation

Figure 4. Acid-base buffering system
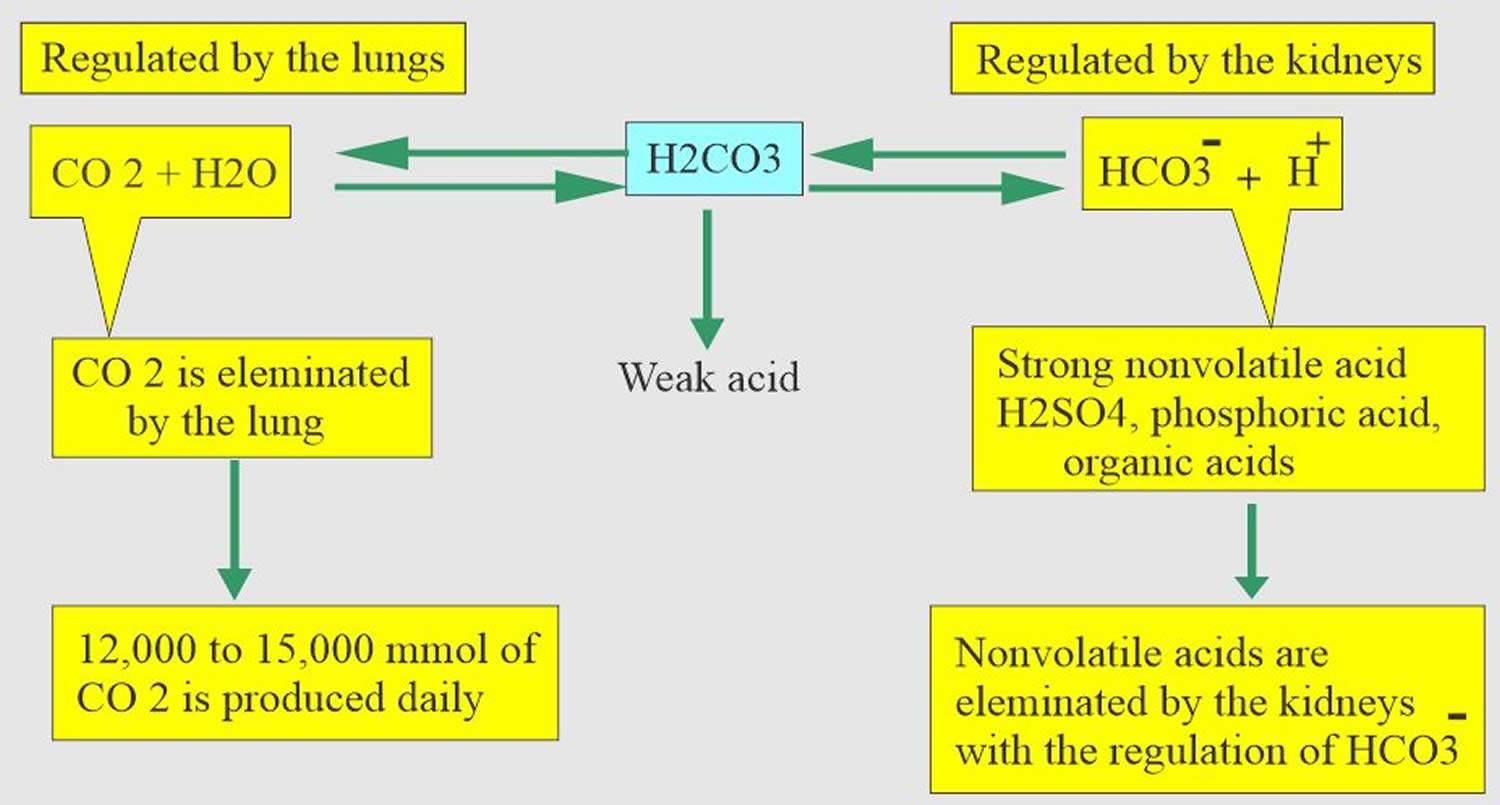
Respiration
The pulmonary system adjusts pH using carbon dioxide (CO2); upon expiration, carbon dioxide (CO2) is projected into the environment. Due to carbon dioxide (CO2) forming carbon dioxide (CO2) in the body when combining with water (H2O), the amount of carbon dioxide (CO2) expired can cause pH to increase or decrease. When the respiratory system is utilized to compensate for metabolic pH disturbances, the effect occurs in minutes to hours 40.
Renal adaptation
The renal system affects pH by reabsorbing bicarbonate (HCO3–) and excreting fixed acids 40, 41. Whether due to pathology or necessary compensation, the kidney excretes or reabsorbs these substances which affect pH. The nephron is the functional unit of the kidney. Blood vessels called glomeruli transport substances found in the blood to the renal tubules so that some can be filtered out while others are reabsorbed into the blood and recycled. This is true for hydrogen ions and bicarbonate. If bicarbonate (HCO3–) is reabsorbed and/or acid is secreted into the urine, the pH becomes more alkaline (pH increases). When bicarbonate (HCO3–) is not reabsorbed or acid is not excreted into the urine, pH becomes more acidic (pH decreases). The metabolic compensation from the renal system takes longer to occur, days rather than minutes or hours.
The renal adaptations are extensive 42:
- Increased urinary excretion of sulfate, phosphate, urate, and chloride;
- Increased urinary excretion of calcium;
- Decreased urinary excretion of citrate;
- Increased urinary excretion of ammonium ions; and
- Kidney vasodilatation and increased glomerular filtration rate.
The kidneys mitigate but do not eliminate all the excess acidity. As the kidneys lose function with aging (when GFR is lower than 30 mL/min/1.73 m²), their ability to excrete acid becomes impaired, which may be another explanation for the loss of bone with aging 43. In fact, counteracting metabolic acidosis helps to preserve muscle mass and to improve bone metabolism 44, 45, 46.
Figure 5. Kidneys control of plasma bicarbonate (HCO3–)
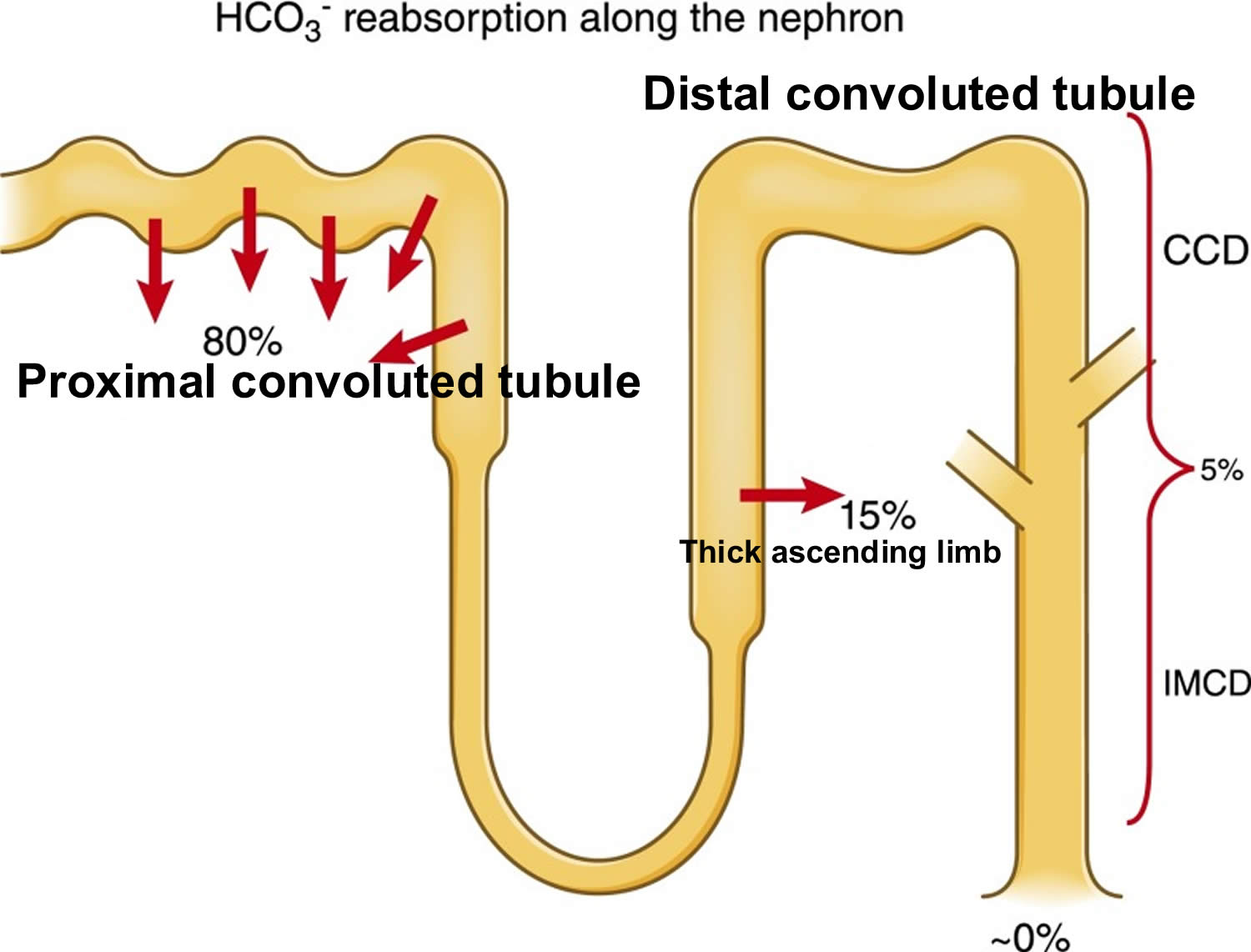
Abbreviations: CCD = cortical collecting duct; IMCD = inner medullary collecting duct
[Source 35 ]Bone for acid buffering
The major reservoir of base is the skeleton (in the form of alkaline salts of calcium), which provides the buffer needed to maintain blood pH and plasma bicarbonate concentrations when renal and respiratory adaptations are inadequate. Acid-promoting diets are associated with increased urinary excretion of both calcium and bone matrix protein and decreased bone density 47. Neutralizing acid intake with diet or alkalinizing supplements decreases urine Calcium and bone matrix protein excretion. Also, to a much smaller degree, skeletal muscle can act as a buffer.
Other buffer systems
Other buffer systems in the human body include the phosphate buffer system, proteins, and hemoglobin. All of these contain bases which accept hydrogen ions which keep the pH from plummeting. The phosphate buffer system, while present globally, is important for the regulation of urine pH. Proteins assist with intracellular pH regulation. Red blood cells use the reaction above to help hemoglobin buffer; carbon dioxide can diffuse across red blood cells and combine with water. This alone would cause an increase in hydrogen ions; however, hemoglobin can bind hydrogen ions. Hemoglobin also can bind carbon dioxide without this reaction. This depends on the amount of oxygen that is bound to hemoglobin. This is called the Haldane effect and the Bohr effect. When hemoglobin is saturated with oxygen, it has a lower affinity for carbon dioxide (CO2) and hydrogen ions and is able to release it.
Metabolic acidosis prevention
You can’t prevent metabolic acidosis. However, you can help reduce your risk by:
- Drinking a lot of water and other fluids.
- Managing your blood sugar levels if you have diabetes.
- Reducing the amount of alcohol that you consume. Moderate alcohol consumption in men is two drinks or fewer per day. In women moderate alcohol consumption is one drink or fewer per day.
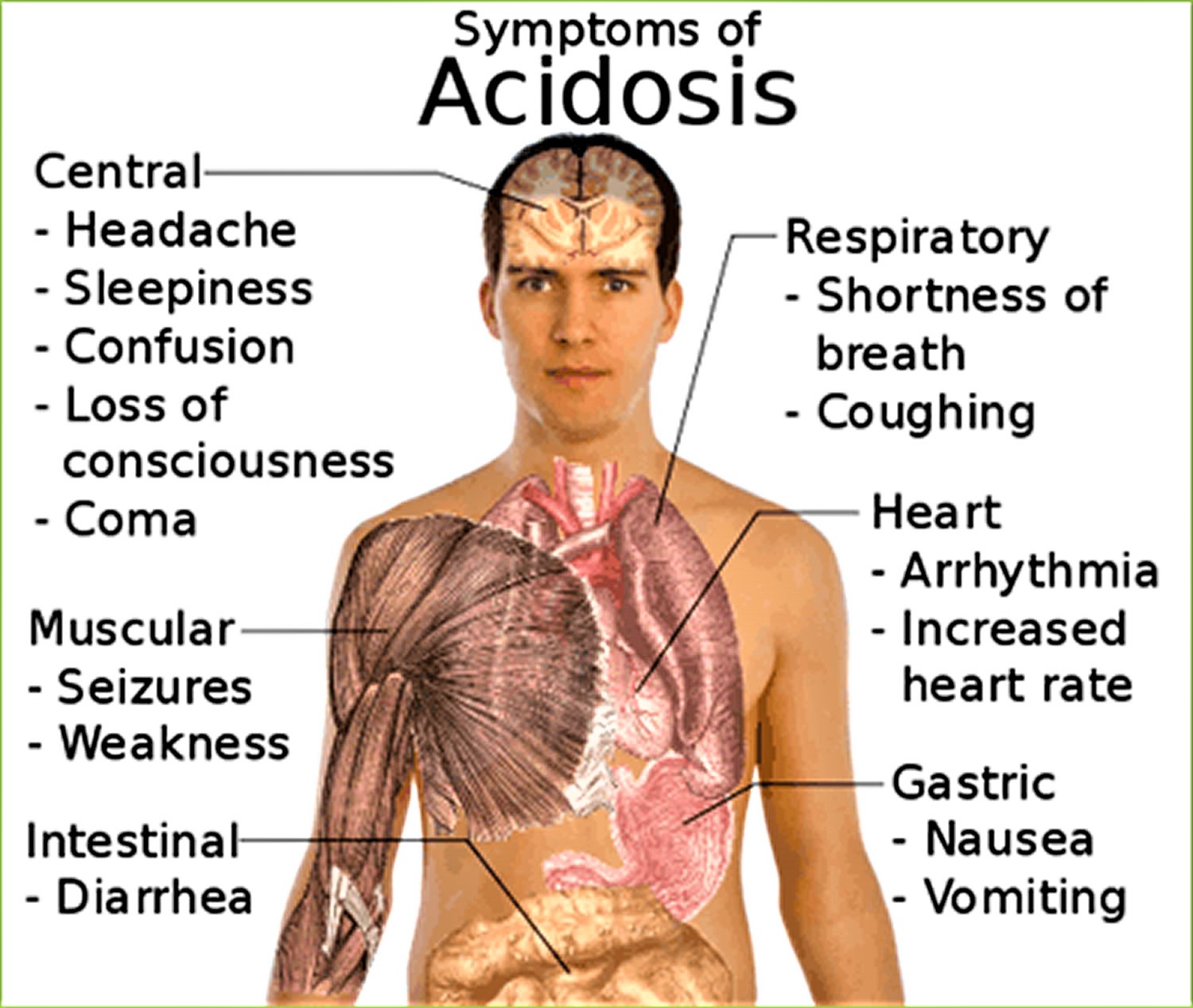
Metabolic acidosis signs and symptoms
Metabolic acidosis symptoms depend on the underlying disease or condition. Acute metabolic acidosis itself causes rapid breathing (an increased rate and depth of breathing). Confusion, headaches or lethargy may also occur. Severe metabolic acidosis can lead to seizures, coma and in some cases death.
Signs and symptoms that can be associated with metabolic acidosis, which is when there is too much acid in the body, include:
- Rapid breathing or long, deep breathing.
- Accelerated heartbeat (tachycardia).
- Shortness of breath
- Mental confusion or dizziness.
- Feeling very tired (fatigue)
- Loss of appetite
- Headache.
- Nausea and vomiting.
- Feeling weak.
- Breath that smells sweet or fruity.
Patients with hyperchloremic acidosis have no effects due to the hyperchloremia necessarily. However, the acidosis can have many poor health effects. A headache, lack of energy, nausea, and vomiting are common complaints, however as acidosis worsens stupor, coma, myocardial instability or arrest may occur. It is expected to see an increase in respiratory rate as the body attempts to decrease CO2 in compensation, however, in long-standing disease this may lead to muscle fatigue and respiratory failure.
A physical exam may show altered mental status, tachycardia, tachypnea, accessory muscle use with respiration, neurological deficits, muscular weakness, cardiac arrhythmias, cardiac murmurs, respiratory wheezing, rales, or rhonchi.
In some situations, metabolic acidosis can be a mild, ongoing (chronic) condition.
Metabolic acidosis diagnosis
Your healthcare provider will perform a physical examination and ask about your symptoms.
These tests can help diagnose acidosis. They can also determine whether the cause is a breathing problem (respiratory acidosis) or a metabolic problem (metabolic acidosis). Tests may include:
- Arterial blood gas (ABG)
- Basic metabolic panel, (a group of blood tests that measure your sodium, potassium, and chloride levels, kidney function, and other chemicals and functions)
- Urine pH
- Urine ketones or blood ketones
- Lactic acid test
- A complete blood count (CBC) to evaluate for an infectious cause with elevated white blood count and fluid body status with hemoglobin and hematocrit values is useful.
Other tests that may be needed to determine the cause of the acidosis include:
- Pulmonary function test to measure breathing and how well the lungs are functioning
- Chest x-ray
- CT abdomen
Physical examination
The best recognized sign of metabolic acidosis is Kussmaul respirations, a form of hyperventilation that serves to increase minute ventilatory volume. This is characterized by an increase in tidal volume rather than respiratory rate and is appreciated as deliberate, slow, deep breathing 48.
Chronic metabolic acidosis in children may be associated with stunted growth and rickets.
Coma and hypotension have been reported with acute severe metabolic acidosis.
Other physical signs of metabolic acidosis are not specific and depend on the underlying cause. Some examples include xerosis, scratch marks on the skin, pallor, drowsiness, fetor, asterixis, and pericardial rub for kidney failure, as well as reduced skin turgor, dry mucous membranes, and fruity breath odor for diabetic ketoacidosis (DKA).
Arterial blood gas (ABG) analysis
Arterial blood gas (ABG) sampling, is a test often performed in an inpatient setting to assess the acid-base status of a patient. A needle is used to draw blood from an artery, often the radial artery, and the blood is analyzed to determine parameters such as the pH, arterial partial pressure of carbon dioxide (PaCO2), arterial partial pressure of oxygen (PaO2), bicarbonate (HCO3–), oxygen saturation (O2 Sat) and more. This allows the physician to understand the status of the patient better. ABGs are especially important in the critically ill. They are the main tool utilized in adjusting to the needs of a patient on a ventilator.
- Arterial partial pressure of carbon dioxide (PaCO2) as carbon dioxide tension, this measures the level of carbon dioxide in your blood.
- Arterial partial pressure of oxygen (PaO2) also known as oxygen tension, this measures how well oxygen is being transferred into your blood.
- Oxygen saturation (O2 Sat) is an assessment of the amount of oxygen in your blood that is based on measuring levels of hemoglobin. Hemoglobin is a protein found inside red blood cells that is responsible for carrying oxygen throughout the body.
- Bicarbonate (HCO3–) concentration: Bicarbonate (HCO3–) is an electrolyte, which is a type of mineral involved in managing your body’s acid-base balance. Most of the carbon dioxide (CO2) in your blood is stored in the form of bicarbonate, so this measurement helps reflect carbon dioxide (CO2) levels.
- Although not universal, some arterial blood gases tests include measurements of hemoglobin as well as altered forms of the hemoglobin protein. Examples of these potential additional measurements include:
- Methemoglobin: Methemoglobin is a form of hemoglobin that has been oxidized, changing its heme iron configuration from the ferrous (Fe2+) to the ferric (Fe3+) state. Unlike normal hemoglobin, methemoglobin does not bind oxygen and as a result cannot deliver oxygen to the tissues.
- Carboxyhemoglobin: Carboxyhemoglobin is a stable complex of carbon monoxide and hemoglobin that forms in red blood cells upon contact with carbon monoxide. This abnormal form of hemoglobin attaches to carbon monoxide and can interfere with oxygen’s ability to travel in the blood.
- Oxyhemoglobin: Oxyhemoglobin represents the fraction of oxygenated hemoglobin in relation to the total hemoglobin present, including non-oxygen-binding hemoglobins. In healthy individuals, oxyhemoglobin and oxygen saturation are approximately equal.
- Deoxyhemoglobin: This is the form of hemoglobin without oxygen in the blood.
The following are the most important Normal Values on an ABG:
- pH = 7.35 to 7.45
- Arterial partial pressure of carbon dioxide (PaCO2) = 35 to 45 mmHg
- Arterial partial pressure of oxygen (PaO2) = 75 to 100 mmHg
- Bicarbonate (HCO3–) = 22 to 26 mEq/L
- O2 Sat = greater than 95%
The ability to quickly and efficiently read an ABG is paramount to quality patient care.
- Look at the pH. Decide whether it is acidotic, alkalotic, or within the physiological range
- Arterial partial pressure of carbon dioxide (PaCO2) level determines respiratory contribution; a high level means the respiratory system is lowering the pH and vice versa.
- Bicarbonate (HCO3–) level denotes metabolic/kidney effect. An elevated bicarbonate (HCO3–) is raising the pH and vice versa.
- If the pH is acidotic, look for the number that corresponds with a lower pH. If it is a respiratory acidosis, the carbon dioxide (CO2) should be high. If the patient is compensating metabolically, the bicarbonate (HCO3–) should be high as well. A metabolic acidosis will be depicted with an bicarbonate (HCO3–) that is low.
- If the pH is alkalotic, again, determine which value is causing this. A respiratory alkalosis will mean the carbon dioxide (CO2) is low; a metabolic alkalosis should lend an bicarbonate (HCO3–) that is high. Compensation with either system will be reflected oppositely; for a respiratory alkalosis the metabolic response should be a low bicarbonate (HCO3–) and for metabolic alkalosis, the respiratory response should be a high carbon dioxide (CO2).
- If the pH level is in the physiological range but the arterial partial pressure of carbon dioxide (PaCO2) and/or bicarbonate (HCO3–) are not within normal limits, there is likely a mixed disorder. Also, compensation does not always occur; this is when clinical information becomes paramount.
- Sometimes it is difficult to ascertain whether a patient has a mixed disorder.
Other tests that are important to perform when analyzing the acid-base status of a patient include those that measure electrolyte levels and renal function. This helps the clinician gather information that can be used to determine the exact mechanism of the acid-base imbalance as well as the factors contributing to the disorders 49, 50.
Urinalysis
Urine pH is normally acidic, at less than 5.0. In acidemia, the urine normally becomes more acidic. If the urine pH is above 5.5 in the face of acidemia, this finding is consistent with a type 1 renal tubular acidosis (RTA). Alkaline urine is typical in salicylate toxicity.
Patients with ethylene glycol toxicity may present with calcium oxalate crystals, which appear needle shaped, in the urine.
Urine Anion Gap
Calculating the urine anion gap is helpful in evaluating some cases of non-anion gap metabolic acidosis. The major measured urinary cations are Na+ and K+, and the major measured urinary anion is Cl-:
- Urine anion gap = Urine Na + Urine K – Urine Cl
In the face of metabolic acidosis, the kidneys increase the amount of NH3 synthesized to buffer the excess H+ and NH4 Cl excretion increases. The increased unmeasured ammonium (NH4+) thus increases the measured anion Cl- in the urine, and the net effect is a negative anion gap, representing a normal response to systemic acidification. The finding of a positive urine anion gap in the face of non-anion gap metabolic acidosis points toward a renal acidification defect (eg, renal tubular acidosis) 51.
Ketone level
Elevations of ketones indicate diabetic, alcoholic, and starvation ketoacidosis 52.
The nitroprusside test is used to detect the presence of ketoacids in the blood and the urine. This test measures only acetoacetate and acetone; therefore, it may underestimate the degree of ketonemia and ketonuria, because it will not detect the presence of beta-hydroxybutyrate. This limitation of the test can be especially problematic in patients with ketoacidosis who cannot convert beta-hydroxybutyrate to acetoacetate because of severe shock or liver failure.
An assay for beta-hydroxybutyrate is unavailable in some hospitals. An indirect method to circumvent this problem is to add a few drops of hydrogen peroxide to a urine specimen. This enzymatically will convert beta-hydroxybutyrate into acetoacetate, which will be detected by the nitroprusside test.
Serum Lactate level
The normal plasma lactate concentration is 0.5-1.5 mEq/L. Lactic acidosis is considered present if the plasma lactate level exceeds 4-5 mEq/L in an acidemic patient.
Most cases of lactic acidosis are due to tissue hypoxia (eg, from shock). Less commonly, underlying disease (eg, diabetic ketoacidosis), drugs, or toxins may be the cause 53.
Salicylate levels and Iron levels
Therapeutic salicylate levels range up to 20-35 mg/dL. Plasma levels exceeding 40-50 mg/dL are in the toxic range.
Plasma levels provide some information as to the severity of intoxication: 40-60 mg/dL is considered mild; 60-100 mg/dL is moderate; and greater than 100 mg/dL is considered severe.
Iron toxicity is associated with lactic acidosis. Iron levels greater than 300 mg/dL are considered toxic.
Special tests
Measuring the transtubular potassium gradient (TTKG) is useful in determining the cause of hyperkalemia or hypokalemia associated with metabolic acidosis.
- Transtubular Potassium Gradient (TTKG) = urine K+ × serum osmolality/serum K+ × urine osmolality
A transtubular potassium gradient (TTKG) of greater than 8 indicates that aldosterone is present and that the collecting duct is responsive to it. A transtubular potassium gradient (TTKG) of less than 5 in the presence of hyperkalemia indicates aldosterone deficiency or resistance. For the test to be interpretable, the urine Na+ level should be greater than 10 mEq/L and the urine osmolality should be greater than or equal to serum osmolality.
Plasma renin activity and plasma aldosterone levels are useful in determining the cause of the hyperkalemia and hypokalemia that accompany metabolic acidosis.
Calculation of fractional excretion of bicarbonate (FEHCO3–) is useful in the diagnosis of proximal renal tubular acidosis (RTA).
The ammonium chloride (NH4Cl) loading test is useful in patients with nephrocalcinosis and/or nephrolithiasis, who may have an incomplete form of distal renal tubular acidosis. These patients may not have a pH less than 7.35 or a drop in serum bicarbonate (HCO3–); metabolic acidosis can be induced by administration of NH4Cl (0.1 g/kg for 3 days). Under these circumstances of induced acidemia, a urine pH greater than 5.3 indicates distal renal tubular acidosis (RTA).
An alternative to the ammonium chloride (NH4Cl) loading test involves the simultaneous oral administration of furosemide to increase distal Na+ delivery and fludrocortisone to increase collecting duct Na+ absorption and proton secretion 54. Under these circumstances, a urine pH greater than 5.3 indicates distal renal tubular acidosis (RTA).
Measuring the urine-blood arterial partial pressure of carbon dioxide (PaCO2) gradient following an bicarbonate (HCO3–) load is useful in some patients with classic distal renal tubular acidosis to differentiate a permeability defect from other defects. This test is useful in patients with nephrocalcinosis in whom distal renal tubular acidosis (RTA) is suspected but urine is acidified appropriately in the face of metabolic acidosis. Some of these patients have a rate-dependent defect in proton secretion, revealed by a low urine-blood PaCO2 gradient following bicarbonate (HCO3–) loading.
Abdominal radiographs (eg, kidneys, ureters, bladder), CT scans, and/or renal ultrasound images may show renal stones or nephrocalcinosis in patients with distal renal tubular acidosis.
Metabolic acidosis treatment
The management of metabolic acidosis should address the cause of the underlying acid-base derangement. For example, adequate fluid resuscitation and correction of electrolyte abnormalities are necessary for sepsis and diabetic ketoacidosis. Other therapies to consider include antidotes for poisoning, dialysis, antibiotics, and intravenous sodium bicarbonate (the chemical in baking soda) administration in certain situations.
Oral sodium bicarbonate (NaHCO3) can be administered in some acute metabolic acidemic states in which correction of metabolic acidosis is unlikely to occur without exogenous alkali administration.
Oral alkali administration is the preferred route of therapy in persons with chronic metabolic acidosis. The most common alkali forms for oral therapy include sodium bicarbonate (NaHCO3) tablets. These are available in 325 and 650 mg strengths (1 g of sodium bicarbonate [NaHCO3] is equal to 11.5 mEq of bicarbonate [HCO3–]). Citrate salts are available in a variety of formulations, as mixtures of citric acid with sodium citrate and/or potassium citrate. These solutions generally contain 1-2 mEq of bicarbonate (HCO3–) per mL. Potassium citrate is useful when the acidosis is accompanied by hypokalemia but should be used cautiously in persons with renal impairment and must be avoided in those with hyperkalemia.
In a 12-month controlled, randomized, interventional trial that included 30 kidney transplant patients with metabolic acidosis, correction of metabolic acidosis with potassium citrate was found to be effective and well tolerated, and was associated with improvements in bone quality, suggesting a beneficial effect of both alkali treatment and restoration of acid/base balance. The researchers concluded that potassium citrate may be superior to sodium bicarbonate, because it lacks volume effects and the obligatory calcium excretion associated with sodium administration 55.
Starvation and alcohol use resulting in acidosis is treated with intravenous glucose, which is administered to stimulate insulin secretion and stop lipolysis and ketosis. For diabetic ketoacidosis (DKA), insulin is administered, usually intravenously, to facilitate cellular uptake of glucose, reduce gluconeogenesis, and halt lipolysis and production of ketone bodies. In addition, normal saline is administered to restore extracellular volume; potassium and phosphate replacement also may be necessary. The acidosis is corrected partly by the metabolism of ketones to bicarbonate (HCO3–), partly by increased H+ secretion by the collecting duct, and partly by H+ excretion as ammonium (NH4+).
In every case of hyperchloremic acidosis, the primary treatment is aimed at identifying and treating the inciting event of pathology. If respiratory fatigue and failure occur, these patients will need to be intubated and placed on mechanical ventilation. Hyperventilation of the patient on ventilator control can help reduce the acid load. In gastrointestinal causes, it is essential to administer intravenous (IV) saline to maintain fluid load as patients will easily dehydrate from diarrhea or suctioning of the intestines. Additionally, electrolytes need to be monitored and replenished as applicable. Of specific importance is the potassium level. The acidosis is moderated by supplementing bicarbonate into the saline fluids until the underlying pathology is repaired. In renal tubular acidosis, large quantities of bicarbonate administration may be necessary. If fluid overload is a concern, diuretics with supplemental potassium may be administered for some effect. If the acidosis is resistant to therapy, it may be necessary to utilize dialysis therapy.
As always, a variety of medications are known to induce hyperchloremic acidosis and should be avoided or used with caution. Gastrointestinal bicarbonate loss is known to occur with calcium chloride, magnesium sulfate, and cholestyramine use. Proximal renal tubular acidosis is associated with streptozotocin, lead, mercury, arginine, valproic acid, gentamicin, ifosfamide, and outdated tetracycline usage. Distal renal tubular acidosis is associated with amphotericin B, toluene, nonsteroidal anti-inflammatory drugs, and lithium use.
Administration of an alkali is the mainstay of treatment for type 1 renal tubular acidosis. Adult patients should be given the amount required to buffer the daily acid load from the diet. This is usually approximately 1-3 mEq/kg/d and can be administered in any form, although the preferred form is as potassium citrate. Correction of acidosis usually corrects the hypokalemia, but K+ supplements may be necessary.
Patients with type 2 renal tubular acidosis typically have hypokalemia and increased urinary K+ wasting. Administration of alkali in those patients leads to more bicarbonate (HCO3–) wasting and can worsen hypokalemia unless K+ is replaced simultaneously. Correcting this form of acidosis with alkali is difficult because a substantial proportion of the administered bicarbonate (HCO3–) is excreted in the urine, and large amounts are needed to correct the acidosis (10-30 mEq/kg/day). Potassium is also required when administering bicarbonate (HCO3–). Correction is essential in children for normal growth, while in adults aggressive correction to a normal level may not be required. Thiazide diuretics can be administered to induce diuresis and mild volume depletion, which, in turn, raises the proximal tubule threshold for bicarbonate (HCO3–) wasting.
Because hyperkalemia is central to the cause of type 4 renal tubular acidosis 56, major treatment goal is to lower the serum K+ level. This can be achieved by placing the patient on a low-potassium diet (1 mEq/kg K+/day) and by withdrawal of drugs that can cause hyperkalemia (eg, angiotensin-converting enzyme [ACE] inhibitors, nonsteroidal anti-inflammatory drugs). Loop diuretics can be helpful in reducing serum potassium levels as long as the patient is not hypovolemic. In resistant cases, fludrocortisone, a synthetic mineralocorticoid, can be used to increase K+ secretion, but this may increase Na+ retention. Alkali therapy is not usually required, because, in many patients, the mild degree of acidosis is corrected by achieving normokalemia. Hyperkalemia and acidosis worsen as kidney function declines further; eventually, the patient develops a high anion gap renal acidosis. Renal replacement therapy should be considered once the measures described fail to control hyperkalemia or acidosis.
Medications are used to treat metabolic acidosis
The over-the-counter (OTC) medications sodium citrate or sodium bicarbonate can help balance the acids in your body. See your healthcare provider before taking any OTC medications to help treat your metabolic acidosis.
Your healthcare provider may also prescribe inotropes. Inotropes help your heart beat stronger, which helps get more oxygen in your body and lowers the amount of acids in your blood. Your healthcare provider can deliver inotropes to your body through an IV in a vein in your arm.
What should I eat or drink if I have metabolic acidosis?
Certain foods and drinks can cause your body to make more acids. Before making any changes to your diet, talk to your healthcare provider. They can guide you on safely incorporating or increasing the right foods or drinks in your diet. They may also refer you to a dietitian who specializes in kidney diseases (renal dietitian).
Foods and drinks that cause your body to make acids include:
- Meats, including poultry and fish.
- Eggs.
- Cheese.
- Grains.
- Alcohol.
Foods or drinks that produce alkali include:
- Fruits.
- Nuts.
- Legumes.
- Vegetables.
- Alkaline water.
Metabolic acidosis prognosis
The prognosis (outlook) of metabolic acidosis is dependent upon the underlying cause and the severity of the acid-base derangement. The prognosis is poor if derangements are large and vitals are unstable. Many cases of metabolic acidosis respond well to treatment after a proper diagnosis. In mild cases, your symptoms may be temporary, and you may not need treatment. Severe cases may involve kidney or other organ failure and death.
In a prospective, observational, cohort study, Maciel and Park 57 looked at differences between survivors and nonsurvivors within a group of 107 patients suffering from metabolic acidosis on admission to an intensive care unit (ICU) 58. The authors found that although acidosis was more severe in nonsurvivors than in survivors, the proportion of acidifying variables was similar on admission between the 2 groups (with hyperchloremia being the primary cause of the acidosis) 58.
The investigators also found that in nonsurviving patients, the degree of metabolic acidosis was similar on the day of death to the level measured when they were admitted to the ICU, but that the proportion of anions had changed 58. Specifically, the chloride levels in the patients had decreased, and the lactate levels had increased 58.
References- Kraut, J., Madias, N. Metabolic acidosis: pathophysiology, diagnosis and management. Nat Rev Nephrol 6, 274–285 (2010). https://doi.org/10.1038/nrneph.2010.33
- Schwalfenberg GK. The alkaline diet: is there evidence that an alkaline pH diet benefits health? J Environ Public Health. 2012;2012:727630. doi: 10.1155/2012/727630
- Ali M., Alam S.P., Kumar S., Anupam Kumar R., Kumar A. Does blood pH change in cancer patients. Int. J. Curr. Res. 2016;8:29543–29544.
- Pandey DG, Sharma S. Biochemistry, Anion Gap. [Updated 2019 Apr 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539757
- Berend K. Review of the Diagnostic Evaluation of Normal Anion Gap Metabolic Acidosis. Kidney Dis (Basel). 2017 Dec;3(4):149-159. doi: 10.1159/000479279
- Gamble JL. Extracellular fluid and its maintenance. N Engl J Med. 1936;250:1150–1152.
- Berend K, de Vries AP, Gans RO. Physiological approach to assessment of acid-base disturbances. N Engl J Med. 2015 Jan 8;372(2):195. doi: 10.1056/NEJMc1413880
- Hopkins E, Sanvictores T, Sharma S. Physiology, Acid Base Balance. [Updated 2022 Sep 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507807
- Kurtz I. Renal Tubular Acidosis: H+/Base and Ammonia Transport Abnormalities and Clinical Syndromes. Adv Chronic Kidney Dis. 2018 Jul;25(4):334-350. doi: 10.1053/j.ackd.2018.05.005
- Renal Tubular Acidosis. https://www.niddk.nih.gov/health-information/kidney-disease/renal-tubular-acidosis
- Pharmacologically-induced metabolic acidosis: a review. Liamis G, Milionis HJ, Elisaf M. Drug Saf. 2010 May 1; 33(5):371-91.
- Acid-base. Gluck SL. Lancet. 1998 Aug 8; 352(9126):474-9.
- Metabolic acidosis-induced insulin resistance and cardiovascular risk. Souto G, Donapetry C, Calviño J, Adeva MM. Metab Syndr Relat Disord. 2011 Aug; 9(4):247-53.
- Metabolic Acidosis. https://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/acid-base-regulation-and-disorders/metabolic-acidosis#sec12-ch157-ch157c-1035
- Ratnam S, Kaeny W, Shapiro JI. Pathogenesis and management of metabolic acidosis and alkalosis. In: Schrier RW, editor. Renal and Electrolyte Disorders. ed 7. Philadelphia: Lippincott, Williams & Wilkins; 2010. pp. 86–121.
- Jean-Louis Vincent; Abraham Edward; Kochanek Patrick (8 July 2011). “Acid-base disorders”. Textbook of Critical Care. Elsevier. ISBN 143771367X.
- Hall MC, Koch MO, McDougal WS. Metabolic consequences of urinary diversion through intestinal segments. Urol Clin North Am. 1991 Nov;18(4):725-35.
- Mahler SA, Conrad SA, Wang H, Arnold TC. Resuscitation with balanced electrolyte solution prevents hyperchloremic metabolic acidosis in patients with diabetic ketoacidosis. Am J Emerg Med. 2011 Jul;29(6):670-4. doi: 10.1016/j.ajem.2010.02.004
- Taylor D, Durward A, Tibby SM, Thorburn K, Holton F, Johnstone IC, Murdoch IA. The influence of hyperchloraemia on acid base interpretation in diabetic ketoacidosis. Intensive Care Med. 2006 Feb;32(2):295-301. doi: 10.1007/s00134-005-0009-1
- Ritzenthaler, T., Grousson, S. and Dailler, F. (2016), Hyperchloremic metabolic acidosis following plasma exchange during myasthenia gravis crisis. J. Clin. Apheresis, 31: 479-480. https://doi.org/10.1002/jca.21432
- Bala I, Dwivedi D, Jain D, Mahajan JK. Hyperchloremic Metabolic Acidosis Following Total Gut Irrigation with Normal Saline in Pediatric Patients: A Rare Occurrence. Indian J Crit Care Med. 2017 Jan;21(1):55-56. doi: 10.4103/0972-5229.198329
- Thongprayoon C, Cheungpasitporn W, Cheng Z, Qian Q. Chloride alterations in hospitalized patients: Prevalence and outcome significance. PLoS One. 2017 Mar 22;12(3):e0174430. doi: 10.1371/journal.pone.0174430
- Kamar FB, McQuillan RF. Hyperchloremic Metabolic Acidosis due to Cholestyramine: A Case Report and Literature Review. Case Rep Nephrol. 2015;2015:309791. doi: 10.1155/2015/309791
- Coe FL. Magnitude of Metabolic Acidosis in Primary Hyperparathyroidism. Arch Intern Med. 1974;134(2):262–265. doi:https://doi.org/10.1001/archinte.1974.00320200072008
- Burger MK, Schaller DJ. Metabolic Acidosis. [Updated 2022 Jul 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482146
- Marano M. Sobre el uso de la fórmula de Winters en la acidosis metabólica crónica [On the use of Winters’ formula in chronic metabolic acidosis]. Rev Psiquiatr Salud Ment. 2015 Jan-Mar;8(1):45-6. Spanish. doi: 10.1016/j.rpsm.2014.07.006
- Sharma S, Hashmi MF, Aggarwal S. Hyperchloremic Acidosis. [Updated 2022 Aug 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482340
- Sharma S, Aggarwal S. Hyperchloremic Acidosis. [Updated 2018 Feb 13]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482340
- Sharma S, Aggarwal S. Hyperchloremic Acidosis. [Updated 2019 Jun 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482340
- Alexander RT, Bitzan M. Renal Tubular Acidosis. Pediatr. Clin. North Am. 2019 Feb;66(1):135-157.
- Pastorekova S, Gillies RJ. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019 Jun;38(1-2):65-77. doi: 10.1007/s10555-019-09799-0
- Ji K, Mayernik L, Moin K, Sloane BF. Acidosis and proteolysis in the tumor microenvironment. Cancer Metastasis Rev. 2019 Jun;38(1-2):103-112. doi: 10.1007/s10555-019-09796-3
- Jin C, Zahid E, Sherazi A, Majumder MR, Bedi P. Cardiac Arrest Due to Benzonatate Overdose. Am J Case Rep. 2019 May 3;20:640-642. doi: 10.12659/AJCR.915151
- Koeppen BM. The kidney and acid-base regulation. Adv Physiol Educ. 2009 Dec;33(4):275-81. doi: 10.1152/advan.00054.2009
- Hamm LL, Nakhoul N, Hering-Smith KS. Acid-Base Homeostasis. Clin J Am Soc Nephrol. 2015 Dec 7;10(12):2232-42. doi: 10.2215/CJN.07400715
- Kisaka T, Cox TA, Dumitrescu D, Wasserman K. CO2 pulse and acid-base status during increasing work rate exercise in health and disease. Respir Physiol Neurobiol. 2015 Nov;218:46-56. doi: 10.1016/j.resp.2015.07.005
- Brinkman JE, Toro F, Sharma S. Physiology, Respiratory Drive. [Updated 2022 Jun 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482414
- Brinkman JE, Sharma S. Respiratory Alkalosis. [Updated 2022 Jul 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482117
- Adamczak M, Surma S. Metabolic Acidosis in Patients with CKD: Epidemiology, Pathogenesis, and Treatment. Kidney Dis (Basel). 2021 Jun 4;7(6):452-467. doi: 10.1159/000516371
- Hopkins E, Sharma S. Physiology, Acid Base Balance. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507807
- Wesson DE, Nathan T, Rose T, Simoni J, Tran RM. Dietary protein induces endothelin-mediated kidney injury through enhanced intrinsic acid production. Kidney Int. 2007 Feb;71(3):210-7. doi: 10.1038/sj.ki.5002036
- Acidosis: An Old Idea Validated by New Research. Integr Med (Encinitas). 2015;14(1):8-12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4566456/
- Frassetto LA, Morris RC, Jr, Sebastian A. Effect of age on blood acid-base composition in adult humans: role of age-related renal functional decline. Am J Physiol. 1996;271(6 Pt 2):F1114–F1122
- Kopple J.D., Kalantar-Zadeh K., Mehrotra R. Risks of chronic metabolic acidosis in patients with chronic kidney disease. Kidney Int. 2005;67:S21–S27. doi: 10.1111/j.1523-1755.2005.09503.x
- Dubey A.K., Sahoo J., Vairappan B., Haridasan S., Parameswaran S., Priyamvada P.S. Correction of metabolic acidosis improves muscle mass and renal function in chronic kidney disease stages 3 and 4: A randomized controlled trial. Nephrol. Dial. Transplant. 2020;35:121–129. doi: 10.1093/ndt/gfy214
- Noce A., Marrone G., Ottaviani E., Guerriero C., Di Daniele F., Pietroboni Zaitseva A., Di Daniele N. Uremic sarcopenia and its possible nutritional approach. Nutrients. 2021;13:147. doi: 10.3390/nu13010147
- Diet acids and alkalis influence calcium retention in bone. Buclin T, Cosma M, Appenzeller M, Jacquet AF, Décosterd LA, Biollaz J, Burckhardt P. Osteoporos Int. 2001; 12(6):493-9.
- Metabolic Acidosis Clinical Presentation. https://emedicine.medscape.com/article/242975-clinical#b3
- Patel S, Sharma S. Respiratory Acidosis. [Updated 2022 Jun 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482430
- Rajkumar P, Pluznick JL. Acid-base regulation in the renal proximal tubules: using novel pH sensors to maintain homeostasis. Am J Physiol Renal Physiol. 2018 Nov 1;315(5):F1187-F1190. doi: 10.1152/ajprenal.00185.2018
- Pereira PC, Miranda DM, Oliveira EA, Silva AC. Molecular pathophysiology of renal tubular acidosis. Curr Genomics. 2009 Mar;10(1):51-9. doi: 10.2174/138920209787581262
- Metabolic Acidosis Workup. https://emedicine.medscape.com/article/242975-workup#c12
- Seheult J, Fitzpatrick G, Boran G. Lactic acidosis: an update. Clin Chem Lab Med. 2017 Mar 1;55(3):322-333. doi: 10.1515/cclm-2016-0438
- Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int. 2007 Jun;71(12):1310-6. doi: 10.1038/sj.ki.5002220
- Starke A, Corsenca A, Kohler T, Knubben J, Kraenzlin M, Uebelhart D, Wüthrich RP, von Rechenberg B, Müller R, Ambühl PM. Correction of metabolic acidosis with potassium citrate in renal transplant patients and its effect on bone quality. Clin J Am Soc Nephrol. 2012 Sep;7(9):1461-72. doi: 10.2215/CJN.01100112
- Harris AN, Grimm PR, Lee HW, Delpire E, Fang L, Verlander JW, Welling PA, Weiner ID. Mechanism of Hyperkalemia-Induced Metabolic Acidosis. J Am Soc Nephrol. 2018 May;29(5):1411-1425. doi: 10.1681/ASN.2017111163
- Maciel AT, Park M. Differences in acid-base behavior between intensive care unit survivors and nonsurvivors using both a physicochemical and a standard base excess approach: a prospective, observational study. J Crit Care. 2009 Dec;24(4):477-83. doi: 10.1016/j.jcrc.2009.01.005





{kind=link}