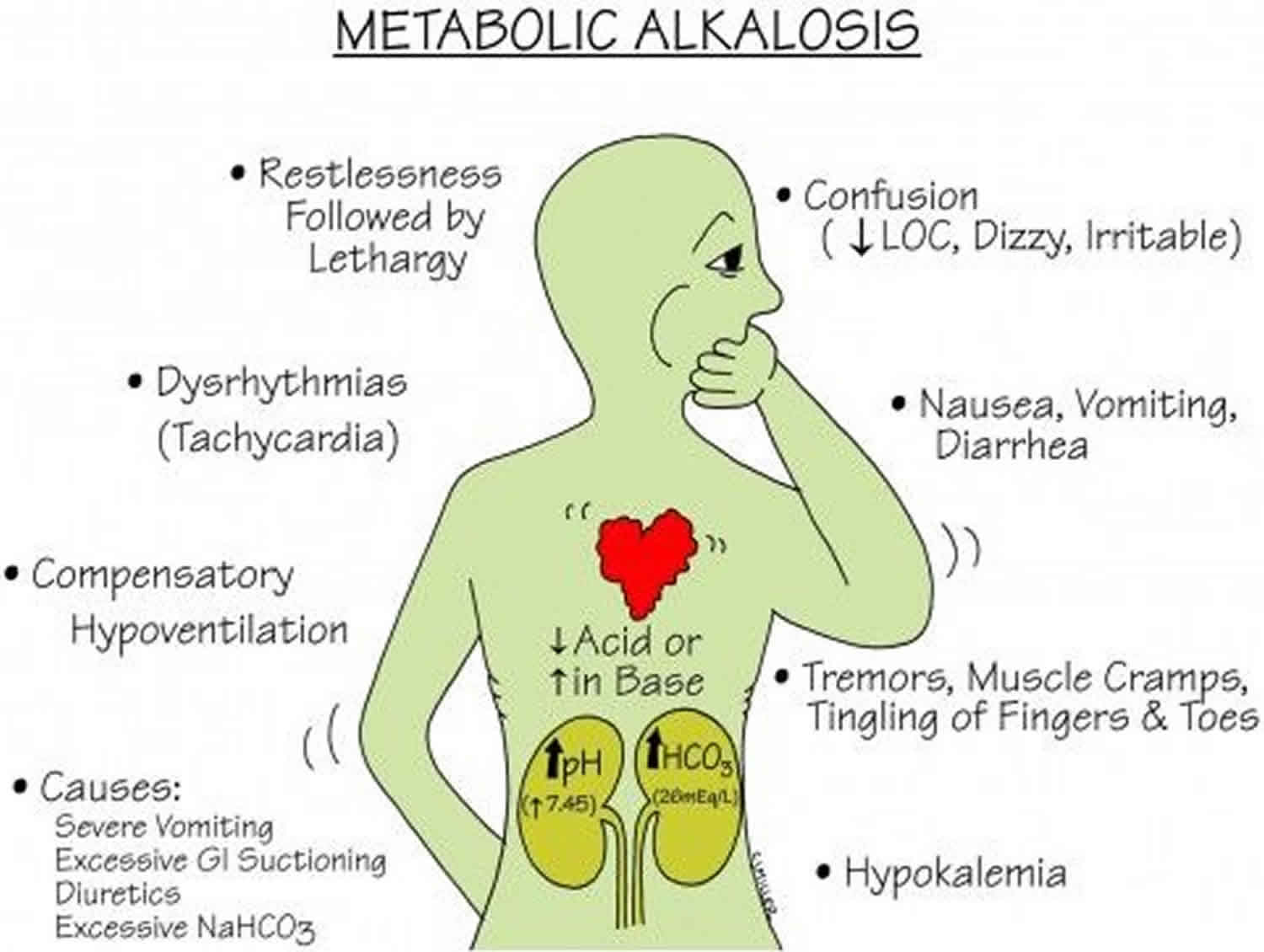
Metabolic alkalosis
Metabolic alkalosis is defined as a disease state that increases the serum bicarbonate concentration (HCO3–) above 30 meq/L causing the arterial blood pH to rise into the alkaline range greater than 7.45 secondary to some metabolic process 1, 2. Metabolic alkalosis is a very common disorder in hospitalized patients, especially in ICU settings 3. The incidence of metabolic alkalosis has shown to be 22.5% to 44.7% in inpatient studies in the United States 4, 5. The pH is a number that shows how acidic or alkaline a substance is. A pH of less than 7 is acidic, and greater than 7 is alkaline. The pH of blood is about 7.4 (a slightly alkaline range of 7.35 to 7.45). Your blood pH is highly stable within a normal range of pH 7.35 to 7.45 and it’s tightly regulated by your kidneys and respiratory system. The primary pH buffering system in the human body is the bicarbonate (HCO3–) and carbon dioxide (CO2). Bicarbonate (HCO3–) functions as an alkalotic substance. Carbon dioxide (CO2) functions as an acidic substance. Therefore, an increase in serum bicarbonate (HCO3–) or a decrease in CO2 (carbon dioxide) will make blood more alkaline. The opposite is also true where decreases in bicarbonate (HCO3–) or an increase in carbon dioxide (CO2) will make blood more acidic. The carbon dioxide (CO2) levels are physiologically regulated by the pulmonary system through respiration, whereas the serum bicarbonate (HCO3–) levels are regulated through your kidneys by two mechanisms: bicarbonate [HCO3–] (a base) reclamation mainly in the proximal tubule and bicarbonate [HCO3–] (a base) generation predominantly in the distal nephron. Elevated pH above 7.45 and elevated plasma bicarbonate (HCO3–) level above 30 meq/L characterize metabolic alkalosis. When bicarbonate (HCO3–) is elevated the arterial partial pressure of carbon dioxide (PaCO2) must also be elevated to maintain pH to its normal range. Therefore with metabolic alkalosis, the compensation is to decrease alveolar ventilation (hypoventilation) in order to increase the arterial partial pressure of carbon dioxide (PaCO2).
To understand acid-base buffering system, it is important to recall that pH is governed by the ratio bicarbonate [HCO3–] (a base)/arterial partial pressure of carbon dioxide (PaCO2) (an acid). So long as the ratio is normal, pH will be normal.
Normal body functions and metabolism generate large quantities of acids that must be neutralized and/or eliminated to maintain blood pH balance. Most of the acid is carbonic acid (H2CO3), which is created from carbon dioxide (CO2) and water (H2O). Carbon dioxide (CO2) is produced as the body uses glucose (sugar) or fat for energy. In its normal state, the body maintains carbon dioxide (arterial partial pressure of carbon dioxide [PaCO2]) in a well-controlled range from 35 to 45 mm Hg by balancing its production and elimination. Lesser quantities of lactic acid, ketoacids, and other organic acids are also produced.
- Carbon dioxide (CO2) + water (H2O) -> H2CO3 (carbonic acid) -> HCO3– + H+
According to the Henderson-Hasselbalch equation (Figure 2), maintaining physiological pH depends on arterial partial pressure of carbon dioxide (PaCO2), which in turn depends on alveolar ventilation (hypoventilation causes acidosis and hyperventilation causes alkalosis). The kidneys participate in maintaining the stable pH by reabsorption of bicarbonate (3,600 mmol of bicarbonate is filtrated in glomeruli during 24 hour) and excretion of hydrogen ions from nonvolatile acids (including sulfur and phosphate) as titratable acidity (0.3 mmol hydrogen ions/kg/day) and in the form of ammonium ion (0.7 mmol hydrogen ions/kg/day) 6, 7.
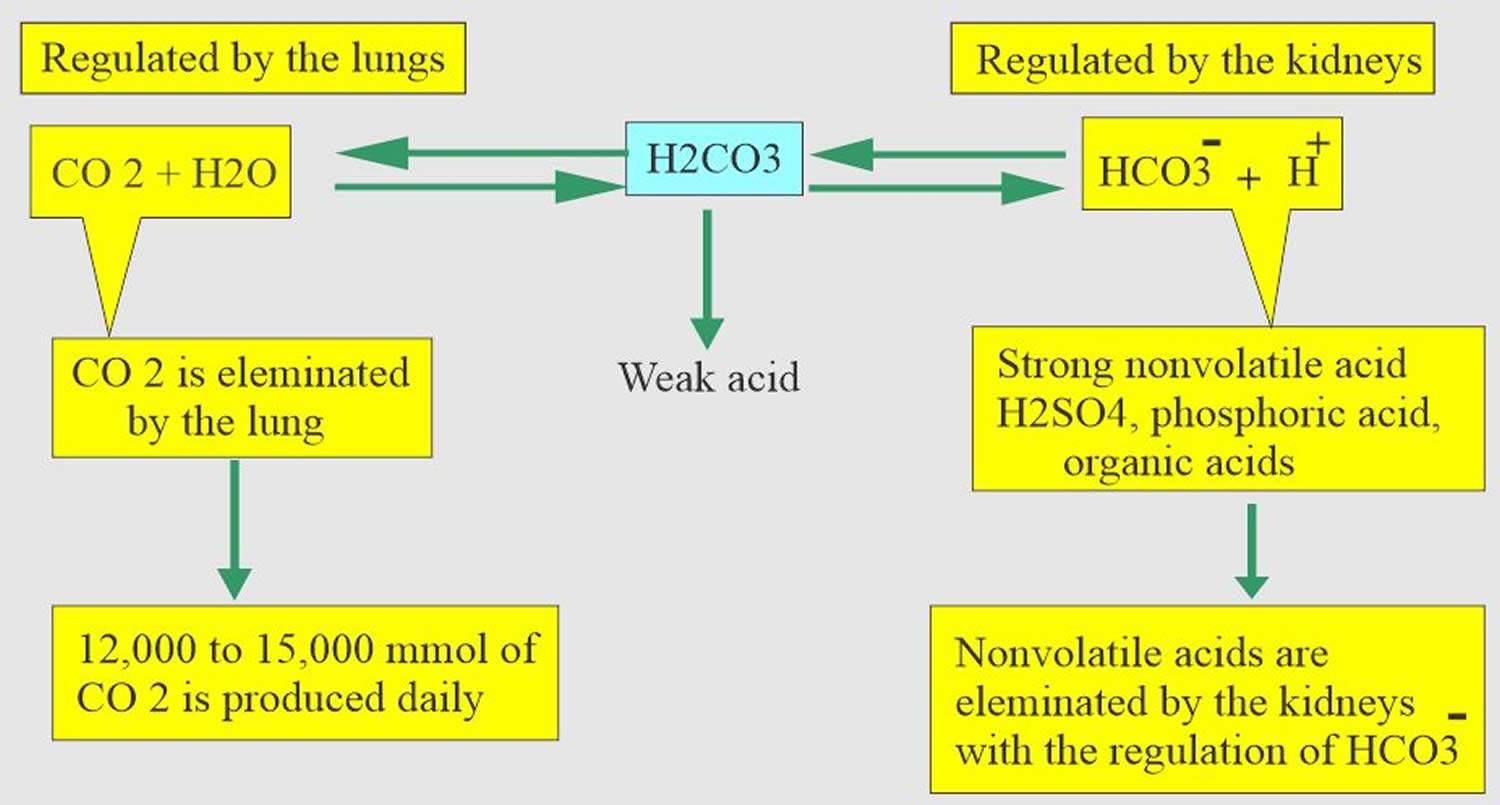
The lungs and kidneys are the major organs involved in regulating blood pH. And to compensate for the metabolic alkalosis, you decrease your breathing rate (hypoventilation) to produce more carbon dioxide (CO2) 8, 9.
- The lungs flush acid out of your body by exhaling carbon dioxide (CO2). Raising and lowering the respiratory rate alters the amount of carbon dioxide (CO2) that is breathed out, and this can affect blood pH within minutes 10.
- The kidneys excrete acids in the urine, and they regulate the concentration of bicarbonate (HCO3–, a base) in blood. Acid-base changes due to increases or decreases in bicarbonate [HCO3–] concentration occur more slowly than changes in carbon dioxide (CO2), taking hours or days. Bicarbonate (HCO3–) reabsorption occurs in the kidneys in every part of the tubules. About 85–90% of the filtered bicarbonate is reabsorbed in the proximal tubules, 10% in the ascending arms of the Henle loop, 6% in the distal tubules, and 4% in the collecting tubules 6, 7.
Both of these processes are always at work, and they keep the blood pH in healthy people tightly controlled. The absolute quantities of acids or bases are less important than the balance between the two and its effect on blood pH.
Buffering systems that resist changes in pH also contribute to the regulation of acid and base concentrations. The main buffers in blood are hemoglobin (in red blood cells), plasma proteins, carbon dioxide (CO2), bicarbonate (HCO3–) and phosphates.
Carbon dioxide (CO2) plays a remarkable role in the human body mainly through pH regulation of the blood. The pH is the primary stimulus to initiate ventilation. In its normal state, the body maintains carbon dioxide (CO2) in a well-controlled range from 38 to 42 mm Hg by balancing its production and elimination. In a state of hypoventilation (breathing that is too shallow or too slow to meet the needs of the body), the body produces more carbon dioxide (CO2) than it can eliminate, causing a net retention of carbon dioxide (CO2). The increased carbon dioxide (CO2) is what leads to an increase in hydrogen ions (H+) and a slight increase in bicarbonate (HCO3–), as seen by a right shift in the following equilibrium reaction of carbon dioxide:
- Carbon dioxide (CO2) + water (H2O) -> H2CO3 (carbonic acid) -> HCO3– + H+
The buffer system created by carbon dioxide consists of the following three molecules in equilibrium: carbon dioxide (CO2), H2CO3 (carbonic acid), and bicarbonate (HCO3–). When hydrogen ions (H+) is high, bicarbonate (HCO3–) buffers the low pH. When hydroxide (OH–) is high, H2CO3 (carbonic acid) buffers the high pH.
Usually metabolic alkalosis indicates an accumulation of “excess” bicarbonate (HCO3–). The source of excess bicarbonate (HCO3–) can be exogenous, endogenous, or both 11.
Metabolic alkalosis the mechanisms of excess bicarbonate (HCO3–) generation 11, 12, 13:
- Increased bicarbonate in the extracellular compartment via ingestion and absorption or infusion of bicarbonate (HCO3–) or alkali (milk-alkali syndrome) or increased parenteral intake of sodium bicarbonate [NaHCO3] or NaHCO3 precursors (i.e., sodium acetate, sodium citrate, sodium gluconate)
- Distal renal tubule bicarbonate (HCO3–) generation through enhanced hydrogen ions (H+) secretion
- Diuretic-induced alkalosis—diuretics (loop diuretic and thiazide diuretic) that block sodium and chloride [NaCl] reabsorption or other sodium salts such as sodium sulfate [Na2SO4] or sodium penicillin can cause increased bicarbonate absorption at the proximal tubule leading to increased serum bicarbonate concentration, also called contraction alkalosis.
- Increased renal reabsorption of bicarbonate (HCO3–) can also cause metabolic alkalosis (severe hypokalemia or potassium (K+) depletion [shifting H+ into cells], primary hyperaldosteronism, Cushing syndrome, Bartter syndrome, Gitelman syndrome, toxic ingestion of licorice, excessive chloruretic diuretic use) 14, 15, 16, 17.
- Excess loss of hydrogen ions (H+) via the removal of hydrochloric acid (HCl) from body this occurs primarily due to gastric losses due to prolonged and severe gastric aspiration, nasogastric suction, excessive vomiting of gastric contents as in pyloric stenosis and congenital chloride-rich diarrhea 18, 19
Exogenous sources are sodium (Na+) or potassium (K+) bicarbonate (HCO3–) salts (e.g., Sodium bicarbonate [NaHCO3] and Potassium bicarbonate [KHCO3]) or salts of precursors (organic anions such as lactate, acetate or citrate, which generate bicarbonate [HCO3–] when completely oxidized). These salts can be ingested/absorbed or infused.
The two potential endogenous sources of large amounts of bicarbonate (HCO3–) are:
- The stomach and
- The kidneys.
Net endogenous bicarbonate (HCO3–) generation requires hydrogen ions (H+) removal from the body. Bicarbonate (HCO3–) is generated when hydrochloric acid (HCl) is secreted into gastric lumen, but net bicarbonate (HCO3–) accumulation in the extracellular fluid (ECF) requires the hydrochloric acid (HCl) to be lost externally, usually as a result of vomiting and/or suction.
Normally, kidney hydrogen ions (H+) excretion into the urine (as ammonium [NH4+] and/or titratable acid) generates bicarbonate (HCO3–) to replace the quantity decomposed by nonvolatile hydrogen ions (H+) derived from dietary intake and metabolism and any bicarbonate (HCO3–) lost in alkaline stool. To the extent kidney bicarbonate (HCO3–) generation exceeds this requirement, “excess” bicarbonate (HCO3–) is generated. This generally occurs when the following conditions coexist: (1) the kidney tubules/ducts beyond the early distal tubule are avidly reabsorbing Na+ (for example, aldosterone activity is high), and (2) the delivery of salt and volume to these sites is relatively large.
Urine chloride is a direct measurement of chloride being excreted into urine. This test is useful to help determine the cause of metabolic alkalosis 20, 21, 22.
The most common causes of metabolic alkalosis are the use of diuretics (water pills tha help rid your body of sodium and water) and the external loss of gastric hydrochloric acid (HCl) secretions 23.
Metabolic alkalosis is further divided into 2 main categories 24, 25, 26, 27, 28, 29, 23:
- Chloride-responsive alkalosis with urine chloride less than 20 mEq/L. Chloride-responsive alkalosis causes include loss of hydrogen via the gastrointestinal tract (vomiting, nasogastric suction), congenital chloride diarrhea syndrome (congenital chloridorrhea), loss of colonic secretions via villous adenoma, contraction alkalosis, diuretic therapy (thiazides and loop diuretics [after discontinuation]), post-hypercapnia syndrome, cystic fibrosis, exogenous alkalotic agent use and laxative abuse are also potential causes.
- Chloride-resistant alkalosis with urine chloride greater than 20 mEq/L.
- Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
- Primary hyperaldosteronism: adrenal adenoma, bilateral adrenal hyperplasia, adrenal carcinoma, glucocorticoid-remediable hyperaldosteronism
- 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2): 11 β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) normally converts active cortisol to inactive cortisone and protects mineralocorticoid receptor (MR) occupied by cortisol. The Apparent Mineralocorticoid Excess syndrome is an ultrarare autosomal recessive disorder caused by 11β-hydroxysteroid dehydrogenase type 2 gene (HSD11B2) resulting in impairment of the enzyme 11β-HSD2 leading to excessive cortisol that is able to activate mineralocorticoid receptor (MR). The Apparent Mineralocorticoid Excess syndrome is typically characterized by hypertension with hypokalemia, metabolic alkalosis, low renin activity, and low aldosterone level. The cause is inherited in autosomal recessive form, mutations in 11β-HSD2 gene or acquired form by ingestion of competitive inhibitors of 11β-HSD2 such as liquorice, chewing tobacco or carbenoxolone. Uncontrolled hypertension and prolong hypokalemia are the leading causes of end organ damages and cardiovascular mortality 30.
- Congenital adrenal hyperplasia (CAH) 11-hydroxylase or 17-hydroxylase deficiency
- Current use of diuretics in hypertension
- Cushing syndrome
- Exogenous mineralocorticoids or glucocorticoids
- Liddle syndrome
- Renovascular hypertension
- Causes of chloride-resistant alkalosis (urine chloride >20 mEq/L) without hypertension include the following:
- Bartter syndrome
- Gitelman syndrome
- Severe potassium depletion (hypokalemia)
- Current use of thiazides and loop diuretics
- Hypomagnesemia
- Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
Other causes include the following:
- Exogenous alkali administration – Sodium bicarbonate therapy in the presence of renal failure, metabolism of lactic acid or ketoacids
- Milk-alkali syndrome
- Hypercalcemia
- Intravenous penicillin
- Refeeding alkalosis
- Massive blood transfusion
The management of metabolic alkalosis depends primarily on the underlying cause and on the patient’s volume status. Managing alkalosis requires an interprofessional team of healthcare professionals, including several physicians in different specialties, laboratory technologists and pharmacists.
Figure 1. Abnormal acid-base compensation

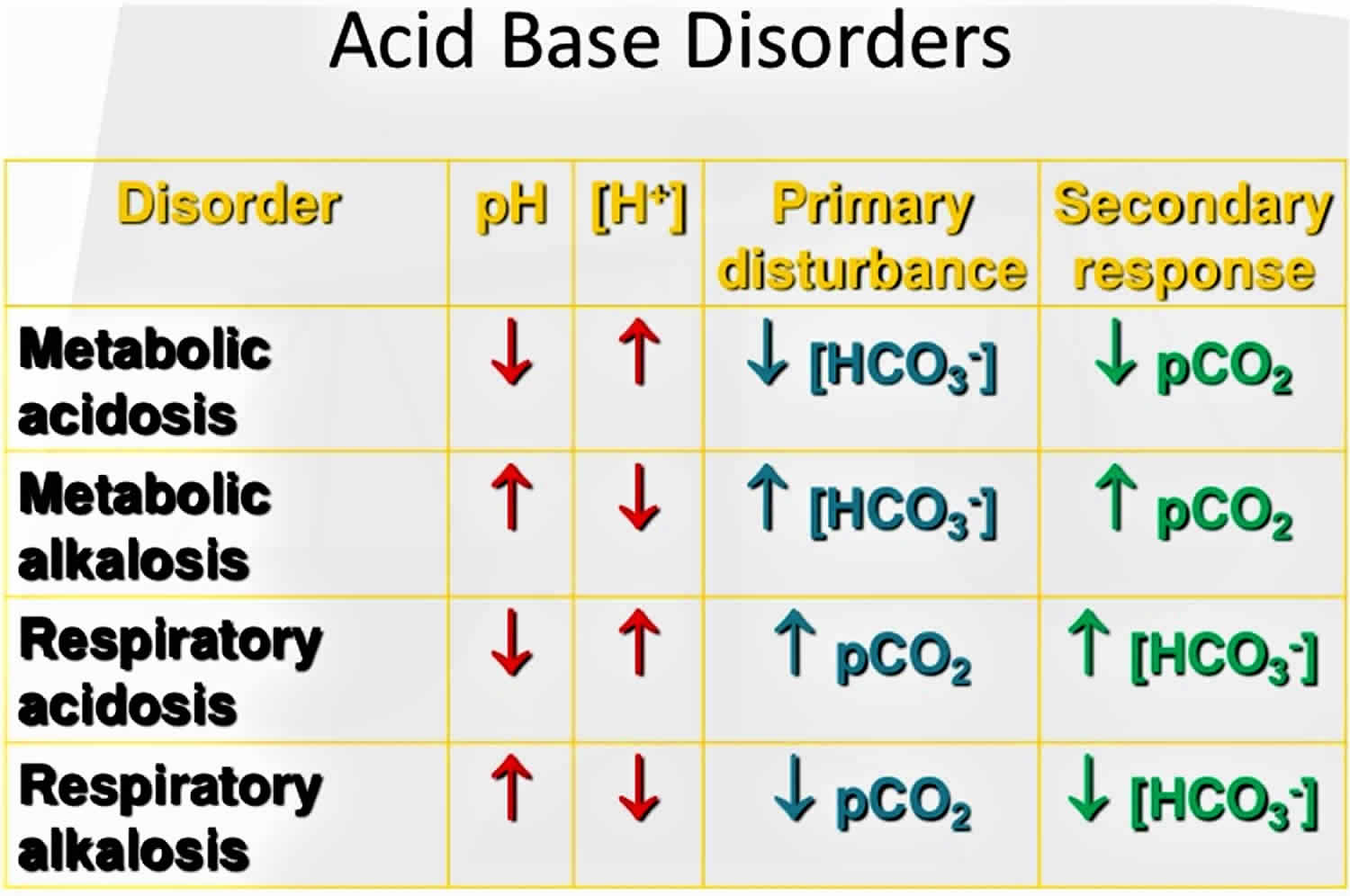
Figure 2. Henderson-Hasselbalch equation
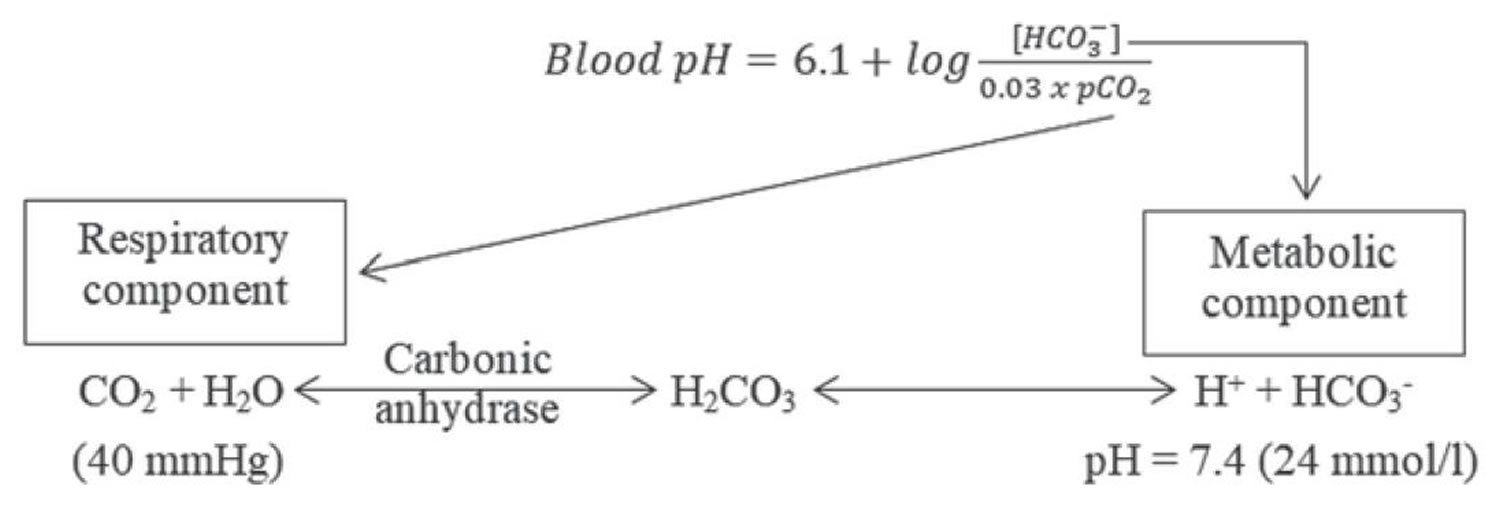
Figure 3. Acid-base buffering system
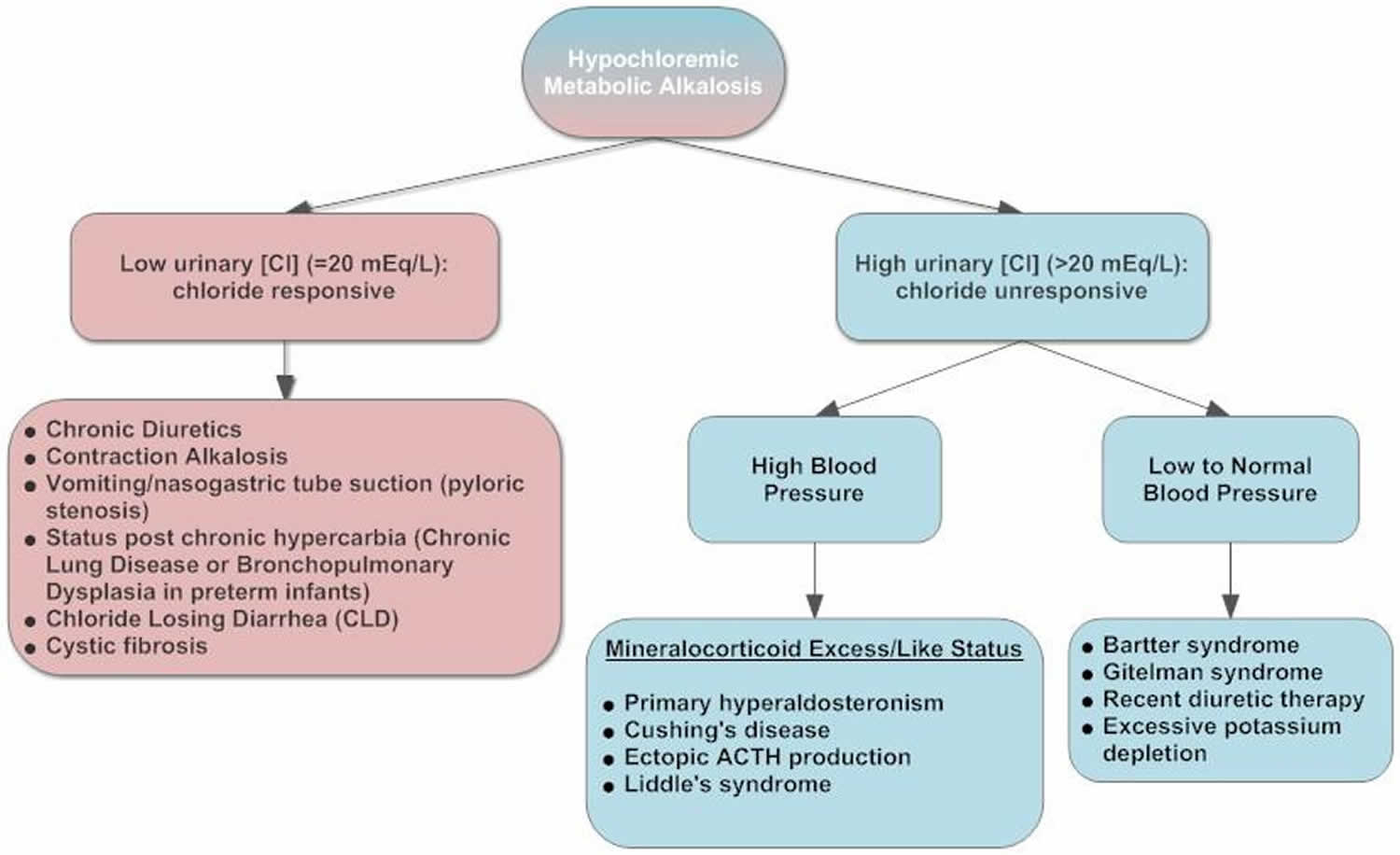
Hypochloremic metabolic alkalosis
Hypochloremia is defined as a serum chloride level of less than 95 mEq/L. Hypochloremic alkalosis results from either low chloride intake or excessive chloride wasting. Whereas low chloride intake is very uncommon, excessive chloride wasting often occurs in hospitalized children, usually as a result of diuretic therapy or nasogastric tube suctioning 32. Hypochloremic alkalosis is also seen in hypertrophic pyloric stenosis where it occurs in only about half the patients 33. Diarrhea, when watery is also highly suggestive of chloride-losing diarrhea.
Hypochloremic alkalosis can be classified into two categories 24, 25, 26, 27, 28, 29, 23:
- Chloride-responsive alkalosis with urine chloride less than 20 mEq/L. Chloride-responsive alkalosis causes include loss of hydrogen via the gastrointestinal tract (vomiting, nasogastric suction), congenital chloride diarrhea syndrome (congenital chloridorrhea), loss of colonic secretions via villous adenoma, contraction alkalosis, diuretic therapy (thiazides and loop diuretics [after discontinuation]), post-hypercapnia syndrome, cystic fibrosis, exogenous alkalotic agent use and laxative abuse are also potential causes.
- Chloride-resistant alkalosis with urine chloride greater than 20 mEq/L.
- Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
- Primary hyperaldosteronism: adrenal adenoma, bilateral adrenal hyperplasia, adrenal carcinoma, glucocorticoid-remediable hyperaldosteronism
- 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2): 11 β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) normally converts active cortisol to inactive cortisone and protects mineralocorticoid receptor (MR) occupied by cortisol. The Apparent Mineralocorticoid Excess syndrome is an ultrarare autosomal recessive disorder caused by 11β-hydroxysteroid dehydrogenase type 2 gene (HSD11B2) resulting in impairment of the enzyme 11β-HSD2 leading to excessive cortisol that is able to activate mineralocorticoid receptor (MR). The Apparent Mineralocorticoid Excess syndrome is typically characterized by hypertension with hypokalemia, metabolic alkalosis, low renin activity, and low aldosterone level. The cause is inherited in autosomal recessive form, mutations in 11β-HSD2 gene or acquired form by ingestion of competitive inhibitors of 11β-HSD2 such as liquorice, chewing tobacco or carbenoxolone. Uncontrolled hypertension and prolong hypokalemia are the leading causes of end organ damages and cardiovascular mortality 30.
- Congenital adrenal hyperplasia (CAH) 11-hydroxylase or 17-hydroxylase deficiency
- Current use of diuretics in hypertension
- Cushing syndrome
- Exogenous mineralocorticoids or glucocorticoids
- Liddle syndrome
- Renovascular hypertension
- Causes of chloride-resistant alkalosis (urine chloride >20 mEq/L) without hypertension include the following:
- Bartter syndrome
- Gitelman syndrome
- Severe potassium depletion (hypokalemia)
- Current use of thiazides and loop diuretics
- Hypomagnesemia
- Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
Replacement of electrolytes with chloride salts is the most important mode of therapy for hypochloremic alkalosis. Nonsteroidal anti-inflammatory drugs (NSAIDs) are used in patients with Bartter syndrome. Hydrochloric acid (HCl) and carbonic anhydrase inhibitors may be used in some acute situations.
In patients with chloride-losing diarrhea, fluid intake should be encouraged so as to prevent renal damage resulting from recurrent dehydration. Patients or caregivers should be instructed to avoid long periods of exposure to hot climates, which may exacerbate dehydration episodes.
Constipation must be treated in patients with Bartter syndrome. Any intercurrent febrile illnesses, especially urinary tract infections, must be treated to prevent further renal damage.
Figure 3. Hypochloremic metabolic alkalosis diagnostic algorithm
Hypochloremic alkalosis signs and symptoms
Prenatal polyhydramnios is present in most patients with congenital forms of metabolic alkalosis, especially chloride-losing diarrhea. Premature birth resulting from polyhydramnios is common in patients with Bartter syndrome and chloride-losing diarrhea. Lack of meconium is highly suggestive of intrauterine diarrhea. Prolonged neonatal jaundice may be present. A history of hypotonia and lethargy without sepsis is significant in patients with early-onset hypochloremia and hypokalemia.
In infants, a history of repeated vomiting may be suggestive of severe gastroesophageal reflux or pyloric stenosis. Failure to thrive is common. Constipation is very common in patients with Bartter syndrome. Diarrhea, when watery (see the image below), is highly suggestive of Chloride-losing diarrhea.
A salty taste upon being kissed may help identify patients with cystic fibrosis. Guidelines for newborn screening for cystic fibrosis have been established by the Centers of Disease Control and Prevention (CDC) 34. Central nervous system (CNS) dysfunctions (eg, lethargy, confusion, or seizure) are observed in patients with severe alkalosis. Neuromuscular symptoms include weakness and muscle cramps.
Central nervous system (CNS) manifestations range from mild to severe, depending on the severity of alkalosis, and may include the following:
- Confusion
- Apathy
- Disorientation
- Excessive sleeping
- Seizure
- Stupor
Other symptoms (eg, abdominal distention, dry skin, apathy, loss of interests, growth retardation 35 and frequent hospital admissions because of recurrent dehydration) are significant diagnostic clues during childhood.
Other history
A family history may be suggestive. Consanguinity, recurrent prematurity, neonatal demise, and psychomotor retardation are helpful clues to familial conditions.
A psychosocial history may reveal loss of interests and behavioral problems, which were reported in patients with chronic hypochloremic alkalosis. Difficulty in school performance may be a consequence of the disorder.
In hospitalized patients with hypochloremic metabolic alkalosis, the physician should always ask about nasogastric tube suctioning and oral secretions. Overzealous use of loop or thiazide diuretics, especially in the intensive care unit (ICU), is another important factor.
Physical examination
Patients with hypochloremic alkalosis commonly are small for their age, lethargic, or apathetic. Signs of chronic dehydration (eg, skin tenting and poor peripheral perfusion) may be evident upon presentation. One study reported that cystic fibrosis was diagnosed in an infant who presented with dehydration and metabolic alkalosis 36.
Weight and height usually fall below the reference range in patients with chronic disease but are not affected in patients with acute disease. In one series, both weight and height were in the lowest 3% in more than 60% of patients with chloride-losing diarrhea 37.
Depending on the cause of the hypochloremic alkalosis, the abdomen may be scaphoid (in Bartter syndrome) or distended (in chloride-losing diarrhea).
Additional abdominal findings that may be present are as follows:
- Peristaltic waves in children with chloride-losing diarrhea
- Exacerbated bowel sounds in patients with chloride-losing diarrhea
- Hard stools in patients with Bartter syndrome
- Hepatomegaly (suggesting cystic fibrosis)
Musculoskeletal findings include muscle wasting, atrophy, and hypotonia. Respiratory findings include shallow breathing and hypopnea in severely affected children.
Hypochloremic metabolic alkalosis complications
Disease-related complications of hypochloremic alkalosis include the following:
- Nephrocalcinosis and nephrolithiasis in patients with Bartter syndrome and in those with chloride-losing diarrhea
- Coexisting electrolyte abnormalities such as hypokalemia, hyponatremia, and hypercalcemia may be present
- Liver damage and recurrent chest infection leading to hepatic and pulmonary failure, respectively, in patients with cystic fibrosis
- End-stage renal disease (ESRD) in patients with poor compliance; ESRD can occur in all conditions mentioned, including Bartter syndrome and chloride-losing diarrhea
Hypochloremic alkalosis diagnosis
The flowchart in Figure 3 depicts a workup approach to hypochloremic alkalosis.
Laboratory studies
Amniocentesis
Amniotic fluid sodium and chloride concentrations may reflect fetal values; these are high in fetuses with chloride-losing diarrhea. Levels may also be elevated in patients with Bartter syndrome. Although testing for alpha1 -fetoprotein is not routine, levels may be elevated.
Blood workup
Serum electrolyte levels may be within the reference range, especially in neonates and treated patients. However, typical findings include low concentrations of serum chloride, sodium, and potassium. Attention must be paid in interpreting the serum potassium level in relation to the state of metabolic alkalosis. For example, the potassium shift from serum into the intracellular compartment increases as the serum pH rises; thus, the potassium level is less than normal by 0.6 mmol/L when measured at a serum pH of 7.5.
Serum pH and bicarbonate, calcium, uric acid, hemoglobin (if patient is not anemic), renin, and aldosterone levels may be elevated. The serum renin level is exponentially high, in line with secondary hyperaldosteronism due to chronic volume depletion, and this finding is supported by low or normal blood pressure measurements.
Urine and stool studies
In patients with Bartter syndrome, urine chloride, sodium, and potassium concentrations are usually measured. Urine calcium-to-creatinine and uric acid–to–creatinine ratios are usually high. Stool electrolytes cannot be measured because of well-formed or hard stool. Fractional excretion (Fex) studies are more reliable than absolute values. Usually, results are higher than reference range values, as follows:
- Fractional excretion (Fex) sodium concentration >1%
- Fractional excretion (Fex) potassium concentration >35%
- Fractional excretion (Fex) chloride concentration >2.5% (2.7% ± 1.1%)
In patients with chloride-losing diarrhea, urine chloride concentration is very low or undetectable (< 10 mmol/L). Stool is usually watery, and electrolyte studies are very helpful and diagnostic, as follows:
- Stool chloride concentration >100 mmol/L
- Stool sodium and potassium concentrations are elevated
- Stool chloride concentration is greater than stool sodium plus potassium concentrations, which is normally less than either; chloride concentrations are lowest in colonic secretions (usually < 35 mmol/L)
- The ratio of stool chloride to combined sodium and potassium concentrations is greater than 0.6
Patients with cystic fibrosis typically demonstrate high sweat chloride and sodium concentrations. Urine chloride concentration is usually very low, and stools are usually not watery, as they are in patients with chloride-losing diarrhea.
Kidney and liver function tests
- Renal function is usually normal. The glomerular filtration rate (GFR) may be low in patients with severe disease.
- Liver function test results are usually within the reference range in patients with chloride-losing diarrhea and Bartter syndrome but may be deranged in patients with cystic fibrosis.
Genetic studies
DNA diagnosis is available for most congenital disorders that cause hypochloremic metabolic alkalosis. For chloride-losing diarrhea, the chloride-losing diarrhea (SLC26A3) locus is on band 7q22-q31.1 38. Bartter syndrome is identified by NKCC2, ROMK, and CLCNKB 39; Bartter syndrome with deafness is identified by BSND; and Bartter syndrome with autosomal dominant hypocalcemia is identified by CASR. For cystic fibrosis, the CFTR locus is on band 7q31.2. For Gitelman syndrome, the NCCT locus is on 16q.
Ultrasonography
Prenatal ultrasonography may be useful in the detection of minimal polyhydramnios and assessment of intestinal fluid content, which is increased in patients with chloride-losing diarrhea.
Postnatal ultrasonography (see the images below) may be useful in the evaluation of a fluid-filled bowel, which is characteristically increased in patients with chloride-losing diarrhea. Ultrasonography may also assist in the evaluation of renal echogenicity, nephrocalcinosis, medullary or diffuse calcinosis, and renal growth.
Physiologic study of renal tubules
Physiologic study of renal tubules by performing maximal free water clearance during hypotonic saline diuresis is indicated.
Oral administration of water 20 mL/kg over 30 minutes is followed by administration of a one-half isotonic sodium chloride solution at a rate of 600 mL/m²/h for 2-3 hours. During this time, urine is collected in aliquots over 30-minute periods for 4-6 aliquots.
These samples are sent for evaluation of creatinine, sodium, potassium, and chloride levels, as well as for osmolality, pH, and volume. Usually, urine is diluted by oral administration of water. Halfway through each collection, a blood sample is obtained for evaluation of creatinine, sodium, potassium, and chloride levels, and for pH and osmolality. The clearance of each substance is calculated, and a ratio is derived by means of the following formula:
- Water clearance/(chloride clearance + water clearance)
Usually, the result of this formula reflects the percentage of distal tubule sodium and chloride reabsorption. Normal values are up to 85-90%, which means that the percentage of chloride and sodium excreted should be 10-15% (corrected to a GFR of 100 mL/min/1.73 m²). In patients with Bartter syndrome, the percentage of chloride and sodium excreted can reach 35% or more.
Other studies
Additional studies that may be considered include the following:
- Wrist radiography – This may be performed to determine bone age in infants with growth failure; it may also help assess bone density and the presence of rickets
- Upper gastrointestinal (GI) series – This helps detect gastroesophageal reflux and pyloric stenosis, which are case-dependent conditions 40
- Computed tomography (CT) of the brain – This is useful for evaluation of brain growth and calcifications
- Magnetic resonance imaging (MRI) of the brain – This is helpful in patients who present with seizures
- Electroencephalography (EEG) – This is also helpful in patients who present with seizures
- Renal nuclear scanning – This may facilitate assessment of renal function but is not useful in all patients
- Renal biopsy – This is not usually indicated, but if it is performed, it may reveal interstitial fibrosis and calcium/urate crystal deposition
Hypochloremic metabolic alkalosis treatment
Hydration status and electrolyte levels must be assessed. Replacement of electrolytes with chloride salts is the most important mode of therapy for hypochloremic alkalosis. A full nutritional assessment should be obtained, energy intake calculated, and adequate energy intake ensured through oral or nasogastric methods.
Nonsteroidal anti-inflammatory drugs (NSAIDs; eg, indomethacin) are used in patients with Bartter syndrome. Hydrochloric acid (HCl) and carbonic anhydrase inhibitors (eg, acetazolamide) may be used in some acute situations. Potential complications of pharmacotherapy include the following:
- Indomethacin-induced nephrotoxicity
- Acetazolamide treatment compromising respiratory function in children with lung disease
Discharge medication instructions should be clearly written, and a supply sufficiently large to last until the patient is seen in the outpatient clinic should be prescribed.
Acute emergency management (6 hours or less)
Initial management includes assessment of dehydration status and severity of hypochloremia, hypokalemia, hyponatremia, and metabolic alkalosis. If the patient is in shock, treatment should be directed toward aggressive resuscitation with isotonic fluid, preferably normal saline. Blood and urine samples for testing of electrolytes should always be obtained before any form of therapy is initiated; this is of great help in differentiating etiologic factors in new cases.
Chronic acid-base disturbances must not be treated too rapidly; more serious complications may be prevented by meticulous and slow correction. For example, consider the case of a child whose initial blood work shows the following results:
- Sodium 120 mmol/L
- Potassium 2 mmol/L
- Chloride 80 mmol/L
- Bicarbonate 40 mmol/L
- pH 7.5
In this child, assessment of cardiac function is indicated. If there is no dysrhythmia, rapid correction of this severe hypokalemia is unnecessary. Administration of 5% dextrose in 0.9 isotonic sodium chloride solution plus potassium chloride 20 mEq/L at a maintenance rate can be a safe measure.
Maintenance management (7-72 hours)
Maintenance therapy depends on how much improvement occurred after 6 hours of initial fluid and electrolyte administration. The aim is to increase the serum potassium concentration very slowly as the serum bicarbonate level drops. This helps prevent a sharp increase in serum potassium concentration and its subsequent detrimental effects on cardiac conductivity.
Long-term management (after 72 hours)
For long-term management, intravenous (IV) fluids can be discontinued. The physician should calculate the average daily amounts of chloride, sodium, and potassium that were required to correct the serum electrolyte levels. The total amounts can then be administered orally in 3-4 divided doses per day. In most patients, the average chloride dose required is 4-10 mEq/kg/day in the form of sodium and potassium salts.
Other management procedures depend on the primary cause of hypochloremic alkalosis.
Surgical or endoscopic intervention
Surgical intervention is usually unnecessary. If ileus is suspected in a child with severe hypokalemia, the appropriate treatment is administration of potassium chloride, not surgical intervention. However, if the cause of hypochloremic alkalosis is an upper gastrointestinal (GI) tract abnormality, such as gastroesophageal reflux or pyloric stenosis, surgical or endoscopic intervention is indicated.
Diet
Kilojoule intake should be appropriate for the patient’s catabolic status, usually 100-150% of the recommended daily allowance (RDA). Additional protein should be ingested to prevent malnutrition. Fat requirements depend on the individual patient. For example, patients with cystic fibrosis have special dietary needs that should be met.
Multivitamins and hematinic agents should be provided as required. Supplemental trace elements (eg, zinc) should be provided to patients with a trace-element deficiency, such as some patients with chloride-losing diarrhea (chloride-losing diarrhea). High sodium and potassium diets are required for all children with chronic metabolic alkalosis secondary to Bartter syndrome or chloride-losing diarrhea.
Activity
Normal activity should be recommended for children with hypochloremic alkalosis unless central nervous system (CNS) damage is severe, in which case special restrictions are required.
Children with refractory severe hypokalemia should avoid extended exposure to heat, especially in hot climates. Exposure to heat may cause dehydration and may exacerbate the condition.
Genetic counseling
Genetic counseling should be considered when prenatal diagnosis is offered to mothers with familial diseases, such as cystic fibrosis, Bartter syndrome, or chloride-losing diarrhea.
Long-term monitoring
Patients should receive regular follow-up examinations by a physician and nurse clinician. Such examinations should take place at least once every month in infants but may be less frequent in older children and children who are more stable.
The preclinic laboratory workup includes a biochemical profile and monitoring of urine electrolytes. The pharmacotherapeutic regimen should be reviewed at each visit. Medications should be refilled and dosages adjusted in accordance with the patient’s clinical status and laboratory results.
Diagnostic imaging studies should be repeated as necessary. For example, kidney ultrasonography may be needed to assess the degree of nephrocalcinosis in children with Bartter syndrome.
Growth parameters should be assessed, and the question of whether growth hormone therapy is needed should be evaluated in consultation with a pediatric endocrinologist. Renal function should be assessed, and every effort should be made to minimize the use of nephrotoxic agents if possible.
Patients with chronic diseases, such as Bartter syndrome, chloride-losing diarrhea and cystic fibrosis, should have lifelong follow-up care.
Future pregnancies in women with a child with hypochloremic alkalosis should be monitored in a tertiary care center so that early diagnosis and intervention are available at delivery.
Hypochloremic metabolic alkalosis prognosis
Hypochloremic alkalosis prognosis is usually good for patients with Bartter syndrome, provided the patient complies well with treatment. Children who receive effective treatment have minimal risk of severe renal damage.
In patients with chloride-losing diarrhea, renal failure and end-stage renal disease (ESRD) may complicate the picture if diagnosis and treatment are delayed.
In patients with cystic fibrosis, prognosis depends on the severity of lung and liver involvement.
Anorexia and polyuria eventually lead to malnutrition and growth failure. Chronic dehydration frequently causes constipation. A small muscle mass and muscle wasting are frequently seen in patients following a late diagnosis or in untreated patients.
Central nervous system (CNS) effects include cerebral dysfunction and defective cognitive function resulting from chronic hypoperfusion in moderate-to-severe metabolic alkalosis due to hypokalemic and hypochloremic states. Hypopnea is due to depression of respiratory drive. CNS calcification occurs in some patients for unclear reasons. Seizure disorder, brain atrophy, and mental retardation are other known complications.
Depending on the renal disorder, complications may include nephrocalcinosis, interstitial nephropathy, hypercalcemia, hyperuricemia, hypertension during the late stages of renal damage, and renal failure. Children with end-stage renal disease require renal replacement therapy in the form of hemodialysis or peritoneal dialysis. Kidney transplantation with the consequences of graft loss due to metabolic derangements adds more morbidity in these patients.
Metabolic alkalosis causes
The most common causes of metabolic alkalosis are the use of diuretics (water pills tha help rid your body of sodium and water) and the external loss of gastric hydrochloric acid (HCl) secretions 23.
Other causes include the following:
- Exogenous alkali administration – Sodium bicarbonate therapy in the presence of renal failure, metabolism of lactic acid or ketoacids
- Milk-alkali syndrome
- Hypercalcemia
- Intravenous penicillin
- Refeeding alkalosis
- Massive blood transfusion
Metabolic alkalosis is further divided into 2 main categories 24, 25, 26, 27, 28, 29, 23:
- Chloride-responsive alkalosis with urine chloride less than 20 mEq/L. Chloride-responsive alkalosis causes include loss of hydrogen via the gastrointestinal tract (vomiting, nasogastric suction), congenital chloride diarrhea syndrome (congenital chloridorrhea), loss of colonic secretions via villous adenoma, contraction alkalosis, diuretic therapy (thiazides and loop diuretics [after discontinuation]), post-hypercapnia syndrome, cystic fibrosis, exogenous alkalotic agent use and laxative abuse are also potential causes.
- Chloride-resistant alkalosis with urine chloride greater than 20 mEq/L.
- Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
- Primary hyperaldosteronism: adrenal adenoma, bilateral adrenal hyperplasia, adrenal carcinoma, glucocorticoid-remediable hyperaldosteronism
- 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2): 11 β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) normally converts active cortisol to inactive cortisone and protects mineralocorticoid receptor (MR) occupied by cortisol. The Apparent Mineralocorticoid Excess syndrome is an ultrarare autosomal recessive disorder caused by 11β-hydroxysteroid dehydrogenase type 2 gene (HSD11B2) resulting in impairment of the enzyme 11β-HSD2 leading to excessive cortisol that is able to activate mineralocorticoid receptor (MR). The Apparent Mineralocorticoid Excess syndrome is typically characterized by hypertension with hypokalemia, metabolic alkalosis, low renin activity, and low aldosterone level. The cause is inherited in autosomal recessive form, mutations in 11β-HSD2 gene or acquired form by ingestion of competitive inhibitors of 11β-HSD2 such as liquorice, chewing tobacco or carbenoxolone. Uncontrolled hypertension and prolong hypokalemia are the leading causes of end organ damages and cardiovascular mortality 30.
- Congenital adrenal hyperplasia (CAH) 11-hydroxylase or 17-hydroxylase deficiency
- Current use of diuretics in hypertension
- Cushing syndrome
- Exogenous mineralocorticoids or glucocorticoids
- Liddle syndrome
- Renovascular hypertension
- Causes of chloride-resistant alkalosis (urine chloride >20 mEq/L) without hypertension include the following:
- Bartter syndrome
- Gitelman syndrome
- Severe potassium depletion (hypokalemia)
- Current use of thiazides and loop diuretics
- Hypomagnesemia
- Causes of chloride-resistant alkalosis (urine chloride > 20 mEq/L) with hypertension include the following:
Chloride-responsive metabolic alkalosis
The principal causes of chloride-responsive metabolic alkalosis are the loss of gastric secretions, ingestion of large doses of nonabsorbable antacids, and use of thiazide or loop diuretics 41. Miscellaneous causes account for the remainder of cases 11.
Gastric secretions are rich in hydrochloric acid (HCl). The secretion of hydrochloric acid (HCl) by the stomach usually stimulates bicarbonate secretion by the pancreas once the hydrochloric acid (HCl) reaches the duodenum. Ordinarily, these pancreatic secretions neutralize the gastric secretions, and no net gain or loss of hydrogen ions or bicarbonate occurs.
When hydrochloric acid (HCl) is lost through vomiting (including purging, in persons with eating disorders) or nasogastric suction, pancreatic secretions are not stimulated and a net gain of bicarbonate into the systemic circulation occurs, generating a metabolic alkalosis 42. Volume depletion maintains alkalosis. In this case, the hypokalemia is secondary to the alkalosis itself and to renal loss of potassium ions from the stimulation of aldosterone secretion.
Ingestion of large doses of non-absorbable antacids (eg, magnesium hydroxide) may generate metabolic alkalosis by a rather complicated mechanism. Upon ingestion of magnesium hydroxide, calcium, or aluminum with base hydroxide or carbonate, the hydroxide anion buffers hydrogen ions in the stomach. The cation binds to bicarbonate secreted by the pancreas, leading to loss of bicarbonate with stools. In this process, both hydrogen ions and bicarbonate are lost, and, usually, no acid-base disturbance occurs. Sometimes, however, not all the bicarbonate binds to the ingested cation, which means that some bicarbonate is reabsorbed in excess of the lost hydrogen ions. This occurs primarily when antacids are administered with a cation-exchange resin (eg, sodium polystyrene sulfonate [Kayexalate]); the resin binds the cation, leaving bicarbonate unbound.
Thiazides and loop diuretics enhance sodium chloride excretion in the distal convoluted tubule and the thick ascending loop, respectively. These agents cause metabolic alkalosis by chloride depletion and by increased delivery of sodium ions to the collecting duct, which enhances potassium ion and hydrogen ion secretion.
Miscellaneous causes of metabolic alkalosis include villous adenomas, which are a rare cause of diarrhea. Villous adenomas usually lead to metabolic acidosis from loss of colonic secretions that are rich in bicarbonate, but occasionally these tumors cause metabolic alkalosis. The mechanism is not well understood. Some authors opine that hypokalemia from these tumors is the cause of the metabolic alkalosis.
Congenital chloridorrhea also known as congenital chloride-rich diarrhea, is a rare form of severe secretory diarrhea that is inherited as an autosomal recessive trait. Mutations in the down-regulated adenoma gene result in defective function of the chloride/bicarbonate exchange in the colon and ileum, leading to increased secretion of chloride and reabsorption of bicarbonate.
During respiratory acidosis, the kidneys reabsorb bicarbonate and secrete chloride to compensate for the acidosis. In the posthypercapnic state, urine chloride is high and can lead to chloride depletion. Once the respiratory acidosis is corrected, the kidneys cannot excrete the excess bicarbonate because of the low luminal chloride.
Infants with cystic fibrosis may develop metabolic alkalosis because of loss of chloride in sweat. These infants are also prone to volume depletion.
Chloride-resistant metabolic alkalosis
Causes of chloride-resistant metabolic alkalosis can be divided into those associated with hypertension and those associated with hypotension or normotension (normal blood pressure). Chloride-resistant metabolic alkalosis with high blood pressure may result from primary hyperaldosteronism, as well as a variety of acquired and hereditary disorders. An adrenal adenoma (most common), bilateral adrenal hyperplasia, or an adrenal carcinoma may cause primary hyperaldosteronism 43.
Another cause of primary hyperaldosteronism is glucocorticoid-remediable aldosteronism, an autosomal dominant disorder, in which ectopic production of aldosterone in the zona fasciculata of the adrenal cortex occurs. In this case, aldosterone production is controlled by adrenocorticotropic hormone (ACTH) rather than angiotensin 2 and potassium, its principal regulators. This type of primary hyperaldosteronism responds to glucocorticoid therapy, which inhibits aldosterone secretion by suppressing ACTH (adrenocorticotropic hormone).
The mineralocorticoid receptor (MR) in the collecting duct usually is responsive to both aldosterone and cortisol. Cortisol has a higher affinity for the MR and circulates at a higher concentration than aldosterone. Under physiological conditions, however, the enzyme 11-beta-hydroxysteroid dehydrogenase type 2 (11B-HSD2) inactivates cortisol to cortisone in the collecting duct, allowing aldosterone free access to its receptor. Deficiency of 11-beta-hydroxysteroid dehydrogenase type 2 (11B-HSD2) enzyme leads to occupation and activation of the mineralocorticoid receptor (MR) by cortisol, which, like aldosterone, then stimulates the epithelial sodium channel (ENaC). Cortisol behaves as a mineralocorticoid under these circumstances.
11-beta-hydroxysteroid dehydrogenase type 2 (11B-HSD2) deficiency may be inherited as an autosomal recessive trait, manifesting as syndrome of apparent mineralocorticoid excess (AME). The enzyme may be inhibited by glycyrrhizic acid, which is found in licorice and chewing tobacco, or carbenoxolone, which is a synthetic derivative of glycyrrhizinic acid. Deficiency or inhibition of 11B-HSD2 causes hypertension with low renin and low aldosterone, hypokalemia, and metabolic alkalosis. Serum cortisol is within the reference range because the negative feedback of cortisol on adrenocorticotropic hormone (ACTH) is intact.
Active use of thiazides or loop diuretics in hypertension is the most common cause of metabolic alkalosis in hypertensive patients.
The enhanced mineralocorticoid effect in Cushing syndrome is caused by occupation of the mineralocorticoid receptor (MR) by the high concentration of cortisol. Hypokalemia and metabolic alkalosis are more common in Cushing syndrome caused by ectopic ACTH production (90%) than in other causes of Cushing syndrome (10%). This difference is related to the higher concentration of plasma cortisol and the defective enzyme 11-beta-hydroxysteroid dehydrogenase (11B-HSD) activity found in ectopic ACTH production.
Liddle syndrome is a rare autosomal dominant disorder arising from a gain-of-function mutation in the beta (SCNN1B) or gamma subunit (SCNN1G) of the epithelial sodium channel (ENaC) in the collecting duct. The epithelial sodium channel (ENaC) behaves as if it is permanently open, and unregulated reabsorption of sodium (Na+) occurs, leading to volume expansion and hypertension. This unregulated sodium (Na+) reabsorption is responsible for secondary renal hydrogen ion and potassium ion losses and persists despite suppression of aldosterone.
Significant unilateral or bilateral renal artery stenosis stimulates the renin-angiotensin-aldosterone system, leading to hypertension and hypokalemic metabolic alkalosis.
Renin- or deoxycorticosterone-secreting tumors are rare. In renin-secreting tumors, excessive amounts of renin are secreted by tumors in the juxtaglomerular apparatus, stimulating aldosterone secretion. In the latter, deoxycorticosterone, rather than aldosterone, is secreted by some adrenal tumors and has mineralocorticoid effects.
Mutation in mineralocorticoid receptor is a form of early-onset hypertension with autosomal dominant inheritance that has now been linked to a specific mutation of the mineralocorticoid receptor (MR). This mutation results in constitutive activation of the mineralocorticoid receptor (MR), making the mineralocorticoid receptor (MR) responsive to progesterone. Activation of mineralocorticoid receptor (MR) leads to unregulated sodium (Na+) ion reabsorption via the collecting duct sodium (Na+) ion channel, with accompanying hypokalemia and alkalosis. The disease is characterized by severe exacerbations of hypertension during pregnancy, and spironolactone can exacerbate hypertension.
Congenital adrenal hyperplasia (CAH) can be caused by deficiency of either 11-beta-hydroxylase or 17-alpha-hydroxylase. Both enzymes are involved in the synthesis of adrenal steroids. Deficiency of either enzyme leads to increased levels of the mineralocorticoid 11-deoxycortisol, while cortisol and aldosterone production is impaired. 11-Hydroxylase deficiency differs from 17-hydroxylase deficiency by the presence of virilization.
Chloride-resistant alkalosis (urine chloride >20 mEq/L) with hypotension or normotension may be a manifestation of Bartter syndrome, an inherited autosomal recessive disorder. In Bartter syndrome, impaired reabsorption of sodium ions and chloride ions in the thick ascending loop of Henle leads to their increased delivery to the distal nephron.
The impaired reabsorption of sodium chloride (NaCl) in the loop of Henle is secondary to loss of function mutations of 1 of several transporters in this site of the nephron: (1) the furosemide-sensitive Na+/K+/2Cl- cotransporter (NKCC2); (2) the basolateral chloride voltage-gated channel Kb (CLCNKB); (3) the inwardly rectifying apical potassium ion channel (ROMK1); (4) barttin (BSND), the beta-subunit of the chloride channels, CLC-Ka and CLC-Kb; and (5) the calcium sensing receptor (CaSR).
Mutations of chloride voltage-gated channel Kb (CLCNKB) gene cause classic Bartter syndrome, while mutations of the other 2 transporters manifest with the antenatal form of Bartter syndrome 44. Edema and hypertension are absent, and hypercalciuria is common because the impaired reabsorption of sodium chloride inhibits the paracellular reabsorption of calcium. Because loop diuretics inhibit the Na+/K+/2Cl- transporter, the electrolyte abnormalities observed in Bartter syndrome and with loop diuretic use are similar.
Gitelman syndrome is an inherited autosomal recessive disorder in which loss of function of the thiazide-sensitive sodium/chloride transporter (NCCT) in the distal convoluted tubule occurs. The subsequent increased distal solute delivery and salt wasting with stimulation of the renin-angiotensin-aldosterone system lead to hypokalemic metabolic alkalosis. Other features of the syndrome are hypocalciuria and hypomagnesemia. The electrolyte abnormalities resemble those caused by thiazide diuretics.
Pure hypokalemia (ie, severe potassium ion depletion) causes mild metabolic alkalosis, but, in combination with hyperaldosteronism, the alkalosis is more severe. Possible mechanisms of alkalosis in hypokalemia are enhanced proximal bicarbonate reabsorption, stimulated renal ammonia genesis, impaired renal chloride reabsorption, reduced GFR (in animals), and intracellular acidosis in the distal nephron with subsequent enhanced hydrogen secretion.
Magnesium depletion (ie, hypomagnesemia) may lead to metabolic alkalosis. The mechanism probably involves hypokalemia, which is usually caused by or associated with magnesium depletion.
Other causes
The kidneys are able to excrete any excess alkali load, whether it is exogenous (eg, infusion of sodium bicarbonate) or endogenous (eg, metabolism of lactate to bicarbonate in lactic acidosis). However, in renal failure or in any condition that maintains the alkalosis, this natural ability of the kidneys to excrete the excess bicarbonate is impaired. Examples include the following:
- Alkali-loading alkalosis
- Hypercalcemia
- Intravenous penicillin
- Hypoproteinemic alkalosis
Milk-alkali syndrome comprises hypercalcemia, renal insufficiency, and metabolic alkalosis. Before the advent of H2-receptor antagonists, milk-alkali syndrome was observed in patients who ingested large amounts of milk and antacids as treatment for peptic ulcers. Currently, the syndrome is observed mainly in people who chronically ingest large doses of calcium carbonate, with or without vitamin D (typically for osteoporosis prevention) 45. The hypercalcemia that develops in some of these persons increases renal bicarbonate reabsorption. Renal insufficiency can occur secondary to nephrocalcinosis or hypercalcemia and contributes to maintaining the metabolic alkalosis.
Patients with end-stage renal disease (ESRD) are dialyzed with a high concentration of bicarbonate in the dialysate to reverse metabolic acidosis (ie, hemodialysis using high bicarbonate dialysate). Sometimes, this high bicarbonate exceeds the amount needed to buffer the acidosis. Because the ability of the kidneys to excrete the excess bicarbonate is absent or severely diminished, the alkalosis persists temporarily. The degree of alkalosis might be severe if the patient also has vomiting.
Metabolic alkalosis has been reported after regional citrate anticoagulation in hemodialysis or in continuous renal replacement therapies. Citrate is infused in the blood inflow line in the hemodialysis circuit, where it prevents clotting by binding calcium. Because the dialyzer does not remove citrate completely, a fraction of the infused citrate might reach the systemic circulation. Citrate in the blood is metabolized to bicarbonate in the liver. The accumulated bicarbonate may lead to metabolic alkalosis.
In an international prospective cohort study involving 17,031 patients receiving thrice-weekly hemodialysis, high dialysate bicarbonate, especially in patients with prolonged exposure, contributed to higher mortality, most likely through development of post-dialysis metabolic alkalosis. The positive association between dialysate bicarbonate concentration and mortality 1.08 per 4 mEq/L higher; adjusted hazard ratio for dialysate bicarbonate ≥38 vs. 33–37 mEq/L, 1.07 was consistent across pre-dialysis session serum bicarbonate levels and between facilities that used one dialysate bicarbonate concentration and those that prescribed different concentrations for each patient 46.
Metabolic alkalosis may be a potential complication of plasmapheresis in patients with renal failure. The source of alkali is the citrate that is used to prevent clotting in the extracorporeal circuit and in the stored blood from which the fresh frozen plasma is prepared. Using heparin as the anticoagulant and using albumin instead of fresh frozen plasma as the replacement solution can prevent the metabolic alkalosis.
Recovery from lactic acidosis or ketoacidosis in the presence of volume depletion or renal failure typically occurs when exogenous bicarbonate is administered to correct the acidosis. When the patient recovers, the beta-hydroxybutyrate and lactate are metabolized to bicarbonate and the original bicarbonate deficit is recovered. The administered bicarbonate now becomes a surplus.
Refeeding with a carbohydrate-rich diet after prolonged fasting results in mild metabolic alkalosis because of enhanced metabolism of ketoacids to bicarbonate.
Massive blood transfusion results in mild metabolic alkalosis as the citrate in the transfused blood is converted to bicarbonate. Metabolic alkalosis is more likely to develop in the presence of renal insufficiency.
Hypercalcemia may cause metabolic alkalosis by volume depletion and enhanced bicarbonate reabsorption in the proximal tubule. However, hypercalcemia from primary hyperparathyroidism is usually associated with a metabolic acidosis.
The intravenous administration of penicillin, carbenicillin, or other semisynthetic penicillins may cause hypokalemic metabolic alkalosis. This occurs because of distal delivery of nonreabsorbable anions with an absorbable cation such as Na+.
Metabolic alkalosis has been reported in patients with hypoproteinemia. The mechanism of alkalosis is not clear, but it may be related to loss of negative charges of albumin. A decrease in plasma albumin of 1 g/dL is associated with an increase in plasma bicarbonate of 3.4 mEq/L.
Metabolic alkalosis pathophysiology
There is a multitude of disease states that induce metabolic alkalosis. In general, the causes can be narrowed down to an intracellular shift of hydrogen ions (H+), gastrointestinal loss of hydrogen ions (H+), excessive renal hydrogen ions (H+) loss, retention or addition of bicarbonate (HCO3–) ions, or volume contraction around a constant amount of extracellular bicarbonate (HCO3–) known as contraction alkalosis. All of which leads to the net result of increased levels of bicarbonate (HCO3–) in the blood. As long as renal function is maintained, excess bicarbonate is excreted in the urine fairly rapidly. As a result, metabolic alkalosis will persevere if the ability to eliminate bicarbonate is impaired due to one of the following causes: hypovolemia, reduced effective arterial blood volume, chloride depletion, hypokalemia, reduced glomerular filtration rate, and/or hyperaldosteronism 13.
Intracellular shift of hydrogen
Anytime that hydrogen ions (H+) are shifted intracellularly, this imbalance in the buffer system has a relative increase in bicarbonate. Processes that drive hydrogen intracellularly include hypokalemia (low potassium in the bloodstream).
Gastrointestinal loss of hydrogen
Stomach fluids hydrochloric acid (HCl) are highly acidic at a pH of approximately 1.5 to 3.5. Hydrogen secretion is accomplished via parietal cells in the gastric mucosa. Therefore, the large volume loss of gastric secretions (hydrochloric acid [HCl]) will correlate as a loss of hydrogen chloride, an acidic substance, leading to a relative increase in bicarbonate in the blood, thus driving alkalosis. Losses can occur pathologically via vomiting or nasogastric suctioning.
Renal loss of hydrogen
Hydrogen is used within the kidneys are an antiporter energy gradient to retain a multitude of other elements. Of interest here, sodium (Na+) is reabsorbed through an exchange for hydrogen ions (H+) in the renal collecting ducts under the influence of aldosterone. Therefore, pathologies that increase the levels of mineralocorticoids or increase the effect of aldosterone, such as Conn syndrome will lead to hypernatremia, hypokalemia, and hydrogen loss in the urine. In a similar vein of thought, loop and thiazide diuretics are capable of inducing secondary hyperaldosteronism by increasing sodium and fluid load to the distal nephron, which encourages the renin-angiotensin-aldosterone system. Genetic defects that lead to decreased expression of ion transporters in the Loop of Henle are possible but less common. These syndromes are known as Bartter syndrome and Gitelman syndrome. The net effect of these genetic defects is akin to the action of loop diuretics.
Retention or addition of bicarbonate
Several causes lead to increases in bicarbonate (HCO3–) within the blood. The simplest of which is an overdose of exogenous sodium bicarbonate [NaHCO3] in a medical setting. Milk-alkali syndrome is a pathology where the patient consumes excessive quantities of oral calcium antacids, which leads to hypercalcemia and varying degrees of renal failure. Additionally, since antacids are neutralizing agents, they add alkaline substances to the body while reducing acid levels thus increasing pH. A pathology that is in line with normal physiology is the body’s natural compensation mechanism for an increase in carbon dioxide (CO2) in the bloodstream (hypercarbia). When a patient hypoventilates, carbon dioxide (CO2) retention occurs in the lungs and subsequently reduces pH. Over time, the renal system compensates by retaining bicarbonate to balance pH. This is a slower process. Once the hypoventilation is corrected, such as with a ventilator-assisted respiratory failure patient carbon dioxide (CO2) levels will quickly decrease, but bicarbonate (HCO3–) levels will lag in reducing. This causes an elevation in the arterial carbon dioxide (CO2) level also called post-hypercapnia metabolic alkalosis, which is self-correcting. It is possible to calculate the expected pCO2 in the setting of metabolic alkalosis to determine if it is a compensatory increase in bicarbonate, or if there is an underlying pathology driving alkalosis using the following equation:
- Expected pCO2 = 0.7 (HCO3–) + 20 mmHg +/- 5
If the expected pCO2 does not match the measured value, an underlying metabolic alkalosis is a likely present. For example, if the patient’s pCO2 is 37, therefore there is inadequate compensation or a concurrent respiratory alkalosis.
Contraction alkalosis
Contraction alkalosis occurs when a large volume of sodium-rich, bicarbonate low fluid is lost from the body. This occurs with diuretic use, cystic fibrosis, congenital chloride diarrhea, among others. The net concentration of bicarbonate increases as a result. This pathology is easily offset by the release of hydrogen from intracellular space to balance the pH in most incidences.
Metabolic alkalosis symptoms
Symptoms of metabolic alkalosis are not specific and can present with a myriad of signs and symptoms depending on the underlying cause. Because hypokalemia is usually present, the patient may experience weakness, muscle aches and pain (myalgia), polyuria (excessive urination defined as a urine output exceeding 3 L/day in adults and 2 L/m² in children), and cardiac arrhythmias 47. Hypoventilation develops because of inhibition of the respiratory center in the medulla. Symptoms of hypocalcemia (eg, jitteriness, perioral tingling, muscle spasms) may be present. Metabolic alkalosis can have central nervous system manifestations ranging from confusion to coma, peripheral neuropathic symptoms of tremor, tingling and numbness, muscle weakness and twitching, and arrhythmias, particularly when associated with hypokalemia and hypocalcemia 48, 49. Nonhypochloremic metabolic alkalosis associates with hypertension and is usually the result of syndromes of excess mineralocorticoid production. These generally correlate with signs of volume expansion, hypertension, and hypokalemia 50. Persistent and projectile, non-bilious vomiting in a two to six week old, otherwise well-appearing infant is a hallmark presentation of pyloric stenosis.
Other symptoms (eg, abdominal distention, dry skin, apathy, loss of interests, growth retardation 35 and frequent hospital admissions because of recurrent dehydration) are significant diagnostic clues during childhood.
Metabolic alkalosis complications
Alkalosis may lead to tetany, seizures, and decreased mental status. Metabolic alkalosis also decreases coronary blood flow and predisposes persons to refractory arrhythmias (atrial and ventricular tachyarrhythmias), especially when it’s associated with hypokalemia and hypocalcemia 51. Metabolic alkalosis causes hypoventilation, which may cause hypoxemia, especially in patients with poor respiratory reserve, and it may impair weaning from mechanical ventilation. By increasing ammonia production, it can precipitate hepatic encephalopathy in susceptible individuals.
Metabolic alkalosis diagnosis
Your healthcare provider will perform a physical examination and ask about your symptoms.
Important points in the history include the following:
- Vomiting or diarrhea – Gastrointestinal (GI) losses of hydrochloric acid (HCl)
- Age of onset and family history of alkalosis – Familial disorders (eg, Bartter syndrome, which starts during childhood)
- Renal failure – Alkali-loading alkalosis develops only when impairment of renal function occurs
- Drug use (eg, loop or thiazide diuretics; licorice; tobacco chewing; carbenoxolone; fludrocortisone; glucocorticoids; antacids [eg, magnesium hydroxide]; calcium carbonate)
- Previous gastrointestinal surgery (eg, ileostomy) 52, 53
These tests can help diagnose metabolic alkalosis. Tests may include:
- Arterial blood gas (ABG). An arterial blood gas is a laboratory test used for the measurement of arterial pH, arterial partial pressure of oxygen (PaO2), arterial partial pressure of carbon dioxide (PaCO2), bicarbonate (HCO3–), base excess, total carbon dioxide (CO2) and oxygen (O2) saturation.
- A venous blood gas test is a laboratory test identical to an arterial blood gas test, except the blood is drawn from a venous site. This results in a slightly more acidic “normal” pH range.
- Basic metabolic panel, (a group of blood tests that measure your sodium, potassium, and chloride levels, kidney function, and other chemicals and functions)
- Urine and stool studies
- Urine chloride is a direct measurement of chloride being excreted into urine. This test is useful to help determine the etiology of metabolic alkalosis 20, 21, 22
- A complete blood count (CBC) to evaluate for an infectious cause with elevated white blood count and fluid body status with hemoglobin and hematocrit values is useful.
- Cultures of blood, sputum, urine, and other sites
- Thyroid testing
- Beta-human chorionic hormone levels
- Drug screens and theophylline and salicylate levels
Other tests that may be needed to determine the cause of the alkalosis include:
- Plasma renin activity and aldosterone level
- Amniocentesis
- Genetic studies
- Physiologic study of renal tubules
- Ultrasonography
- Pulmonary function test to measure breathing and how well the lungs are functioning
- Chest x-ray
- CT abdomen
Additional studies that may be considered include the following:
- Wrist radiography – This may be performed to determine bone age in infants with growth failure; it may also help assess bone density and the presence of rickets
- Upper gastrointestinal (GI) series – This helps detect gastroesophageal reflux and pyloric stenosis, which are case-dependent conditions 40
- Computed tomography (CT) of the brain – This is useful for evaluation of brain growth and calcifications
- Magnetic resonance imaging (MRI) of the brain – This is helpful in patients who present with seizures
- Electroencephalography (EEG) – This is also helpful in patients who present with seizures
- Renal nuclear scanning – This may facilitate assessment of renal function but is not useful in all patients
- Renal biopsy – This is not usually indicated, but if it is performed, it may reveal interstitial fibrosis and calcium/urate crystal deposition
Physical examination
The physical signs of metabolic alkalosis are not specific and depend on the severity of the alkalosis. Because metabolic alkalosis decreases ionized calcium concentration, signs of hypocalcemia (eg, tetany, Chvostek sign, Trousseau sign), change in mental status, or seizures may be present.
Physical examination is helpful to establish the cause of metabolic alkalosis. Important aspects of the physical examination include the evaluation of hypertension and of volume status.
Hypertension accompanies several causes of metabolic alkalosis. Volume status assessment includes evaluation of orthostatic changes in blood pressure and heart rate, mucous membranes, presence or absence of edema, skin turgor, weight change, and urine output. Volume depletion usually accompanies chloride-responsive alkalosis, while volume expansion accompanies chloride-resistant alkalosis.
Arterial blood gas (ABG) analysis
Arterial blood gas (ABG) sampling, is a test often performed in an inpatient setting to assess the acid-base status of a patient. A needle is used to draw blood from an artery, often the radial artery, and the blood is analyzed to determine parameters such as the pH, arterial partial pressure of carbon dioxide (PaCO2), arterial partial pressure of oxygen (PaO2), bicarbonate (HCO3–), oxygen saturation (O2 Sat) and more. This allows the physician to understand the status of the patient better. ABGs are especially important in the critically ill. They are the main tool utilized in adjusting to the needs of a patient on a ventilator.
- Arterial partial pressure of carbon dioxide (PaCO2) as carbon dioxide tension, this measures the level of carbon dioxide in your blood.
- Arterial partial pressure of oxygen (PaO2) also known as oxygen tension, this measures how well oxygen is being transferred into your blood.
- Oxygen saturation (O2 Sat) is an assessment of the amount of oxygen in your blood that is based on measuring levels of hemoglobin. Hemoglobin is a protein found inside red blood cells that is responsible for carrying oxygen throughout the body.
- Bicarbonate (HCO3–) concentration: Bicarbonate (HCO3–) is an electrolyte, which is a type of mineral involved in managing your body’s acid-base balance. Most of the carbon dioxide (CO2) in your blood is stored in the form of bicarbonate, so this measurement helps reflect carbon dioxide (CO2) levels.
- Although not universal, some arterial blood gases tests include measurements of hemoglobin as well as altered forms of the hemoglobin protein. Examples of these potential additional measurements include:
- Methemoglobin: Methemoglobin is a form of hemoglobin that has been oxidized, changing its heme iron configuration from the ferrous (Fe2+) to the ferric (Fe3+) state. Unlike normal hemoglobin, methemoglobin does not bind oxygen and as a result cannot deliver oxygen to the tissues.
- Carboxyhemoglobin: Carboxyhemoglobin is a stable complex of carbon monoxide and hemoglobin that forms in red blood cells upon contact with carbon monoxide. This abnormal form of hemoglobin attaches to carbon monoxide and can interfere with oxygen’s ability to travel in the blood.
- Oxyhemoglobin: Oxyhemoglobin represents the fraction of oxygenated hemoglobin in relation to the total hemoglobin present, including non-oxygen-binding hemoglobins. In healthy individuals, oxyhemoglobin and oxygen saturation are approximately equal.
- Deoxyhemoglobin: This is the form of hemoglobin without oxygen in the blood.
The following are the most important Normal Values on an ABG:
- pH = 7.35 to 7.45
- Arterial partial pressure of carbon dioxide (PaCO2) = 35 to 45 mmHg
- Arterial partial pressure of oxygen (PaO2) = 75 to 100 mmHg
- Bicarbonate (HCO3–) = 22 to 26 mEq/L
- O2 Sat = greater than 95%
The ability to quickly and efficiently read an ABG is paramount to quality patient care.
- Look at the pH. Decide whether it is acidotic, alkalotic, or within the physiological range
- Arterial partial pressure of carbon dioxide (PaCO2) level determines respiratory contribution; a high level means the respiratory system is lowering the pH and vice versa.
- Bicarbonate (HCO3–) level denotes metabolic/kidney effect. An elevated bicarbonate (HCO3–) is raising the pH and vice versa.
- If the pH is acidotic, look for the number that corresponds with a lower pH. If it is a respiratory acidosis, the carbon dioxide (CO2) should be high. If the patient is compensating metabolically, the bicarbonate (HCO3–) should be high as well. A metabolic acidosis will be depicted with an bicarbonate (HCO3–) that is low.
- If the pH is alkalotic, again, determine which value is causing this. A respiratory alkalosis will mean the carbon dioxide (CO2) is low; a metabolic alkalosis should lend an bicarbonate (HCO3–) that is high. Compensation with either system will be reflected oppositely; for a respiratory alkalosis the metabolic response should be a low bicarbonate (HCO3–) and for metabolic alkalosis, the respiratory response should be a high carbon dioxide (CO2).
- If the pH level is in the physiological range but the arterial partial pressure of carbon dioxide (PaCO2) and/or bicarbonate (HCO3–) are not within normal limits, there is likely a mixed disorder. Also, compensation does not always occur; this is when clinical information becomes paramount.
- Sometimes it is difficult to ascertain whether a patient has a mixed disorder.
Other tests that are important to perform when analyzing the acid-base status of a patient include those that measure electrolyte levels and renal function. This helps the clinician gather information that can be used to determine the exact mechanism of the acid-base imbalance as well as the factors contributing to the disorders 54, 55.
Serum Anion Gap
The calculation of the serum anion gap:
- Serum anion gap = (Na+) – [(HCO3– + Cl–)]
Where Na+ is plasma sodium concentration, HCO3– is plasma bicarbonate concentration, and Cl– is plasma chloride concentration. The anions are negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]. The anion gap is the difference between measured cations (positively charged ions like sodium [Na+] and potassium [K+]) and measured anions (negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]) 56. Calculation of the serum anion gap may help to differentiate between primary metabolic alkalosis and metabolic compensation for respiratory acidosis. The anion gap is frequently elevated to a modest degree in metabolic alkalosis because of the increase in the negative charge of albumin and the enhanced production of lactate. A normal serum anion gap is measured to be 5 to 16 mEq/L, with autoanalyzers using an ion-selective electrode. However, the anion gap value is dependent on the type of instrument used to measure its components 57. Therefore, you should know the reference range of the analyzer used and, if known, the patient’s baseline anion gap, too. In any event, the only definitive way to diagnose metabolic alkalosis is with a simultaneous blood gases analysis that shows elevation of both pH and arterial partial pressure of carbon dioxide (PaCO2) and increased calculated bicarbonate.
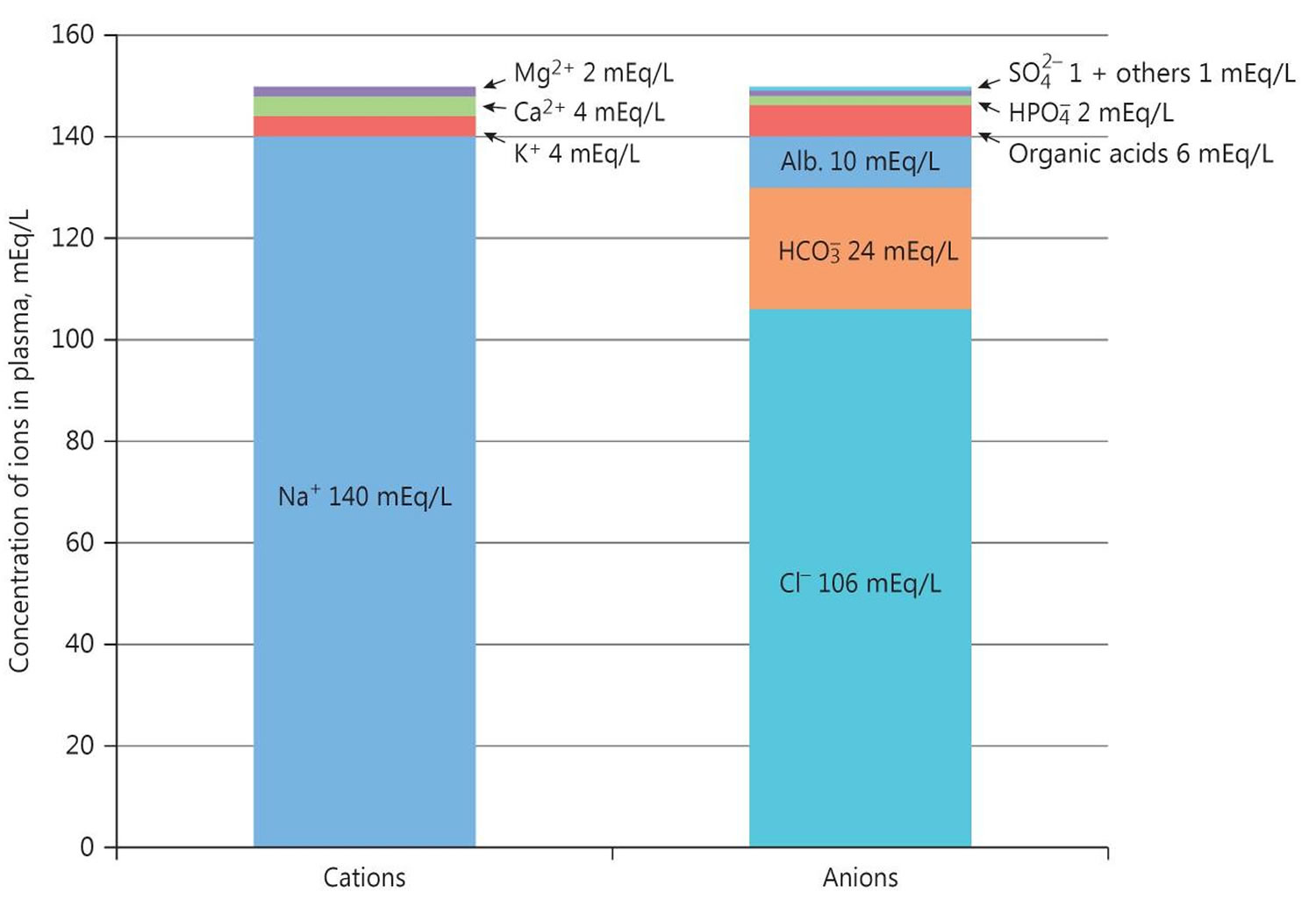
The anion gap is a calculation to determine the quantity of ionically active components within your blood that are not routinely measured. Serum anion gap is affected by the concentrations of all anions and cations which are not included in its calculations: i.e., albumin, globulin, potassium, calcium, magnesium, and organic and inorganic acids (see Figure 1). Because of the narrow extracellular concentration, most ions are omitted from the anion gap calculation. Since there are always components not directly measured, we expect this value to not equal 0. Most of this number is due to albumin (Alb); this anion is not accounted for in the anion gap formula, which is a large reason why the anion gap is not closer to zero. According to James Gamble 58, electrical neutrality in solution demands that the sum of the cations is equal to the sum of the anions (Figure 1). Sodium, chloride, bicarbonate, and albumin are quantitatively the major ions in the extracellular fluid compartment and are therefore used to calculate the anion gap 57. A true “ion gap,” however, does not exist in vivo which makes the anion gap a fundamental tool to evaluate acid-base disorders 59. Albumin is normally 4 mg/dL. Because of the large effect of albumin on anion gap, if a patient’s albumin level is abnormal, their expected anion gap will not be accurate 22. This can be corrected using simple math. The correction factor for albumin is 2.3–2.5 × [albumin], in g/dL 57. Therefore, each g/dL albumin decline will decrease the anion gap with about 2.5 mEq/L. To appreciate these facts, the anion gap formula should be: [Na+] − [Cl−] − [HCO3−] − 2.5 [albumin, in g/dL]. This equation is about zero in health, to stress the balance of ions, and also shows the relevance of albumin as a negative ion 57.
Serum bicarbonate (HCO3–) concentration can be calculated from a blood gas sample using the Henderson-Hasselbalch equation, as follows (see Figure 2 above):
- pH = 6.10 + log (HCO3– ÷ 0.03 × PaCO2)
- Alternatively, bicarbonate (HCO3–) = 24 × PaCO2 ÷ [H+]
Because pH and arterial partial pressure of carbon dioxide (PaCO2) are directly measured, bicarbonate (HCO3–) can be calculated.
Another means of assessing serum bicarbonate (HCO3–) concentration is with the total carbon dioxide content in serum, which is routinely measured with serum electrolytes obtained from venous blood. In this method, a strong acid is added to serum, which interacts with bicarbonate in the serum sample, forming carbonic acid. Carbonic acid dissociates to carbon dioxide and water; then, carbon dioxide is measured.
Note that the carbon dioxide measured includes bicarbonate and dissolved carbon dioxide. The contribution of dissolved carbon dioxide is quite small (0.03 × PaCO2) and is usually ignored, although it accounts for a difference of 1-3 mEq/L between the measured total carbon dioxide content in venous blood and the calculated bicarbonate in arterial blood. Thus, at an arterial partial pressure of carbon dioxide (PaCO2) of 40, a total carbon dioxide (CO2) content of 25 means a true bicarbonate concentration of 23.8 (ie, 25 – 0.03 × 40).
The Henderson-Hasselbalch equation may fail to account for acid-base findings in critically ill patients. An alternative method of acid-base analysis, known as the quantitative, or strong ion, approach, determines pH on the basis of the following 3 independent variables 60:
- Strong ion difference: Ions almost completely dissociated at physiologic pH (the cations Na+, K+, Ca+, and Mg+, and the anions Cl- and lactate)
- Total weak acid concentration: Ions that can be dissociated or associated at physiologic pH (albumin and phosphate)
- pCO2 (mm Hg)
In a study that compared the conventional Henderson-Hasselbalch equation with the strong ion approach, carried out in 100 patients with trauma who were admitted to a surgical intensive care unit, the investigators concluded that the strong ion approach provides a more accurate means of diagnosing acid-base disorders, including metabolic alkalosis and tertiary disorders 61.
Figure 4. Normal anion gap levels
Plasma renin activity and aldosterone level
Measuring the plasma renin activity and aldosterone level may help in finding the etiology of metabolic alkalosis, especially in patients with hypertension, hypokalemic metabolic alkalosis, and renal potassium wasting without diuretic use. Low renin activity and high plasma aldosterone levels are found in primary hyperaldosteronism, including glucocorticoid-remediable hyperaldosteronism.
Low plasma renin activity and aldosterone levels are found in the following circumstances:
- Cushing syndrome
- Exogenous steroid use
- Congenital adrenal hyperplasia (CAH)
- 11-beta-hydroxysteroid dehydrogenase (11B-HSD) deficiency
- Deoxycorticosterone (DOC)-secreting tumors
- Liddle syndrome
High plasma renin activity and aldosterone levels are found in the following circumstances:
- Renal artery stenosis
- Diuretic use
- Renin-secreting tumors
- Bartter syndrome
- Gitelman syndrome
Amniocentesis
Amniotic fluid sodium and chloride concentrations may reflect fetal values; these are high in fetuses with chloride-losing diarrhea. Levels may also be elevated in patients with Bartter syndrome. Although testing for alpha1 -fetoprotein is not routine, levels may be elevated.
Blood workup
Serum electrolyte levels may be within the reference range, especially in neonates and treated patients. However, typical findings include low concentrations of serum chloride, sodium, and potassium. Attention must be paid in interpreting the serum potassium level in relation to the state of metabolic alkalosis. For example, the potassium shift from serum into the intracellular compartment increases as the serum pH rises; thus, the potassium level is less than normal by 0.6 mmol/L when measured at a serum pH of 7.5.
Serum pH and bicarbonate, calcium, uric acid, hemoglobin (if patient is not anemic), renin, and aldosterone levels may be elevated. The serum renin level is exponentially high, in line with secondary hyperaldosteronism due to chronic volume depletion, and this finding is supported by low or normal blood pressure measurements.
Urine and stool studies
In patients with Bartter syndrome, urine chloride, sodium, and potassium concentrations are usually measured. Urine calcium-to-creatinine and uric acid–to–creatinine ratios are usually high. Stool electrolytes cannot be measured because of well-formed or hard stool. Fractional excretion (Fex) studies are more reliable than absolute values. Usually, results are higher than reference range values, as follows:
- Fractional excretion (Fex) sodium concentration >1%
- Fractional excretion (Fex) potassium concentration >35%
- Fractional excretion (Fex) chloride concentration >2.5% (2.7% ± 1.1%)
In patients with chloride-losing diarrhea, urine chloride concentration is very low or undetectable (< 10 mmol/L). Stool is usually watery, and electrolyte studies are very helpful and diagnostic, as follows:
- Stool chloride concentration >100 mmol/L
- Stool sodium and potassium concentrations are elevated
- Stool chloride concentration is greater than stool sodium plus potassium concentrations, which is normally less than either; chloride concentrations are lowest in colonic secretions (usually < 35 mmol/L)
- The ratio of stool chloride to combined sodium and potassium concentrations is greater than 0.6
Patients with cystic fibrosis typically demonstrate high sweat chloride and sodium concentrations. Urine chloride concentration is usually very low, and stools are usually not watery, as they are in patients with chloride-losing diarrhea.
Kidney and liver function tests
- Renal function is usually normal. The glomerular filtration rate (GFR) may be low in patients with severe disease.
- Liver function test results are usually within the reference range in patients with chloride-losing diarrhea and Bartter syndrome but may be deranged in patients with cystic fibrosis.
Genetic studies
DNA diagnosis is available for most congenital disorders that cause hypochloremic metabolic alkalosis. For chloride-losing diarrhea, the chloride-losing diarrhea (SLC26A3) locus is on band 7q22-q31.1 38. Bartter syndrome is identified by NKCC2, ROMK, and CLCNKB 39; Bartter syndrome with deafness is identified by BSND; and Bartter syndrome with autosomal dominant hypocalcemia is identified by CASR. For cystic fibrosis, the CFTR locus is on band 7q31.2. For Gitelman syndrome, the NCCT locus is on 16q.
Ultrasonography
Prenatal ultrasonography may be useful in the detection of minimal polyhydramnios and assessment of intestinal fluid content, which is increased in patients with chloride-losing diarrhea.
Postnatal ultrasonography (see the images below) may be useful in the evaluation of a fluid-filled bowel, which is characteristically increased in patients with chloride-losing diarrhea. Ultrasonography may also assist in the evaluation of renal echogenicity, nephrocalcinosis, medullary or diffuse calcinosis, and renal growth.
Physiologic study of renal tubules
Physiologic study of renal tubules by performing maximal free water clearance during hypotonic saline diuresis is indicated.
Oral administration of water 20 mL/kg over 30 minutes is followed by administration of a one-half isotonic sodium chloride solution at a rate of 600 mL/m²/h for 2-3 hours. During this time, urine is collected in aliquots over 30-minute periods for 4-6 aliquots.
These samples are sent for evaluation of creatinine, sodium, potassium, and chloride levels, as well as for osmolality, pH, and volume. Usually, urine is diluted by oral administration of water. Halfway through each collection, a blood sample is obtained for evaluation of creatinine, sodium, potassium, and chloride levels, and for pH and osmolality. The clearance of each substance is calculated, and a ratio is derived by means of the following formula:
- Water clearance/(chloride clearance + water clearance)
Usually, the result of this formula reflects the percentage of distal tubule sodium and chloride reabsorption. Normal values are up to 85-90%, which means that the percentage of chloride and sodium excreted should be 10-15% (corrected to a GFR of 100 mL/min/1.73 m²). In patients with Bartter syndrome, the percentage of chloride and sodium excreted can reach 35% or more.
Other studies
Additional studies that may be considered include the following:
- Wrist radiography – This may be performed to determine bone age in infants with growth failure; it may also help assess bone density and the presence of rickets
- Upper gastrointestinal (GI) series – This helps detect gastroesophageal reflux and pyloric stenosis, which are case-dependent conditions 40
- Computed tomography (CT) of the brain – This is useful for evaluation of brain growth and calcifications
- Magnetic resonance imaging (MRI) of the brain – This is helpful in patients who present with seizures
- Electroencephalography (EEG) – This is also helpful in patients who present with seizures
- Renal nuclear scanning – This may facilitate assessment of renal function but is not useful in all patients
- Renal biopsy – This is not usually indicated, but if it is performed, it may reveal interstitial fibrosis and calcium/urate crystal deposition
Metabolic alkalosis treatment
The management of metabolic alkalosis depends primarily on the underlying etiology and on the patient’s volume status 62. In the case of vomiting, administer antiemetics, if possible. If continuous gastric suction is necessary, gastric acid secretion can be reduced with H2-blockers or more efficiently with proton pump inhibitors. In patients who are on thiazide or loop diuretics, the dose can be reduced or the drug can be stopped if appropriate. Alternatively, a potassium-sparing diuretic or acetazolamide can be added.
Acetazolamide also appears safe and effective in patients with metabolic alkalosis following treatment of respiratory acidosis from exacerbations of chronic obstructive pulmonary disease (COPD) 63, 64. One randomized trial found that the duration of mechanical ventilation in patients with COPD or obesity-hypoventilation syndrome with metabolic alkalosis was not significantly reduced in patients who received early administration of acetazolamide, compared with placebo 65.
However, a systematic review and meta-analysis of that trial and five other randomized controlled studies concluded that in patients with respiratory failure and metabolic alkalosis, therapy with acetazolamide or other carbonic anhydrase inhibitors may have favorable effects on blood gas parameters. In mechanically ventilated patients, carbonic anhydrase inhibitor therapy may decrease the duration of mechanical ventilation 66.
Hydrochloric acid
Intravenous hydrochloric acid (HCl) is indicated in severe metabolic alkalosis (pH >7.55) or when sodium or potassium chloride cannot be administered because of volume overload or advanced renal failure. Hydrochloric acid (HCl) may also be indicated if rapid correction of severe metabolic alkalosis is warranted (eg, cardiac arrhythmias, hepatic encephalopathy, digoxin cardiotoxicity) 67. Seek the advice of a nephrologist when severe alkalosis is present and HCl therapy or dialysis is contemplated.
Dialysis
Both peritoneal dialysis and hemodialysis can be used with certain modifications of the dialysate to correct metabolic alkalosis. The main indication of dialysis in metabolic alkalosis is in patients with advanced renal failure, who usually have volume overload and are resistant to acetazolamide.
With hemodialysis, metabolic alkalosis may be corrected by using a low-bicarbonate dialysate (bicarbonate can be as low as 18 mmol/L). Otherwise, acetate-free biofiltration (buffer-free dialysate), in which bicarbonate is not present in the dialysate but is infused separately as needed, may be used. In peritoneal dialysis, dialysis can be performed using isotonic sodium chloride solution as the dialysate.
Chloride-responsive metabolic alkalosis treatment
If chloride-responsive metabolic alkalosis occurs with volume depletion, treat the alkalosis with an intravenous infusion of isotonic sodium chloride solution. Because this type of alkalosis is usually associated with hypokalemia, also use potassium chloride to correct the hypokalemia.
If chloride-responsive alkalosis occurs in the setting of edematous states (eg, congestive heart failure [CHF]), use potassium chloride instead of sodium chloride to correct the alkalosis and avoid volume overload. If diuresis is needed, a carbonic anhydrase inhibitor (eg, acetazolamide) or a potassium-sparing diuretic (eg, spironolactone, amiloride, triamterene) can be used to correct the alkalosis.
Chloride-resistant metabolic alkalosis treatment
Management of chloride-resistant metabolic alkalosis is based on the specific cause.
Primary hyperaldosteronism
Metabolic alkalosis is corrected with the aldosterone antagonist spironolactone or with other potassium-sparing diuretics (eg, amiloride, triamterene). If the cause of primary hyperaldosteronism is an adrenal adenoma or carcinoma, surgical removal of the tumor should correct the alkalosis. In glucocorticoid-remediable hyperaldosteronism, metabolic alkalosis and hypertension are responsive to dexamethasone.
Cushing syndrome
Potassium-sparing diuretics should correct the alkalosis until surgical therapy is performed. Definitive therapy includes transsphenoidal microresection of adrenocorticotropic hormone (ACTH)–producing pituitary adenomas and adrenalectomy for adrenal tumors.
Syndrome of apparent mineralocorticoid excess
Metabolic alkalosis in the syndrome of apparent mineralocorticoid excess (AME) may be treated with potassium-sparing diuretics. On the other hand, dexamethasone may be used to suppress cortisol production by inhibiting ACTH. Unlike cortisol and some synthetic glucocorticoids, dexamethasone does not activate the mineralocorticoid receptor.
Licorice ingestion
Discontinuation of licorice ingestion corrects the alkalosis; however, because full recovery of the enzyme 11B-HSD may take as long as 2 weeks following long-term licorice use, potassium-sparing diuretics can be used during this interval.
Bartter syndrome and Gitelman syndrome
Metabolic alkalosis can be corrected partially with the following:
- Potassium supplementation
- Potassium-sparing diuretics
- Nonsteroidal anti-inflammatory drugs
- ACE inhibitors
Indomethacin is the most prescribed NSAID for the treatment of Bartter syndrome; in patients with Gitelman synrome it has shown to be more effective than eplerenone or amiloride for treating associated hypokalemia 68.
Liddle syndrome
Metabolic alkalosis can be treated with amiloride or triamterene but not with spironolactone. Both amiloride and triamterene inhibit the apical sodium ion channel in the collecting duct. Spironolactone, which is a mineralocorticoid receptor antagonist that works upstream of the defective sodium ion channel, does not correct the alkalosis or the hypertension.
Metabolic alkalosis prognosis
Metabolic alkalosis prognosis depends on associated problems of volume depletion, electrolyte, and hormonal disturbances and varies based on the primary cause of the alkalosis. Patients with metabolic alkalosis have been found to have increased ICU duration of stay, more days on mechanical ventilation, and higher hospital mortality. An increase of 5-mEq/L in the serum bicarbonate level over 30 mEq/L correlated with an odds ratio of 1.21 for hospital mortality. The association between metabolic alkalosis and mortality occurs independently of the etiology of alkalosis 67.
References- Hasan, A. (2013). Metabolic Alkalosis. In: Handbook of Blood Gas/Acid-Base Interpretation. Springer, London. https://doi.org/10.1007/978-1-4471-4315-4_10
- Kraut JA, Lew V, Madias NE. Re-Evaluation of Total CO2 Concentration in Apparently Healthy Younger Adults. Am J Nephrol. 2018;48(1):15-20. doi: 10.1159/000489965
- Maehle K, Haug B, Flaatten H, Nielsen E: Metabolic alkalosis is the most common acid-base disorder in ICU patients. Critical Care 28[14]: 420–420, 2014. 10.1186/cc13802
- Hodgkin JE, Soeprono FF, Chan DM. Incidence of metabolic alkalemia in hospitalized patients. Crit Care Med. 1980 Dec;8(12):725-8. doi: 10.1097/00003246-198012000-00005
- Mazzara JT, Ayres SM, Grace WJ. Extreme hypocapnia in the critically ill patient. Am J Med. 1974 Apr;56(4):450-6. doi: 10.1016/0002-9343(74)90475-6
- Koeppen BM. The kidney and acid-base regulation. Adv Physiol Educ. 2009 Dec;33(4):275-81. doi: 10.1152/advan.00054.2009
- Hamm LL, Nakhoul N, Hering-Smith KS. Acid-Base Homeostasis. Clin J Am Soc Nephrol. 2015 Dec 7;10(12):2232-42. doi: 10.2215/CJN.07400715
- Kisaka T, Cox TA, Dumitrescu D, Wasserman K. CO2 pulse and acid-base status during increasing work rate exercise in health and disease. Respir Physiol Neurobiol. 2015 Nov;218:46-56. doi: 10.1016/j.resp.2015.07.005
- Brinkman JE, Toro F, Sharma S. Physiology, Respiratory Drive. [Updated 2022 Jun 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482414
- Brinkman JE, Sharma S. Respiratory Alkalosis. [Updated 2022 Jul 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482117
- Emmett M. Metabolic Alkalosis: A Brief Pathophysiologic Review. Clin J Am Soc Nephrol. 2020 Dec 7;15(12):1848-1856. doi: 10.2215/CJN.16041219
- Sur M, Shah AD. Alkalosis. [Updated 2022 May 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK545269
- Brinkman JE, Sharma S. Physiology, Metabolic Alkalosis. [Updated 2022 Jul 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482291
- Medarov BI. Milk-alkali syndrome. Mayo Clin Proc. 2009 Mar;84(3):261-7. doi: 10.1016/S0025-6196(11)61144-0
- Koni I, Takeda R. [Acid-base disturbance in adrenal and parathyroid diseases]. Nihon Rinsho. 1992 Sep;50(9):2191-8. Japanese.
- Khanna A, Kurtzman NA. Metabolic alkalosis. Respir Care. 2001 Apr;46(4):354-65.
- Mamalis D, Stratigou T, Vallianou NG, Ioannidis GG, Apostolou T. Persistent hypokalemia due to a rare mutation in gitelman’s syndrome. Saudi J Kidney Dis Transpl. 2020 Jan-Feb;31(1):259-262. doi: 10.4103/1319-2442.279949
- STRODER J, BLENNEMANN H. METABOLIC ALKALOSIS IN PYLORIC STENOSIS. Lancet. 1964 Oct 24;2(7365):914-5. doi: 10.1016/s0140-6736(64)90785-8
- C T Howe, L P Le Quesne, Pyloric stenosis: The metabolic effects, British Journal of Surgery, Volume 51, Issue 12, December 1964, Pages 923–932, https://doi.org/10.1002/bjs.1800511215
- Stimson L, Reynolds T. Differential diagnosis for chronic hypokalaemia. BMJ Case Rep. 2018 Jun 5;2018:bcr2017223680. doi: 10.1136/bcr-2017-223680
- Galla JH. Metabolic alkalosis. J Am Soc Nephrol. 2000 Feb;11(2):369-375. doi: 10.1681/ASN.V112369
- Hopkins E, Sanvictores T, Sharma S. Physiology, Acid Base Balance. [Updated 2022 Sep 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507807
- Metabolic Alkalosis. https://emedicine.medscape.com/article/243160-overview#a6
- Siegler JC, Marshall P. The effect of metabolic alkalosis on central and peripheral mechanisms associated with exercise-induced muscle fatigue in humans. Exp Physiol. 2015 Apr 20;100(5):519-30. doi: 10.1113/EP085054
- Khanna A, Kurtzman NA. Metabolic alkalosis. J Nephrol. 2006 Mar-Apr;19 Suppl 9:S86-96.
- Seldin DW, Rector FC Jr. Symposium on acid-base homeostasis. The generation and maintenance of metabolic alkalosis. Kidney Int. 1972 May;1(5):306-21. doi: 10.1038/ki.1972.43
- Oppersma E, Doorduin J, van der Hoeven JG, Veltink PH, van Hees HWH, Heunks LMA. The effect of metabolic alkalosis on the ventilatory response in healthy subjects. Respir Physiol Neurobiol. 2018 Feb;249:47-53. doi: 10.1016/j.resp.2018.01.002
- Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 2018 Apr;93(4):893-902. doi: 10.1016/j.kint.2017.10.023
- Galla JH, Gifford JD, Luke RG, Rome L. Adaptations to chloride-depletion alkalosis. Am J Physiol. 1991 Oct;261(4 Pt 2):R771-81. doi: 10.1152/ajpregu.1991.261.4.R771
- Yau M, Haider S, Khattab A, Ling C, Mathew M, Zaidi S, Bloch M, Patel M, Ewert S, Abdullah W, Toygar A, Mudryi V, Al Badi M, Alzubdi M, Wilson RC, Al Azkawi HS, Ozdemir HN, Abu-Amer W, Hertecant J, Razzaghy-Azar M, Funder JW, Al Senani A, Sun L, Kim SM, Yuen T, Zaidi M, New MI. Clinical, genetic, and structural basis of apparent mineralocorticoid excess due to 11β-hydroxysteroid dehydrogenase type 2 deficiency. Proc Natl Acad Sci U S A. 2017 Dec 26;114(52):E11248-E11256. doi: 10.1073/pnas.1716621115
- Adamczak M, Surma S. Metabolic Acidosis in Patients with CKD: Epidemiology, Pathogenesis, and Treatment. Kidney Dis (Basel). 2021 Jun 4;7(6):452-467. doi: 10.1159/000516371
- Hypochloremic Alkalosis. https://emedicine.medscape.com/article/945263-overview
- Breaux CW Jr, Hood JS, Georgeson KE. The significance of alkalosis and hypochloremia in hypertrophic pyloric stenosis. J Pediatr Surg. 1989;24(12):1250-1252. doi:10.1016/s0022-3468(89)80561-5
- [Guideline] Grosse SD, Boyle CA, Botkin JR, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep. 2004 Oct 15. 53:1-36.
- Querfeld U, Lechner S, Janecke AR. Hypochloremic metabolic alkalosis and failure to thrive: answer. Pediatr Nephrol. 2011 Jun. 26 (6):895-6.
- Aranzamendi RJ, Breitman F, Asciutto C, Delgado N, Castanos C. [Dehydration and metabolic alkalosis: an unusual presentation of cystic fibrosis in an infant]. Arch Argent Pediatr. 2008 Oct. 106(5):443-6.
- Al-Abbad A, Nazer H, Sanjad SA, Al-Sabban E. Congenital chloride diarrhea: A single center experience with ten patients. Ann Saudi Med. 1995 Sep. 15(5):466-9.
- Makela S, Kere J, Holmberg C. SLC26A3 mutations in congenital chloride diarrhea. Hum Mutat. 2002 Dec. 20(6):425-38.
- Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997 Oct. 17(2):171-8.
- Hulka F, Campbell TJ, Campbell JR, Harrison MW. Evolution in the recognition of infantile hypertrophic pyloric stenosis. Pediatrics. 1997 Aug. 100(2):E9.
- Gillion V, Jadoul M, Devuyst O, Pochet JM. The patient with metabolic alkalosis. Acta Clin Belg. 2019 Feb;74(1):34-40. doi: 10.1080/17843286.2018.1539373
- Mehler, P.S. and Walsh, K. (2016), Electrolyte and acid-base abnormalities associated with purging behaviors. Int. J. Eat. Disord., 49: 311-318. https://doi.org/10.1002/eat.22503
- Vaidya A, Mulatero P, Baudrand R, Adler GK. The Expanding Spectrum of Primary Aldosteronism: Implications for Diagnosis, Pathogenesis, and Treatment. Endocr Rev. 2018 Dec 1;39(6):1057-1088. doi: 10.1210/er.2018-00139
- Gollasch B, Anistan YM, Canaan-Kühl S, Gollasch M. Late-onset Bartter syndrome type II. Clin Kidney J. 2017 Oct;10(5):594-599. doi: 10.1093/ckj/sfx033
- Patel AM, Adeseun GA, Goldfarb S. Calcium-alkali syndrome in the modern era. Nutrients. 2013 Nov 27;5(12):4880-93. doi: 10.3390/nu5124880
- Tentori F, Karaboyas A, Robinson BM, Morgenstern H, Zhang J, Sen A, Ikizler TA, Rayner H, Fissell RB, Vanholder R, Tomo T, Port FK. Association of dialysate bicarbonate concentration with mortality in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2013 Oct;62(4):738-46. doi: 10.1053/j.ajkd.2013.03.035
- Metabolic Alkalosis Clinical Presentation. https://emedicine.medscape.com/article/243160-clinical
- Dhondup T, Qian Q. Acid-Base and Electrolyte Disorders in Patients with and without Chronic Kidney Disease: An Update. Kidney Dis (Basel). 2017 Dec;3(4):136-148. doi: 10.1159/000479968
- Espay AJ. Neurologic complications of electrolyte disturbances and acid-base balance. Handb Clin Neurol. 2014;119:365-82. doi: 10.1016/B978-0-7020-4086-3.00023-0
- Seifter JL, Chang HY. Disorders of Acid-Base Balance: New Perspectives. Kidney Dis (Basel). 2017 Jan;2(4):170-186. doi: 10.1159/000453028
- Salanova-Villanueva L, Bernis-Carro C, Alberto-Blazquez L, Sanchez-Tomero JA. Severe arrhythmia due to hypokalemia. Influence from diuretic substances. Nefrologia. 2015;35(3):334-6. English, Spanish. doi: 10.1016/j.nefro.2015.05.008
- Gennari FJ, Weise WJ. Acid-base disturbances in gastrointestinal disease. Clin J Am Soc Nephrol. 2008 Nov;3(6):1861-8. doi: 10.2215/CJN.02450508
- Weise WJ, Serrano FA, Fought J, Gennari FJ. Acute electrolyte and acid-base disorders in patients with ileostomies: a case series. Am J Kidney Dis. 2008 Sep;52(3):494-500. doi: 10.1053/j.ajkd.2008.04.015
- Patel S, Sharma S. Respiratory Acidosis. [Updated 2022 Jun 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482430
- Rajkumar P, Pluznick JL. Acid-base regulation in the renal proximal tubules: using novel pH sensors to maintain homeostasis. Am J Physiol Renal Physiol. 2018 Nov 1;315(5):F1187-F1190. doi: 10.1152/ajprenal.00185.2018
- Pandey DG, Sharma S. Biochemistry, Anion Gap. [Updated 2019 Apr 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539757
- Berend K. Review of the Diagnostic Evaluation of Normal Anion Gap Metabolic Acidosis. Kidney Dis (Basel). 2017 Dec;3(4):149-159. doi: 10.1159/000479279
- Gamble JL. Extracellular fluid and its maintenance. N Engl J Med. 1936;250:1150–1152.
- Berend K, de Vries AP, Gans RO. Physiological approach to assessment of acid-base disturbances. N Engl J Med. 2015 Jan 8;372(2):195. doi: 10.1056/NEJMc1413880
- Stewart PA. Modern quantitative acid-base chemistry. Can J Physiol Pharmacol. 1983 Dec;61(12):1444-61. doi: 10.1139/y83-207
- Kaplan LJ, Cheung NH, Maerz L, Lui F, Schuster K, Luckianow G, Davis K. A physicochemical approach to acid-base balance in critically ill trauma patients minimizes errors and reduces inappropriate plasma volume expansion. J Trauma. 2009 Apr;66(4):1045-51. doi: 10.1097/TA.0b013e31819a04be
- Metabolic Alkalosis Treatment & Management. https://emedicine.medscape.com/article/243160-treatment
- Fontana V, Santinelli S, Internullo M, Marinelli P, Sardo L, Alessandrini G, Borgognoni L, Ferrazza AM, Bonini M, Palange P. Effect of acetazolamide on post-NIV metabolic alkalosis in acute exacerbated COPD patients. Eur Rev Med Pharmacol Sci. 2016;20(1):37-43. https://www.europeanreview.org/wp/wp-content/uploads/37-43.pdf
- Faisy C, Meziani F, Planquette B, et al. Effect of Acetazolamide vs Placebo on Duration of Invasive Mechanical Ventilation Among Patients With Chronic Obstructive Pulmonary Disease: A Randomized Clinical Trial. JAMA. 2016;315(5):480–488. doi:10.1001/jama.2016.0019
- Rialp Cervera G, Raurich Puigdevall JM, Morán Chorro I, Martín Delgado MC, Heras la Calle G, Mas Serra A, Vallverdú Perapoch I. Effects of early administration of acetazolamide on the duration of mechanical ventilation in patients with chronic obstructive pulmonary disease or obesity-hypoventilation syndrome with metabolic alkalosis. A randomized trial. Pulm Pharmacol Ther. 2017 Jun;44:30-37. doi: 10.1016/j.pupt.2017.03.002
- Tanios BY, Omran MO, Noujeim C, Lotfi T, Mallat SS, Bou-Khalil PK, Akl EA, Itani HS. Carbonic anhydrase inhibitors in patients with respiratory failure and metabolic alkalosis: a systematic review and meta-analysis of randomized controlled trials. Crit Care. 2018 Oct 29;22(1):275. doi: 10.1186/s13054-018-2207-6
- Guffey JD, Haas CE, Crowley A, Connor KA, Kaufman DC. Hydrochloric Acid Infusion for the Treatment of Metabolic Alkalosis in Surgical Intensive Care Unit Patients. Ann Pharmacother. 2018 Jun;52(6):522-526. doi: 10.1177/1060028018754389
- Nuñez-Gonzalez L, Carrera N, Garcia-Gonzalez MA. Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Bartter and Gitelman Syndromes: A Primer for Clinicians. Int J Mol Sci. 2021 Oct 22;22(21):11414. doi: 10.3390/ijms222111414





{kind=link}