
What is mitochondrial disease
Mitochondrial disease refers to a group of disorders caused by genetic mutations that affect the mitochondria (the structures in each cell of your body, except your red blood cells, that are responsible for making energy). Mitochondrial disease is an inherited chronic illness that can be present at birth or develop later in life. Although mitochondrial disease primarily affects children, adult onset is becoming more common 1. It is estimated that 1 in 4,000 people has mitochondrial disease. Mitochondrial disease is progressive and there is no cure. People with mitochondrial diseases can present at any age with almost any affected body system; however, the brain, muscles, heart, liver, nerves, eyes, ears and kidneys are the organs and tissues most commonly affected. Mitochondrial disease symptom severity can also vary widely 2. Mitochondrial disease causes debilitating physical, developmental, and cognitive disabilities with symptoms including poor growth; loss of muscle coordination; muscle weakness and pain; seizures; vision and/or hearing loss; gastrointestinal issues; learning disabilities; and organ failure.
Mitochondrial diseases can be caused by changes (mutations) in either the mitochondrial DNA or nuclear DNA that lead to dysfunction of the mitochondria and inadequate production of energy. Those caused by mutations in mitochondrial DNA are transmitted by maternal inheritance, while those caused by mutations in nuclear DNA may follow an autosomal dominant, autosomal recessive, or X-linked pattern of inheritance 2. Treatment varies based on the specific type of condition and the signs and symptoms present in each person 2.
The incidence of mitochondrial disease is about 1:4000 individuals in the US. This is similar to the incidence of cystic fibrosis of caucasian births in the U.S. 3. Every 30 minutes, a child is born who will develop a mitochondrial disease by age 10 4. Each year, 1,000 to 4,000 children in the United states are born with a mitochondrial disease. While exact numbers of children and adults suffering from mitochondrial disease are hard to determine because so many people who suffer from mitochondrial disease are frequently misdiagnosed 4, experts now know mitochondrial disease is approaching the frequency of childhood cancers. Many are misdiagnosed with atypical cerebral palsy, various seizure disorders, childhood diseases and diseases of aging. Still others aren’t diagnosed until after death.
Treatment for mitochondrial diseases varies significantly based on the specific type of condition and the signs and symptoms present in each person. The primary aim of treatment is to alleviate symptoms and slow the progression of the condition. For example, a variety of vitamins and other supplements have been used to treat people affected by mitochondrial conditions with varying degrees of success. Other examples of possible interventions include medications to treat diabetes mellitus, surgery for cataracts, and cochlear implantation for hearing loss 5.
Mitochondrial disease in adults
Many adults are diagnosed with adult-onset mitochondrial disease. Some of these individuals have been ill their whole lives but went undiagnosed. Others have carried the genetic mutation that causes mitochondrial disease since birth but did not show any symptoms until a severe illness brought them on. Adult mitochondrial patients are affected in a similar manner to the children who are affected.
Secondary mitochondrial dysfunction in human diseases
Mitochondrial dysfunction is also seen in a number of different genetic disorders, including ethylmalonic aciduria (caused by mutation of ETHE1) 6, Friedreich ataxia (FXN) 7, hereditary spastic paraplegia 7 (SPG7) 8, and Wilson disease (ATP7B) 9, and is also seen as part of the aging process. These are not strictly mitochondrial diseases. The term mitochondrial disorder usually refers to primary disorders of mitochondrial metabolism affecting oxidative phosphorylation.
What is mitochondria
You have mitochondria present in every cell of your body except red blood cells. Mitochondria are membrane-bound cell organelles (mitochondrion, singular) that generate most of the chemical energy needed to power the cell’s biochemical reactions. The mitochondria in the cells throughout your body are responsible for creating more than 90% of the energy needed by your body to sustain life and support organ function. When mitochondria fail, less and less energy is generated within the cell. Cell injury and even cell death follow. If this process is repeated throughout the body, whole organ systems begin to fail – people get sick, and even die. The parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Chemical energy produced by the mitochondria is stored in a small molecule called adenosine triphosphate (ATP). Mitochondria contain their own small chromosomes. Generally, mitochondria, and therefore mitochondrial DNA, are inherited only from the mother. Problems with mitochondria, the structures that produce energy for all cells, have been linked to the development of Parkinson’s disease.
Mitochondria play a fundamental role in cell physiology; mitochondria organelles are involved in a variety of processes, including bioenergetics, various metabolic pathways, including crucial anabolic and catabolic reactions, such as ATP (adenosine triphosphate) synthesis, the tricarboxylic acid cycle (citric acid cycle or Kreb cycle), and biosynthetic processes, and govern fundamental cellular actions, including proliferation, immunity, and autophagy. Mitochondrial damage and malfunction have been related to the pathogenesis of a large number of human pathologies, such as mitochondrial diseases, neurodegenerative diseases, cancer, cardiovascular diseases, metabolic disorders, and aging. The participation of mitochondria in the redox equilibrium and redox signaling of the cell is also pivotal. Modification of the redox state and increased reactive oxygen species (ROS) production within mitochondria have major consequences for both mitochondrial and extramitochondrial processes and, ultimately, modulate fundamental cellular phenomena such as autophagy and apoptosis.
In people with mitochondrial disease, the parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected 10. Based upon recent epidemiological studies, mitochondrial disorders affect at least 1 in 8000 of the general population 11. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Figure 1. Mitochondria cell
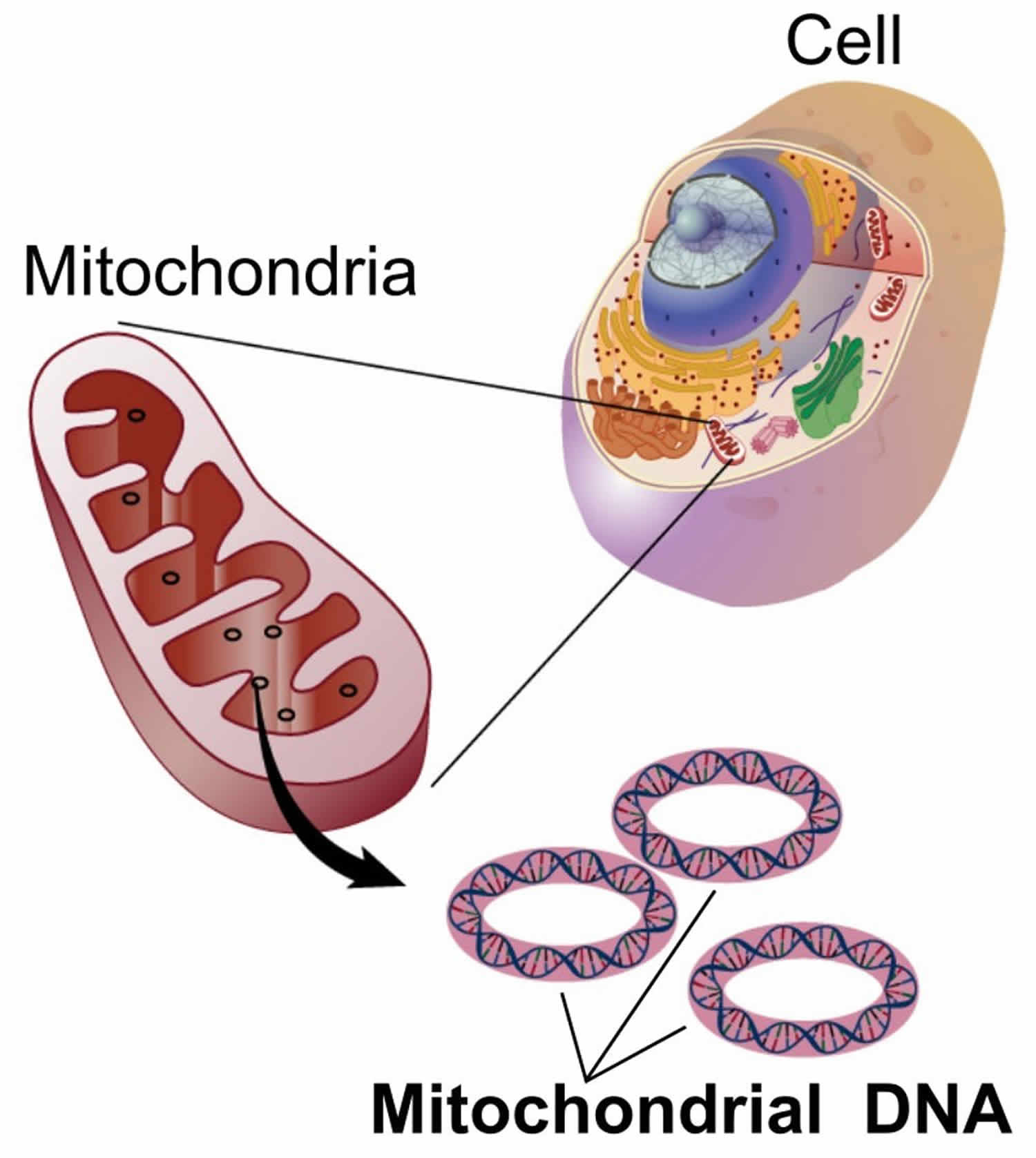
Mitochondrial diseases are the result of either inherited or spontaneous mutations in mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) which lead to altered functions of the proteins or RNA molecules that normally reside in mitochondria. Problems with mitochondrial function, however, may only affect certain tissues as a result of factors occurring during development and growth that scientists do not yet understand. Even when tissue-specific isoforms of mitochondrial proteins are considered, it is difficult to explain the variable patterns of affected organ systems in the mitochondrial disease syndromes seen clinically.
Because mitochondria perform so many different functions in different tissues, there are literally hundreds of different mitochondrial diseases. Each disorder produces a spectrum of abnormalities that can be confusing to both patients and physicians in early stages of diagnosis. Because of the complex interplay between the hundreds of genes and cells that must cooperate to keep our metabolic machinery running smoothly, it is a hallmark of mitochondrial diseases that identical mtDNA mutations may not produce identical diseases. Genocopies are diseases that are caused by the same mutation but which may not look the same clinically.
The converse is also true: different mutations in mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) can lead to the same diseases. In genetics, these are known as phenocopies. A good example is Leigh syndrome, which can be caused by about a dozen different gene defects. Leigh syndrome, originally a neuropathological description of the brain of one affected child, was described by Denis Leigh, the distinguished British physician, in 1951. It is characterized by bilaterally symmetrical MRI abnormalities in the brain stem, cerebellum, and basal ganglia, and often accompanied by elevated lactic acid levels in the blood or cerebrospinal fluid. Leigh syndrome may be caused by the NARP mutation, the MERRF mutation, complex I deficiency, cytochrome oxidase (COX) deficiency, pyruvate dehydrogenase (PDH) deficiency, and other unmapped DNA changes. Not all children with these DNA abnormalities will go on to develop Leigh syndrome, however.
Mitochondrial diseases are even more complex in adults because detectable changes in mitochondrial DNA (mtDNA) occur as we age and, conversely, the aging process itself may result from deteriorating mitochondrial function. There is a broad spectrum of metabolic, inherited and acquired disorders in adults in which abnormal mitochondrial function has been postulated or demonstrated.
What causes mitochondrial disease
Mitochondrial genetic disorders can be caused by changes (mutations) in either the mitochondrial DNA or nuclear DNA that lead to dysfunction of the mitochondria. Most DNA (hereditary material that is passed from parent to child) is packaged within the nucleus of each cell (known as nuclear DNA). However, mitochondria (the structures in each cell that produce energy) contain a small amount of their own DNA, which is known as mitochondrial DNA 3.
When the mitochondria are not working properly, the body does not have enough energy to carry out its normal functions. This can lead to the variety of health problems associated with mitochondrial genetic disorders 12.
Mitochondrial disease prognosis
That is a tough question to answer because the prognosis depends upon the severity of mitochondrial disease and other criteria. As more research dollars are raised to find more effective treatments and ultimately a cure, some of the affected children and adults are living fairly normal lives with mitochondrial disease. In other cases, children may not be able to see, hear, talk or walk. Affected children may not survive beyond their teenage years. Adult onset can result in drastic changes from an active lifestyle to a debilitating illness is a short amount of time.
Is mitochondrial disease fatal?
Some people live a normal life and are minimally affected; others can be severely compromised with mitochondrial disease. It is completely individualized and mitochondrial disease outcome is unpredictable.
Mitochondrial disease life expectancy
The life expectancy depends upon the type and the severity of mitochondrial disease.
Types of mitochondrial disease
- Alpers Disease (Progressive Infantile Poliodystrophy)
- Symptoms: seizures, dementia, spasticity, blindness, liver dysfunction, and cerebral degeneration.
- Treatment is limited to symptom management and supportive care and family education should be addressed as soon as the family is able to absorb the diagnosis.
- Autosomal Dominant Optic Atrophy: Autosomal Dominant Optic Atrophy is an autosomally inherited disease that affects the optic nerves. Autosomal Dominant Optic Atrophy causes reduced visual acuity and is a contributing factor of blindness, vision loss or impairment, beginning in childhood. Autosomal Dominant Optic Atrophy is due to mitochondrial dysfunction mediating the death of optic nerve fibers. In complicated cases of Autosomal Dominant Optic Atrophy, in addition to bilateral optic neuropathy, several other neurological signs of neurological involvement can be observed:peripheral neuropathy, deafness, cerebellar ataxia, spastic paraparesis and myopathy.
- Barth Syndrome / LIC (Lethal Infantile Cardiomyopathy)
- Symptoms: skeletal myopathy, cardiomyopathy, short stature, and neutropenia.
- Cause: X-linked recessive.
- Early and accurate diagnosis is key to survival for affected individuals. Historically, boys died of heart failure or infection by three years of age, but today, with improved diagnosis, treatment, and management, the survival rate and future of these boys is much brighter.
- There are no specific treatments for Barth syndrome. Not all patients exhibit all of the symptoms at any one time, therefore heart symptoms, infections, and nutrition problems are treated as they arise. Careful attention and monitoring for symptoms is advised.
- Beta-oxidation Defects
- Treatment: High carbohydrate-low fat diet, administration of medium-chain triglyceride oil, and diet supplementation with carnitine and/or riboflavin. Avoidance of fasting.
- Carnitine-Acyl-Carnitine Deficiency
- Symptoms: Seizures, apnea, bradycardia, vomiting, lethargy, coma, enlarged liver, limb weakness, myoglobin in the urine, Reye-like symptoms triggered by fasting.
- Cause: Autosomal recessive.
- Carnitine Deficiency
- Symptoms: Seizures, apnea, bradycardia, vomiting, lethargy, coma, enlarged liver, limb weakness, myoglobin in the urine, Reye-like symptoms triggered by fasting.
- Cause: Autosomal recessive.
- Creatine Deficiency Syndromes
- Additional names: Cerebral Creatine Deficiency Syndromes (CCDS) Includes: Guanidinoaceteate Methyltransferase Deficiency (GAMT Deficiency), L-Arginine:Glycine Amidinotransferase Deficiency (AGAT Deficiency), and SLC6A8-Related Creatine Transporter Deficiency (SLC6A8 Deficiency).
- Symptoms: general: mental retardation, seizures, speech delay. Additional possible symptoms: GAMT – behavioral disorder – including autistic behaviors; movement disorders SLC6A8 – growth retardation; (males) mild to severe mental retardation; (females) learning and behavior problems.
- Cause: GAMT and AGAT – autosomal recessive; SLC6A8 – X-linked.
- Co-Enzyme Q10 Deficiency
- Symptoms: Encephalomyopathy, mental retardation, exercise intolerance, ragged-red fibers, and recurrent myoglobin in the urine.
- Cause: Probably autosomal recessive.
- Treatment: Administration of Co-enzyme Q10
- Complex I Deficiency (NADH dehydrogenase (NADH-CoQ reductase) deficiency)
- Inside the mitochondrion is a group of proteins that carry electrons along four chain reactions (Complexes I-IV), resulting in energy production. This chain is known as the Electron Transport Chain. A fifth group (Complex V) churns out the ATP. Together, the electron transport chain and the ATP synthase form the respiratory chain and the whole process is known as oxidative phosphorylation or OXPHOS. Complex I, the first step in this chain, is the most common site for mitochondrial abnormalities, representing as much as one third of the respiratory chain deficiencies. Often presenting at birth or in early childhood, Complex I deficiency is usually a progressive neuro-degenerative disorder and is responsible for a variety of clinical symptoms, particularly in organs and tissues that require high energy levels, such as brain, heart, liver, and skeletal muscles. A number of specific mitochondrial disorders have been associated with Complex I deficiency including: Leber’s hereditary optic neuropathy (LHON), MELAS, MERRF, and Leigh Syndrome (LS).
- There are three major forms of Complex I deficiency:
- 1. Myopathy (muscle disease) – starting in childhood or adulthood, and characterized by weakness or exercise intolerance.
- 2. Mitochondrial encephalomyopathy (brain and muscle disease) – beginning in childhood or adulthood and involving variable symptom combinations which may include: eye muscle paralysis, pigmentary retinopathy (retinal color changes with loss of vision), hearing loss, sensory neuropathy (nerve damage involving the sense organs), seizures, dementia, ataxia (abnormal muscle coordination), and involuntary movements. This form of Complex I deficiency may cause Leigh Syndrome and MELAS.
- 3.Fatal infantile multisystem disorder – characterized by poor muscle tone, developmental delay, heart disease, lactic acidosis, and respiratory failure.
- Most cases of Complex I deficiency result from autosomal recessive inheritance (combination of defective nuclear genes from both the mother and the father). Less frequently, the disorder is maternally inherited or sporadic and the genetic defect is in the mitochondrial DNA.
- Treatment: As with all mitochondrial diseases, there is no cure for Complex I deficiency. A variety of treatments, which may or may not be effective, can include such metabolic therapies as: riboflavin, thiamine, biotin, co-enzyme Q10, carnitine, and the ketogenic diet. Therapies for the infantile multisystem form have been unsuccessful.
- The clinical course and prognosis for Complex I patients is highly variable and may depend on the specific genetic defect, age of onset, organs involved, and other factors.
- Complex II Deficiency (Succinate dehydrogenase deficiency)
- Symptoms: Encephalomyopathy and various manifestations, including failure to thrive, developmental delay, hyoptonia, lethargy, respiratory failure, ataxia, myoclonus. Lactic acidosis common. May cause Leigh Syndrome.
- Cause: Probably autosomal recessive.
- Complex III Deficiency (Ubiquinone-cytochrome c oxidoreductase deficiency)
- Symptoms: Four major forms:
- Fatal infantile encephalomyopathy, congenital lactic acidosis, hypotonia, dystrophic posturing, seizures, and coma. Ragged-red fibers common.
- Encephalomyopathies of later onset (childhood to adult life): various combinations of weakness, short stature, ataxia, dementia, hearing loss, sensory neuropathy, pigmentary retinopathy, and pyramidal signs. Ragged-red fibers common. Possible lactic acidosis.
- Myopathy, with exercise intolerance evolving into fixed weakness. Ragged-red fibers common. Possible lactic acidosis.
- Infantile histiocytoid cardiomyopathy.
- Cause: Probably autosomal recessive.
- Symptoms: Four major forms:
- Complex IV Deficiency / COX Deficiency (Cytochrome c oxidase deficiency)
- Cytochrome c oxidase deficiency is caused by a defect in Complex IV of the respiratory chain.
- Symptoms: Two major forms:
- Encephalomyopathy: Typically normal for the first 6 to 12 months of life and then show developmental regression, ataxia, lactic acidosis, optic atrophy, ophthalmoplegia, nystagmus, dystonia, pyramidal signs, and respiratory problems. Frequent seizures. May cause Leigh Syndrome
- Myopathy: Two main variants:
- Fatal infantile myopathy: may begin soon after birth and accompanied by hypotonia, weakness, lactic acidosis, ragged-red fibers, respiratory failure, and kidney problems.
- Benign infantile myopathy: may begin soon after birth and accompanied by hypotonia, weakness, lactic acidosis, ragged-red fibers, respiratory problems, but (if the child survives) followed by spontaneous improvement.
- Cause: Probably autosomal recessive.
- Complex V Deficiency (ATP synthase deficiency)
- Symptoms: Slow, progressive myopathy.
- Chronic Progressive External Ophthalmoplegia Syndrome (CPEO)
- Symptoms: Similar to those of KSS plus: visual myopathy, retinitis pigmentosa, dysfunction of the central nervous system.
- Cause: Single mitochondrial DNA deletions. Mitochondrial DNA point mutations: A3243G (most common)
- CPT I Deficiency
- Symptoms: Enlarged liver and recurrent Reye-like episodes triggered by fasting or illnesses.
- Causes: Autosomal recessive.
- Treatment: Medium-chain triglycerides.
- CPT II Deficiency
- Symptoms – Myopathic: Exercise intolerance, fasting intolerance, muscle pain, muscle stiffness, and myoglobin in the urine.
- Infantile form: Reye-like syndrome, enlarged liver, hypoglycemia, enlarged heart, and cardiac arrhythmia.
- Causes: Autosomal recessive.
- Treatment – Non-Infantile: High carbohydrate, low-fat diet.
- Symptoms – Myopathic: Exercise intolerance, fasting intolerance, muscle pain, muscle stiffness, and myoglobin in the urine.
- Kearns-Sayre Syndrome (KSS)
- Kearns-Sayre Syndrome is a rare disorder. It is usually caused by a single large deletion (loss) of genetic material within the DNA of the mitochondria (mtDNA), rather than in the DNA of the cell nucleus. These deletions, of which there are over 150 species, typically arise spontaneously. Less frequently, the mutation is transmitted by the mother.
- Kearns-Sayre Syndrome is a slowly progressive multi-system mitochondrial disease that often begins with drooping of the eyelids (ptosis). Other eye muscles eventually become involved, resulting in paralysis of eye movement. Degeneration of the retina usually causes difficulty seeing in dimly lit environments.
- Kearns-Sayre Syndrome is characterized by three main features:
- typical onset before age 20 although may occur in infancy or adulthood
- paralysis of specific eye muscles (called chronic progressive external ophthalmoplegia – CPEO)
- degeneration of the retina causing abnormal accumulation of pigmented (colored)material (pigmentary retinopathy).
- In addition, one or more of the following conditions is present:
- block of electrical signals in the heart (cardiac conduction defects)
- elevated cerebrospinal fluid protein
- incoordination of movements (ataxia).
- Patients with Kearns-Sayre Syndrome may also have such problems as deafness, dementia, kidney dysfunction, and muscle weakness. Endocrine abnormalities including growth retardation, short stature, or diabetes may also be evident.
- As with all mitochondrial diseases, there is no cure for Kearns-Sayre Syndrome. Treatments are based on the types of symptoms and organs involved, and may include: Coenzyme Q10, insulin for diabetes, cardiac drugs, and a cardiac pacemaker which may be life-saving. Surgical intervention for drooping eyelids may be considered but should be undertaken by specialists in ophthalmic surgical centers.
- KSS is slowly progressive and the prognosis varies depending on severity. Death is common in the third or fourth decade and may be due to organ system failures.
- Cause: Most cases are due to large mitochondria DNA deletions.
- Lactic Acidosis
- Cause: The accumulation of lactic acid due to its production exceeding its use. Chronic lactic acidosis is a common symptom of mitochondrial disease.
- LBSL – Leukodystrohpy
- Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation (LBSL) is a result of a DARS2 gene mutation and is characterized by slowly progressive cerebellar ataxia and spasticity with dorsal column dysfunction (decreased position and vibration sense). The neurologic dysfunction involves the legs more than the arms. The tendon reflexes are retained. Deterioration of motor skills usually starts in childhood or adolescence, but occasionally not until adulthood. Dysarthria develops over time. Occasional findings include epilepsy; learning problems; cognitive decline; and reduced consciousness, neurologic deterioration, and fever following minor head trauma. Many affected individuals become wheelchair dependent in their teens or twenties. LBSL is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk family members and prenatal testing for pregnancies at increased risk are possible if the disease-causing mutations have been identified in the family.
- Long-Chain Acyl-CoA Dehydrongenase Deficiency (LCAD)
- Symptoms: Usually causes a fatal syndrome, in infants, typified by failure to thrive, enlarged liver, enlarged heart, metabolic encephalopathy, and hypotonia.
- Cause: Autosomal recessive.
- LCHAD
- Symptoms: Encephalopathy, liver dysfunction, cardiomyopathy, and myopathy. Also pigmentary retinopathy and peripheral neuropathy.
- Cause: Autosomal recessive.
- Treatment: High carbohydrate-low fat diet, administration of medium-chain triglyceride oil, and diet supplementation with carnitine and/or riboflavin. Avoidance of fasting.
- Leigh Disease or Syndrome: Leigh’s Disease is a progressive neurometabolic disorder with a general onset in infancy or childhood, often after a viral infection, but can also occur in teens and adults. It is characterized on MRI by visible necrotizing (dead or dying tissue) lesions on the brain, particularly in the midbrain and brainstem. The child often appears normal at birth but typically begins displaying symptoms within a few months to two years of age, although the timing may be much earlier or later. Initial symptoms can include the loss of basic skills such as sucking, head control, walking and talking. These may be accompanied by other problems such as irritability, loss of appetite, vomiting and seizures. There may be periods of sharp decline or temporary restoration of some functions. Eventually, the child may also have heart, kidney, vision, and breathing complications.
- Treatments: There is no cure for Leigh’s Disease. Treatments generally involve variations of vitamin and supplement therapies, often in a “cocktail” combination, and are only partially effective. Various resource sites include the possible usage of: thiamine, coenzyme Q10, riboflavin, biotin, creatine, succinate, and idebenone. Experimental drugs, such as dichloroacetate (DCA) are also being tried in some clinics. In some cases, a special diet may be ordered and must be monitored by a dietitian knowledgeable in metabolic disorders.
- Prognosis: The prognosis for Leigh’s Disease is poor. Depending on the defect, individuals typically live anywhere from a few years to the mid-teens. Those diagnosed with Leigh-like syndrome or who did not display symptoms until adulthood tend to live longer.
- LHON (Leber’s Hereditary Optic Neuropathy): Leber’s Hereditary Optic Neuropathy is extremely rare genetic disorder which can cause sudden, profound, painless loss of central vision. While symptoms can begin at any age and in men or women, it is most common among men around age 20. Symptoms usually begin as painless blurriness in one eye quickly progressing to severe central vision loss, typically followed within a few months by similar symptoms in the second eye. At this time, experts are unable to tell which, if any family members will develop symptoms, though on average 50% of men and 15% of women with a LHON mutation will lose vision in their lifetime.
- LHON Plus (Leber’s Hereditary Optic Neuropathy Plus extraocular symptoms). Vision loss is typically the only symptom of LHON, LHON PLUS is a rare disease that occurs when a patient has a LHON genetic mutation and also has extraocular symptoms (issues other than vision-related). A patient with LHON PLUS may experience this multi-system involvement, with symptoms involving the central nervous system, ears, endocrinological organs, heart, bone marrow, arteries, kidneys, or the peripheral nervous system.
- Luft Disease
- Symptoms: Hypermetabolism, with fever, heat intolerance, profuse perspiration, polyphagia, polydipsia, ragged-red fibers, and resting tachycardia. Exercise intolerance with mild weakness.
- Cause: Unknown inheritance.
- Multiple Acyl-CoA Dehydrogenase Deficiency (MAD) / Glutaric Aciduria Type II
- Cause: Defects of the flavoproteins responsible for transferring electrons (ETF or ETF-dehydrogenase) therefor affecting the function of all six ETF-funneling acyl-CoA dehydrogenases.
- Medium-Chain Acyl-CoA Dehydrongenase Deficiency (MCAD)
- Symptoms: Afflicts infants or young children with episodes of encephalopathy, enlarged and fatty degeneration of the liver, and low carnitine in the blood.
- Cause: Autosomal recessive.
- Treatment: See Beta-oxidation Defects.
- Mitochondrial Encephalomyopathy Lactic Acidosis and Strokelike Episodes (MELAS)
- MELAS – Mitochondrial Myopathy (muscle weakness), Encephalopathy (brain and central nervous system disease), Lactic Acidosis (buildup of a cell waste product), and Stroke-like Episodes (partial paralysis, partial vision loss, or other neurological abnormalities)
- MELAS is a progressive neurodegenerative disorder with typical onset between the ages of 2 and 15, although it may occur in infancy or as late as adulthood. Initial symptoms may include stroke-like episodes, seizures, migraine headaches, and recurrent vomiting. Usually, the patient appears normal during infancy, although short stature is common. Less common are early infancy symptoms that may include developmental delay, learning disabilities or attention-deficit disorder. Exercise intolerance, limb weakness, hearing loss, and diabetes may also precede the occurrence of the stroke-like episodes. Stroke-like episodes, often accompanied by seizures, are the hallmark symptom of MELAS and cause partial paralysis, loss of vision, and focal neurological defects. The gradual cumulative effects of these episodes often result in variable combinations of loss of motor skills (speech, movement, and eating), impaired sensation (vision loss and loss of body sensations), and mental impairment (dementia). MELAS patients may also suffer additional symptoms including: muscle weakness, peripheral nerve dysfunction, diabetes, hearing loss, cardiac and kidney problems, and digestive abnormalities. Lactic acid usually accumulates at high levels in the blood, cerebrospinal fluid, or both.
- MELAS is maternally inherited due to a defect in the DNA within mitochondria. There are at least 17 different mutations that can cause MELAS. By far the most prevalent is the A3243G mutation, which is responsible for about 80% of the cases. The incidence is unknown, although the epidemiological studies of the MELAS-3243 mtDNA mutation have estimated the prevalence to be 1-16/100,000 in the adult population.
- There is no cure or specific treatment for MELAS. Although clinical trials have not proven their efficacy, general treatments may include such metabolic therapies as: CoQ10, creatine, phylloquinone, and other vitamins and supplements. Drugs such as seizure medications and insulin may be required for additional symptom management. Some patients with muscle dysfunction may benefit from moderate supervised exercise. In select cases, other therapies that may be prescribed include dichloroacetate (DCA) and menadione, though these are not routinely used due to their potential for having harmful side effects.
- The prognosis for MELAS is poor. Typically, the age of death is between 10 to 35 years, although some patients may live longer. Death may come as a result of general body wasting due to progressive dementia and muscle weakness, or complications from other affected organs such as heart or kidneys.
- Symptoms: Short statue, seizures, stroke-like episodes with focused neurological deficits, recurrent headaches, cognitive regression, disease progression, ragged-red fibers.
- Cause: Mitochondrial DNA point mutations: A3243G (most common)
- Mitochondrial Enoyl CoA Reductase Protein Associated Neurodegeneration (MEPAN)
- MEPAN is caused by 2 mutations in the gene MECR ( which encodes the protein mitochondrial trans-2-enoyl-coenzyme A-reductase), a newly described mitochondrial disease, which includes optic atrophy, childhood-onset dystonia (a movement disorder involving high tone and posturing); children may develop difficulty walking, with speech articulation and with vision loss.
- MRI lesions in the basal ganglia can mimic Leigh syndrome as well as one of the NBIA disorders (neurodegneration with brain iron accumulation).
- Common symptoms: movement disorder (dystonia, spasticity, chorea, ataxia) in childhood, reduced vision, difficulty with speech (dysarthria), balance issues
- Myoclonic Epilepsy and Ragged-Red Fiber Disease (MERRF)
- MERRF is a progressive multi-system syndrome usually beginning in childhood, but onset may occur in adulthood. The rate of progression varies widely. Onset and extent of symptoms can differ among affected siblings.
- The classic features of MERRF include:
- Myoclonus (brief, sudden, twitching muscle spasms) – the most characteristic symptom
- Epileptic seizures
- Ataxia (impaired coordination)
- Ragged-red fibers (a characteristic microscopic abnormality observed in muscle biopsy of patients with MERRF and other mitochondrial disorders) Additional symptoms may include: hearing loss, lactic acidosis (elevated lactic acid level in the blood), short stature, exercise intolerance, dementia, cardiac defects, eye abnormalities, and speech impairment.
- Although a few cases of MERRF are sporadic, most cases are maternally inherited due to a mutation within the mitochondria. The most common MERRF mutation is A8344G, which accounted for over 80% of the cases (GeneReview article). Four other mitochondrial DNA mutations have been reported to cause MERRF. While a mother will transmit her MERRF mutation to all of her offspring, some may never display symptoms.
- As with all mitochondrial disorders, there is no cure for MERRF. Therapies may include coenzyme Q10, L-carnitine, and various vitamins, often in a “cocktail” combination. Management of seizures usually requires anticonvulsant drugs. Medications for control of other symptoms may also be necessary.
- The prognosis for MERRF varies widely depending on age of onset, type and severity of symptoms, organs involved, and other factors.
- Symptoms: Myoclonus, epilepsy, progressive ataxia, muscle weakness and degeneration, deafness, and dementia.
Cause: Mitochondrial DNA point mutations: A8344G, T8356C
- Mitochondrial Recessive Ataxia Syndrome (MIRAS)
- Symptoms: encephalopathy, balance problems, ataxia, epilepsy, cognitive impairment, psychiatric symptoms, eye movement disorders, involuntary movements, peripheral neuropathy.
- Cause: POLG mutation, Recessive inheritance: Many sporadic cases.
- Mitochondrial Cytopathy
- Mitochondrial DNA Depletion
- Symptoms: Three forms:
- Congenital myopathy: Neonatal weakness, hypotonia requiring assisted ventilation, possible renal dysfunction. Severe lactic acidosis. Prominent ragged-red fibers. Death due to respiratory failure usually occurs prior to one year of age.
- Infantile myopathy: Following normal early development until one year old, weakness appears and worsens rapidly, causing respiratory failure and death typically within a few years.
- Hepatopathy: Enlarged liver and intractable liver failure, myopathy. Severe lactic acidosis. Death is typical within the first year.
- Cause: Probably autosomal recessive.
- Symptoms: Three forms:
- Mitochondrial Encephalopathy
- Includes: Encephalomyopathy, Encephalomyelopathy
- Myoneurogastointestinal Disorder and Encephalopathy (MNGIE)
- Symptoms: Progressive external ophthalmoplegia, limb weakness, peripheral neuropathy, digestive tract disorders, leukodystrophy, lactic acidosis, ragged red fibers.
- Neuropathy, Ataxia, and Retinitis Pigmentosa (NARP)
- Cause: Mitochondrial DNA point mutations in genes associated with Complex V: T8993G, (also T8993C by some researchers). Leigh Syndrome may result if the percentage of mutation is high enough.
- Pearson Syndrome
- Symptoms: Bone marrow and pancreas dysfunction.
- Cause: Single mitochondrial DNA deletions. Inheritance is usually sporadic. Those who survive infancy usually develop Kearns-Sayre Syndrome (KSS).
- Pyruvate Carboxylase Deficiency
- Symptoms: Lactic acidosis, hypoglycemia, severe retardation, failure to thrive.
- Common symptoms: seizures and spasticity.
- Cause: Autosomal recessive.
- Pyruvate Dehydrogenase Deficiency
- Symptoms: Lactic acidosis, ataxia, pyruvic acidosis, spinal and cerebellar degeneration.
- Less common: Agenesis of the corpus callosum and lesions in the basal ganglia, cerebelum, and brain stem.
- Also: growth delay, hypotonia, seizures, and polyneuropathy. Sometimes found to be the cause of Leigh Syndrome.
- Cause: Varies.
- Pyruvate Dehydrogenase Complex Deficiency. Pyruvate dehydrogenase complex deficiency (PDCD) is an inherited inborn error of metabolism. This means that children born with this disorder can’t convert some of the food they eat into energy. The inability of the body to break down carbohydrates into energy produces a potentially dangerous chemical called lactic acid. High lactic acid can make symptoms worse, as well as cause low blood pressure, vomiting, high heart rate, and rapid breathing.
- Symptoms:
- Newborns
- Lactic Acidosis (excessive blood lactate level)
- Respiratory Failure (ventilator dependence)
- Lethargy or Coma
- Infants/Toddlers
- Developmental Delay leading to intellectual disability
- Seizures
- Brain Malformations (enlarged ventricles, atrophy of brain, corpus callosum malformations)
- Small head circumference
- Hypotonia (low muscle tone, especially trunk – seen in most individuals)
- Hypertonia (increased muscle tone, especially arms and legs – seen in many, and may accompany hypotonia of trunk)
- Older
- Leigh’s Syndrome (lesions in brainstem or basal ganglia of brain, with loss of motor skills, eye movement problems, breathing problems, etc.)
- Ataxia (loss of balance)
- Dystonia (abnormal posture of limbs)
- Hypotonia (low muscle tone, especially trunk – seen in most individuals)
- Hypertonia (increased muscle tone, especially arms and legs – seen in many, and may accompany hypotonia of trunk)
- Peripheral Neuropathy (nerve damage, numbness/tingling)
- Newborns
- Treatment: Ketogenic Diet is the main treatment for pyruvate dehydrogenase complex deficiency currently. About half of children with pyruvate dehydrogenase complex deficiency will experience seizures. Because children present differently with different types and severity of seizures, anti-epileptic (anti-seizure) medications are tailored to each individual child. Your doctor will work with you and your child to determine the best medication course to control your child’s seizures.
- Symptoms:
- POLG Mutations
- Short-Chain Acyl-CoA Dehydrogenase Deficiency (SCAD)
- Symptoms: Failure to thrive, developmental delay, and hypoglycemia.
- Cause: Autosomal recessive.
- Treatment: High carbohydrate-low fat diet, administration of medium-chain triglyceride oil, and diet supplementation with carnitine and/or riboflavin. Avoidance of fasting.
- SCHAD
- Symptoms: Encephalopathy and possibly liver disease or cardiomyopathy.
- Cause: Autosomal recessive.
- Treatment: High carbohydrate-low fat diet, administration of medium-chain triglyceride oil, and diet supplementation with carnitine and/or riboflavin. Avoidance of fasting.
- Very Long-Chain Acyl-CoA Dehydrongenase Deficiency (VLCAD)
- Symptoms: Various manifestations, ranging from fatal infantile encephalopathy to recurrent myoglobin in the urine, similar to the myopathic form of CPT II deficiency.
- Cause: Autosomal recessive.
- Treatment: High carbohydrate-low fat diet, administration of medium-chain triglyceride oil, and diet supplementation with carnitine and/or riboflavin. Avoidance of fasting.
Mitochondrial disease symptoms
The child or adult is at highest risk for neurological and organ damage during and for the two weeks following an illness. Therefore even a simple flu or cold virus can have devastating effects on the patient, even death. Any illness must be treated immediately with medical interventions, like IV fluids and IV antibiotics.
The severity of mitochondrial disease symptoms is different from person to person.
The most common mitochondrial disease symptoms are:
- Poor growth
- Loss of muscle coordination, muscle weakness
- Neurological problems, seizures
- Autism, autistic spectrum, autistic-like features
- Visual and/or hearing problems
- Developmental delays, learning disabilities
- Heart, liver or kidney disease
- Gastrointestinal disorders, severe constipation
- Diabetes
- Increased risk of infection
- Thyroid and/or adrenal dysfunction
- Autonomic dysfunction
- Neuropsychological changes characterized by confusion, disorientation, and memory loss.
Mitochondrial Disease should be suspected when three or more organ systems are involved.
Brain
- Developmental delays
- Migraines
- Seizures
- Dementia
- Autistic Features
- Atypical cerebral palsy
- Neuro-psychiatric disturbances
- Intellectual Disabilities
- Strokes
Nerves
- Weakness (may be intermittent)
- Fainting
- Absent reflexes
- Neuropathic pain
- Dysautonomia
- Temperature Instability
Muscles
- Weakness
- Irritable bowel syndrome
- Gastroesophogeal reflux
- Cramping
- Diarrhea or constipation
- Hypotonia
- Gastrointestinal problems
- Pseudo-obstruction
- Dysmotility
Kidneys
- Renal tubular acidosis or wasting
Heart
- Cardiac conduction defects (heart blocks)
- Cardiomyopathy
Liver
- Hypoglycemia (low blood sugar)
- Liver failure
Ears and eyes
- Visual loss and blindness
- Optic atrophy
- Acquired strabismus
- Ptosis
- Ophthalmoplegia
- Retinitis pigmentosa
- Hearing loss
- Deafness
Pancreas and other glands
- Diabetes
- Exocrine
- Pancreatic
- Failure
- Parathyroid failure
Systemic
- Failure to gain weight
- Unexplained vomiting
- Respiratory problems
- Fatigue
- Short stature
Symptoms an undiagnosed individual exhibit
The child or adult may have seizures, severe vomiting, failure to thrive, heat/cold intolerance, poor muscle tone, delayed achievement of milestones, severe diarrhea/constipation, feeding problems, unable to fight typical childhood infections or repeated infections and fevers without a known origin. A “red flag” for mitochondrial disease is when a child or adult has more than three organ systems with problems or when a “typical” disease exhibits atypical qualities.
Mitochondrial disease diagnosis
For many patients, mitochondrial disease is an inherited genetic condition. An uncertain percentage of patients acquire symptoms due to other factors, including mitochondrial toxins. Unfortunately, mitochondrial genetic disorders can be difficult to diagnose. Referral to an appropriate research center is critical 13. If experienced physicians are involved, however, diagnoses can be made through a combination of clinical observations, laboratory evaluation, cerebral imaging, and muscle biopsies. Despite these advances, many cases do not receive a specific diagnosis. Mitochondrial diseases are often suspected in people who have a condition that effects multiple, unrelated systems of the body. In some cases, the pattern of symptoms may be suggestive of a specific mitochondrial condition. If the mitochondrial disease-causing gene(s) associated with the particular condition is known, the diagnosis can then be confirmed with genetic testing 13.
It is important to determine which type of mitochondrial disease inheritance is present in order to predict the risk of recurrence for future children. The types of mitochondrial disease inheritance include:
- DNA (DNA contained in the nucleus of the cell) inheritance. Also called autosomal inheritance.
- If this gene trait is recessive (one gene from each parent), often no other family members appear to be affected. There is a 25 percent chance of the trait occurring in other siblings.
- If this gene trait is dominant (a gene from either parent), the disease often occurs in other family members. There is a 50 percent chance of the trait occurring in other siblings.
- MtDNA (DNA contained in the mitochondria) inheritance.
- There is a 100 percent chance of the trait occurring in other siblings, since all mitochondria are inherited from the mother, although symptoms might be either more or less severe.
- Combination of mtDNA and nDNA defects:
- Relationship between nDNA and mtDNA and their correlation in mitochondrial formation is unknown.
If a mitochondrial genetic disorder is suspected but the signs and symptoms do not suggest a specific diagnosis, a more extensive work-up may be required. In these cases, a physician may start by evaluating the levels of certain substances in a sample of blood or cerebrospinal fluid. Other tests that can support a diagnosis include 2:
- Exercise testing
- Magnetic resonance spectroscopy and exercise testing may also be of use to detect an elevated lactate level in brain or muscle at rest, or a delay in the recovery of the ATP peak in muscle after exercise. Magnetic resonance spectroscopy (MRS) and exercise testing (with measurement of blood concentration of lactate) may be used to detect evidence of abnormal mitochondrial function non-invasively.
- Imaging studies of the brain such as MRI or CT scan. CT may show basal ganglia calcification and/or diffuse atrophy. MRI may show focal atrophy of the cortex or cerebellum, or high signal change on T2-weighted images, particularly in the occipital cortex 14. There may also be evidence of a generalized leukoencephalopathy 15. Cerebellar atrophy is a prominent feature in children 14.
- Electroencephalography (EEG): Electroencephalography (EEG) is indicated in individuals with suspected encephalopathy or seizures. Encephalopathy may be associated with generalized slow wave activity on the EEG. Generalized or focal spike and wave discharges may be seen in individuals with seizures.
- Peripheral neurophysiologic studies are indicated in individuals with limb weakness, sensory symptoms, or areflexia. Electromyography (EMG) is often normal but may show myopathic features. Nerve conduction velocity (NCV) may be normal or may show a predominantly axonal sensorimotor polyneuropathy.
- Tests that evaluate the heart including electrocardiography and echocardiography. Both electrocardiography and echocardiography may indicate cardiac involvement (cardiomyopathy or atrioventricular conduction defects).
- Muscle biopsy
- Plasma/CSF Lactate/pyruvate:
- Measurement of plasma lactate concentration is indicated in individuals with features of a myopathy or CNS disease. Fasting blood lactate concentrations above 3 mm/L support a diagnosis of mitochondrial disease.
- Measurement of CSF lactate concentration is indicated in individuals with suspected CNS disease. Fasting CSF lactate concentrations above 1.5 mm/L support a diagnosis of mitochondrial disease.
- Note: Normal plasma or CSF lactic acid concentration does not exclude the presence of a mitochondrial disorder.
When possible, confirming a diagnosis with genetic testing can have important implications for family members. Identifying the disease-causing gene(s) will give the family information about the inheritance pattern and the risk to other family members. It will also allow other at-risk family members to undergo genetic testing 2.
Mitochondrial disease treatment
At this time, there are no cures for mitochondrial disease. The management of mitochondrial disease is largely supportive 16.
The goals of mitochondrial disease treatment:
- To alleviate symptoms
- To slow down the progression of the disease
Management issues may include early diagnosis and treatment of diabetes mellitus, cardiac pacing, ptosis correction, intraocular lens replacement for cataracts, and cochlear implantation for sensorineural hearing loss.
A variety of vitamins and co-factors have been used in individuals with mitochondrial disorders, although a Cochrane systematic review has shown that evidence supporting their use is lacking 17.
Food supplements such as ubiquinone (coenzyme Q10, ubidecarenone) are generally well tolerated and some individuals report a subjective benefit on treatment.
Individuals with complex I and/or complex II deficiency may benefit from oral administration of riboflavin.
The role of exercise therapy in mitochondrial myopathy is currently being evaluated 18.
Coenzyme Q10 is specifically indicated in persons with defects of CoQ10 biosynthesis.
Idebenone shows promise for the treatment of Leber hereditary optic neuropathy.
Some secondary causes of mitochondrial dysfunction, such as ethylmalonic aciduria, may have specific treatments 6.
The possibility of nuclear transfer as a means of preventing transmission of pathogenic mtDNA variants is currently being explored 19.
Those with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) may benefit from hematopoietic stem cell transplantation.
The benefits of treatment and effectiveness of therapies vary:
- Sometimes, treatment may be beneficial and noted immediately in some disorders.
- Sometimes, the benefits of treatment may take a few months to notice.
- Sometimes, the benefits of treatment may never be noticed, but the treatment may be effective in delaying or stopping the progression of the disease.
- Sometimes, some patients may not benefit from therapy.
Key points of treatment:
- Never forget that for some symptoms, there is already a standard treatment (anticonvulsant medication for epilepsy, physical therapy for motor problems, etc.)
- Dietary
- Vitamins and supplements
- Avoidance of stressful factors
- Treatment must be tailored by the patient’s physician to meet that patient’s need. Many of these therapies are totally ineffective in some mitochondrial disorders and would be a waste of time, money and effort. In some cases, the treatment could be dangerous.
How effective is treatment?
- Effectiveness varies from patient to patient, depending on the exact disorder and the severity of the disorder.
- As a general rule, those with mild disorders tend to respond to treatment better than those with severe disorders.
- In some circumstances, the treatment can be tailored specifically to the patient, and that treatment is effective, whereas in other circumstance, the treatment is “empiric”, meaning that the treatment makes sense, but that the benefit of treatment is not obvious or proven to be effective.
- Treatment will not reverse the damage already sustained, such as brain malformations.
Dietary Therapy
Many patients, including young children or mentally impaired persons have already “self-adjusted” their diet, because they know what foods their body seem to tolerate. The points below are not meant to be suggested therapies for all patients with oxidative phosphorylation (OXPHOS) disorders, and some of the points are dangerous for patients with other disorders. Do not make any of these dietary changes without consulting a physician. A dietitian experienced in metabolic disorders may be helpful.
Avoid fasting. This is perhaps the most important part of the treatment for most people with metabolic disorders. Fasting means “not eating” and avoiding fasting means avoid prolonged periods without a meal (even an overnight “fast” from 8 pm to 8 am may be dangerous in some patients). This also means that some patients should not intentionally try to lose weight by decreasing their food intake.
In some patients, an unintended fast resulting from an illness that causes vomiting or loss of appetite (like the flu) should be hospitalized to ensure continuous nutrition (intravenous glucose, for example).
Small frequent meals may be better than a typical 3-meal-a-day routine for some patients.
A snack before bedtime may be helpful for some patients. This snack should not be mainly “sugar,” like a candy bar, Jello or sweetened cereal. It is usually best if the snack consists of a complex carbohydrate. Cornstarch is the best complex carbohydrate, but this is not very tasty. Theoretically, the best snack would be a homemade low-sugar rice pudding thickened with a lot of cornstarch. Pasta, a peanut butter sandwich, bread and butter, unsweetened cereal (oatmeal) or a sandwich are acceptable. Many patients benefit by being woken up in the middle of the night for a small meal.
In order to ensure adequate frequent nutrition, sometimes a feeding tube needs to be placed in order for the person to receive feeding at night.
Fat
a) There is conflicting evidence regarding high-fat meals in patients with electron transport chain disorders. In patients that seem to gain weight and thrive on a high-fat diet, it makes sense to continue the treatment. The extra fat can also be in the form of MCT (medium chain triglyceride oil), which is easier to metabolize (4c).
b) In other patients with oxidative phosphorylation (OXPHOS) disorders, reducing fat may be helpful. This includes reducing added oil, butter, & margarine, and cutting down on cheese and fatty meats. This recommendation is not meant to avoid fats altogether. A defect in oxidative phosphorylation (OXPHOS) can create an “energy backup”, as the respiratory chain cannot handle the flow of electrons coming into it. This backup may result in the formation of excess free fatty acids (fats waiting to be burned) , which can poison the enzyme (adenosine nucleotide translocase) that exchanges the low-energy ADP located outside the mitochondria for the high-energy ATP formed at complex v. If you take the approach of limiting fats, extra effort needs to be made to increase the total carbohydrate (in the form of complex carbohydrates) in the diet.
c) In some patients, adding fat in the form of medium chain triglycerides (MCT), may be helpful. Medium chain triglycerides of 8 to 10 carbons long are easier to metabolize (turn into energy) than the longer chain triglycerides (those with 12-18 carbons) because they do not require carnitine to be transported into the mitochondria. MCT Oil© is mainly made of 8 and 10 carbon triglycerides and this type of oil does not occur in nature, but is made from coconut oil. MCT Oil© is made by the baby formula company Mead-Johnson. It comes in quart bottles, available by prescription and runs about $70 a quart. It can be added like oil over pasta and rice. You can cook with it, but this is a light oil and burns easily. The special rules are explained in a recipe book that you can request from the pharmacist. Depending on the situation, a patient may benefit from a few teaspoons to a few tablespoons a day. There are oils sold in health food stores called “MCT Oil” or “medium chain triglyceride oil”. These are much less expensive ($25 per quart), but make sure there is a certified analysis on the label, stating that the vast majority of the oil is C-8 and C-1 0 (and not C-12 or higher).
Iron
Iron generates free radicals under certain conditions, which is especially bad in mitochondrial diseases because the free radicals injure mitochondrial DNA and “poke holes” in the mitochondria, making a bad problem worse. Therefore, excess iron is theoretically harmful.
In people with mitochondrial disease, there is no routine need to give supplemental iron, nor is there a reason to eat foods rich in iron, such as extra red meat, for the purpose of eating foods rich in iron. This does not mean that the person should not eat red meat, especially if they enjoy it. There is no reason to take vitamins with added iron. There is the rare instance when iron is needed, but this is not common.
In addition, vitamin C enhances the absorption of iron from the intestines, and vitamin C should not be given around a meal rich in iron. This is important to remember because some experts feel that vitamin C is a good antioxidant, and also may be helpful in some disorders of oxidative phosphorylation (OXPHOS).
Supportive Therapies
Some mitochondrial disease patients may need additional supportive therapies such as physical therapy, speech therapy or respiratory therapy. While these therapies will not reverse the disease process, they may preserve or even improve the patient’s existing functioning, mobility and strength.
Toxins
Avoid Alcohol and Cigarettes
Alcohol has been know to hasten the progression of some mitochondrial disorders. Cigarette smoke, probably due to the carbon monoxide, is known to hasten the progression of some conditions. Remember that carbon monoxide kills by inhibiting complex IV of oxidative phosphorylation (OXPHOS), why make it worse? Cigarette smoke will make it worse.
Avoid MSG
MSG (monosodium glutamate) has for years been known to cause migraine headaches in otherwise healthy individuals, and may trigger these events in susceptible people with mitochondrial disease. MSG is frequently added to Chinese (and other Asian) foods, and is also found in high levels of dried and canned soup. Read the label and avoid MSG if there is any sensitivity.
Vitamins and Cofactors
Vitamins and cofactors are compounds that are required in order for the chemical reactions, which make energy, to run efficiently. By definition, a cofactor can be made by the body, whereas a vitamin cannot, and therefore must be eaten. For most people, a regular diet contains all the vitamins one could possibly need and their bodies can make as much of any specific cofactor that it needs. For those with mitochondrial disorders, added vitamins and cofactors may be useful.
The use of supplemental vitamins and cofactors is largely unproven and their use is therefore controversial in patients with mitochondrial diseases. For disorders of oxidative phosphorylation (OXPHOS), coenzyme Q10 is considered as a generally accepted effective therapy, although it may not ultimately be effective for an individual patient. Other treatments may be effective in one disorder but not in others. Because of the varied nature of mitochondrial diseases some therapies may be helpful in many, but not in all patients and therefore cannot be considered as “proven and effective.” Some treatments should only be undertaken under the specific guidance of your physician. For specific information about the controversy, as it relates to your or your child’s situation, ask your physician. Most of these vitamins can be purchased from many sources, including the drugstore.
These supplemental compounds can serve two functions:
- possibly enhance enzyme function and result in improved efficiency of energy generation
- serve as antioxidants, which may slow the progression of the disease
Table 1. Vitamins and Supplements That May be Helpful
| First Tier Supplements | |
| Supplement | Dose Range |
| CoQ10 | 5 – 15 mg/kg/day |
| Levo-carnitine (Carnitor) | Variable, starting dose of 30 mg/kg/day, typical maximum of 100 mg/kg/day |
| Riboflavin (B2) | 100 – 400 mg a day |
| Second Tier Supplement | |
| Supplement | Dose Range |
| Acetyl-L-Carnitine | 250 – 1000 mg per day |
| Thiamine (B1) | 50 – 100 mg a day |
| Niacin (B3) | 50 – 100 mg a day |
| Vitamin E | 200 – 400 IU; 1 – 3 times a day |
| Vitamin C | 100 – 500 mg; 1 – 3 times a day |
| Lipoic Acid (a -lipoate) | 60 – 200 mg; 3 times a day |
| Selenium | 25 – 50 micrograms a day |
| b -carotene | 10,000 IU; every other day to daily |
| Biotin | 2.5 – 10 mg a day |
| Folic Acid | 1 – 10 mg a day |
Physiologic Stress
Physiologic stress is triggered by external factors that may result in worsening the metabolic situation, which may result in temporary or permanent worsening of the condition. It is impossible to avoid all physiologic stressful conditions, so one should not attempt to do so. However, recognizing what may be stressful for patients allows one to adjust the lifestyle. Many patients and their parents have already identified these stresses, despite not knowing why the stresses were important, and avoid them.
Cold Stress is extremely important. Thermal regulation (temperature control) is not always normal in people with mitochondrial diseases and exposure to cold can result in severe heat loss and trigger an energy crisis. When going out into the cold, all exposed body parts should be covered, and exposure to extreme cold should be avoided for anything more than a short period. Over bundling can be a problem too (see below).
Heat Stress can be a problem in some people. This is especially true of those with an inability to sweat normally. Heat exhaustion and heat stroke may occur on hot days. It is typical for parents to describe that their child seems to “wilt” in situations like hot classrooms or direct sunlight, whereas the other children function normally. Light clothing is important. Patients should avoid direct sunlight on hot days and stay indoors if it is too warm outside. An air-conditioned environment may be needed.
Starvation — avoid fasting.
Lack of sleep may possibly be harmful.
References- What is Mitochondrial Disease? https://www.umdf.org/what-is-mitochondrial-disease/
- Chinnery PF. Mitochondrial Disorders Overview. 2000 Jun 8 [Updated 2014 Aug 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1224
- About Mitochondrial Disease – Mito FAQ. http://www.mitoaction.org/mito-faq
- Mitochondrial disease Frequently Asked Questions https://www.umdf.org/faq-page-1/
- Treatments & Therapies. https://www.umdf.org/what-is-mitochondrial-disease/treatments-therapies/
- Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M, Zeviani M. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–5.
- Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–7.
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Dürr A, Fontaine B, Ballabio A. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–83.
- Lutsenko S, Cooper MJ. Localization of the Wilson’s disease protein product to mitochondria. Proc Natl Acad Sci U S A. 1998;95:6004–9.
- Schapira AH. Mitochondrial disease. Lancet 2006;368(9529):70‐82.
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG. Prevalence of mitochondrial DNA disease in adults. Annals of Neurology 2008;63(1):35‐9.
- Chinnery PF. Mitochondrial Disorders Overview. 2000 Jun 8 [Updated 2014 Aug 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1224/
- Getting a Diagnosis. https://www.umdf.org/what-is-mitochondrial-disease/getting-a-diagnosis/
- Scaglia F, Wong LJ, Vladutiu GD, Hunter JV. Predominant cerebellar volume loss as a neuroradiologic feature of pediatric respiratory chain defects. AJNR Am J Neuroradiol. 2005;26:1675–80.
- Barragán-Campos HM, Vallee JN, Lo D, Barrera-Ramirez CF, Argote-Greene M, Sanchez-Guerrero J, Estanol B, Guillevin R, Chiras J. Brain magnetic resonance imaging findings in patients with mitochondrial cytopathies. Arch Neurol. 2005;62:737–42.
- Chinnery PF, Turnbull DM. Epidemiology and treatment of mitochondrial disorders. Am J Med Genet. 2001;106:94–101
- Chinnery P, Majamaa K, Turnbull D, Thorburn D. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2006;1:CD004426
- Murphy JL, Blakely EL, Schaefer AM, He L, Wyrick P, Haller RG, Taylor RW, Turnbull DM, Taivassalo T. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain. 2008;131:2832–40
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, Herbert M, Turnbull DM. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–5
- Mitochondrial disease Treatments & Therapies https://www.umdf.org/what-is-mitochondrial-disease/treatments-therapies/





{kind=link}