Occipital horn syndrome
Occipital horn syndrome previously known as Ehlers–Danlos syndrome type IX or X-linked cutis laxa, is a rare X-linked recessive connective tissue disorder of copper metabolism caused by pathogenic variants in ATP7A, encoding a copper transporter 1. The mutated ATP7A gene encodes a copper-transporting ATPase, localized in the trans-Golgi membrane of the cell, and results in abnormal metabolism and distribution of copper 2. Occipital horn syndrome represents a milder form of Menkes disease, characterized by low seric concentrations of normal ATPase and less aggressive phenotype 3.
Occipital horn syndrome is characterized by “occipital horns,” distinctive wedge-shaped calcium deposits at the sites of attachment of the trapezius muscle and the sternocleidomastoid muscle to the occipital bone (Figure 2) 4. Occipital horns may be clinically palpable or observed on skull radiographs. Individuals with occipital horn syndrome also have sagging and non-stretchy skin (cutis laxa) and loose joints, coarse hair, bony exostoses, bladder diverticula, inguinal hernias, and vascular tortuosity, attributed to a decreased activity of lysyl oxidase (LOX), a cupro-enzyme involved in collagen crosslinking. Individuals with occipital horn syndrome are said to have normal or slightly reduced intelligence 5.
The absence of large case series and natural history studies precludes efficient diagnosis and management of occipital horn syndrome patients 1.
Occipital horn syndrome may be considered a “difficult-to-diagnosis” entity, not only because of its rarity, but also because it has a variable, heterogeneous and sometimes unspecific clinical involvement, whose severity levels can also be irregularly defined. A correct and early diagnosis is important in order to start an appropriate follow-up, preventing severe complications.
Laboratorial studies revealing low copper and ceruloplasmin levels in the blood and high copper levels in cultured fibroblasts are suggestive of occipital horn syndrome 6. However, genetics has nowadays the main role in this diagnostic process, with the identification of the ATP7A gene mutations enabling the definitive diagnosis 7.
Figure 1. Occipital horn syndrome
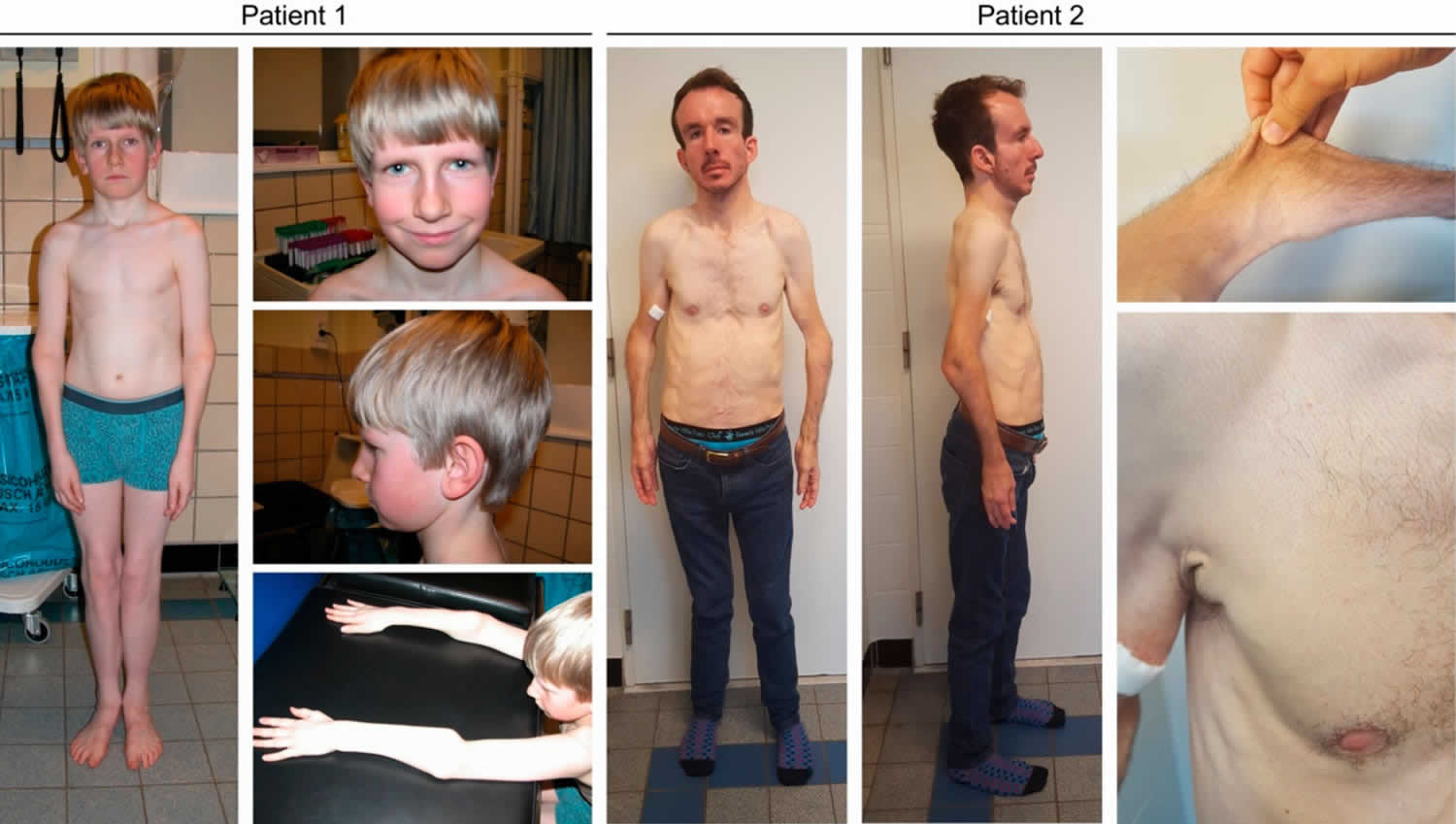
Footnote: Clinical characteristics in subjects Patient 1 and Patient 2. Subject Patient 1 displays mild facial dysmorphism with a flat face, deep-set eyes, a long and narrow nose, large ears and mild webbing of the neck. Craniofacial features in Patient 2 are more pronounced and include dolichocephaly, a high forehead, biparietal narrowing, downslanting eyes, a convex nasal ridge, midfacial hypoplasia, micrognathia and low-set ears. No hair abnormalities were observed. Both have lax and hyperextensible skin, marked joint hypermobility and deformities of the proximal radius.
[Source 1 ]Figure 2. Occipital horn syndrome occipital exostoses
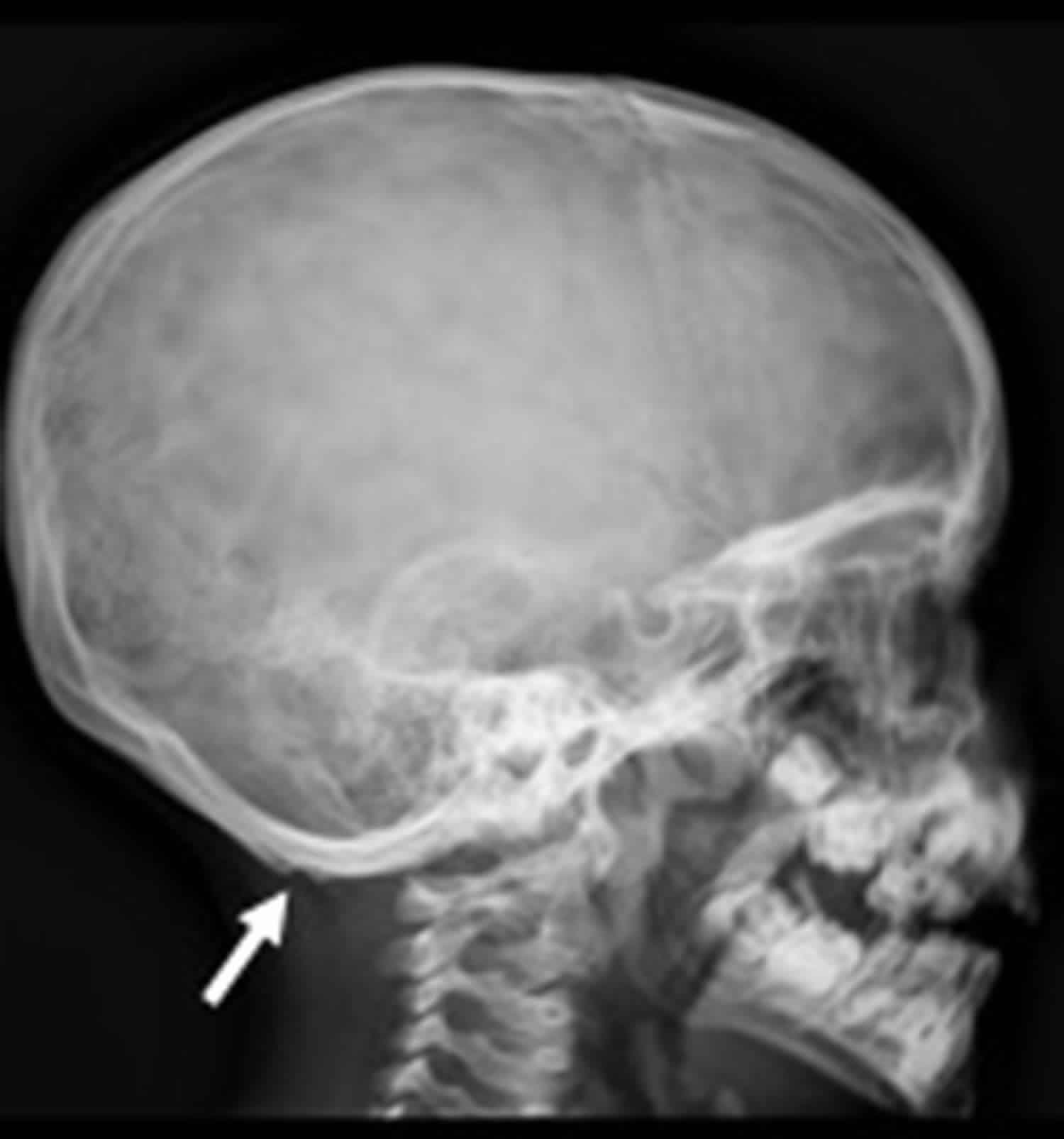
Footnote: Lateral skull X-ray showing bilateral occipital exostoses (arrow).
[Source 1 ]Occipital horn syndrome female
Females who are heterozygous for an ATP7A pathogenic variant are typically asymptomatic, in some instances because of favorably skewed X-chromosome inactivation 8. In theory, unfavorably skewed X-chromosome inactivation in some heterozygous females could be associated with neurologic or other clinical findings related to the disorders.
About 50% of females who are obligate heterozygotes for an ATP7A pathogenic variant demonstrate regions of pili torti 9.
Occipital horn syndrome causes
Occipital horn syndrome is caused by mutations in the ATP7A gene, and it is inherited in an x-linked recessive pattern. The ATP7A gene provides instructions for making a protein that is important for regulating copper levels in the body. Copper is necessary for many cellular functions, but it is toxic when present in excessive amounts. The ATP7A protein is found throughout the body, except in liver cells. In the small intestine, this protein helps control the absorption of copper from food. In other cells, the ATP7A protein has a dual role and shuttles between two cellular locations. The protein normally resides in a cell structure called the Golgi apparatus, which modifies newly produced proteins, including enzymes. In the Golgi apparatus, the ATP7A protein supplies copper to certain enzymes that are critical for the structure and function of bone, skin, hair, blood vessels, and the nervous system. If copper levels in the cell environment are elevated, however, the ATP7A protein moves to the cell membrane and eliminates excess copper from the cell.
Occipital horn syndrome inheritance pattern
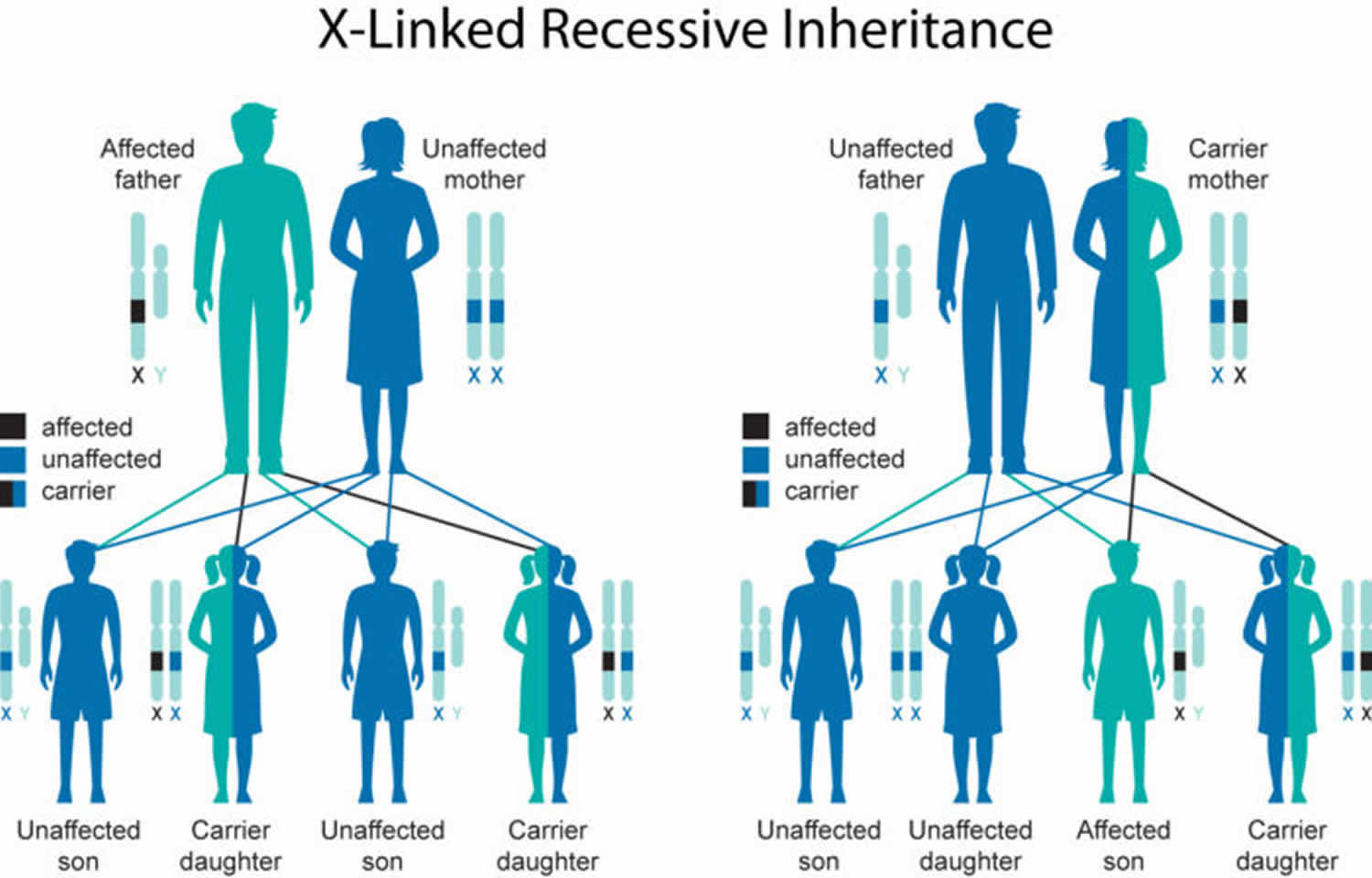
Occipital horn syndrome ATP7A-related copper transport disorders is inherited in an X-linked manner. Approximately one third of affected males have no family history of occipital horn syndrome. If the mother is a heterozygote, the risk of transmitting the ATP7A pathogenic variant is 50% in each pregnancy: a male who inherits the pathogenic variant will be affected with the disorder present in his brother; females who inherit the pathogenic variant will be heterozygotes and will not be affected. Males with occipital horn syndrome will pass the pathogenic variant to all of their daughters and none of their sons. When the pathogenic variant has been identified in an affected family member, heterozygote testing for at-risk female relatives, prenatal testing for pregnancies at increased risk, and preimplantation genetic diagnosis are possible. Prenatal testing for Menkes disease is technically possible by copper transport studies in cultured chorionic villus cells or amniocytes, although its availability is limited.
Figure 3. Occipital horn syndrome X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Occipital horn syndrome symptoms
Occipital horn syndrome is a rare X-linked recessive connective tissue disorder characterized by prominent connective tissue abnormalities including cutis laxa, inguinal hernias, joint laxity, coarse hair, bony exostoses, vascular tortuosity and bladder diverticula, and pathognomonic exostoses with clinically palpable or observed on skull radiographs “occipital horns” (downward pointing exostoses situated in the tendinous insertions of the sternocleidomastoid and trapezius muscles). In addition, patients may show mild to moderate intellectual disability 10.
The clinical characteristics of occipital horn syndrome are listed in Table 1. Craniofacial features include mild facial dysmorphism with a long face, large ears and sagging cheeks in about half of reported patients. Hair abnormalities, including coarse hair and pili torti, are frequently present. Ubiquitous connective tissue manifestations in occipital horn syndrome include increased skin laxity and inguinal hernia.
Concerning the skeletal features, the pathognomonic occipital horns are present in all patients but one, which is probably an age-dependent discrepancy 11. Other less specific skeletal abnormalities include hammer-shaped claviculae and radial/tibial exostoses. Scoliosis, pectus deformities, coxa and genua valga, joint luxations, mainly of the radial heads and general joint hypermobility are frequent but non-specific. These skeletal problems may limit daily activities and often require multiple surgical interventions.
Urological complications, mostly giant bladder diverticula and vesicourethal reflux, are found in over 80% of subjects. Secondary problems such urinary tract infections, urinary retention, bladder rupture, or vesicoureteral reflux-mediated renal failure, which are all related to the bladder diverticula, are of great concern. Surgical interventions removing the diverticula often fail, with frequent relapse of the diverticula. Self-catheterization, vesicostomy, or continent diversion are often required for proper emptying of the bladder.
Previous reports describe a mild neurological phenotype limited to slight generalized muscle weakness and symptoms of dysautonomia 10, however, more detailed case studies reveal some conspicuous observations including developmental delay and intellectual disability in two-thirds of subjects and documented seizures in five individuals. Nevertheless, neurological affliction remains rather mild and normal intelligence is possible. Symptoms of dysautonomia, including chronic diarrhea, temperature instability, and orthostatic hypotension are present in almost 90% of patients and can be disabling.
Brain imaging studies are rarely performed in occipital horn syndrome patients and are often reported as normal. However, intracranial arterial tortuosity are noted in approximately two thirds of patients. Extracranial arterial tortuosity is less frequently observed, but has been reported in the cervical, splenic and hepatic vasculature. Aneurysm formation may complicate pre-existing tortuosity and may affect the arterial and venous circulation.
Table 1. Clinical characteristics of occipital horn syndrome
| Initial Presentation | Number of Patients |
|---|---|
| Neurological | 11 |
| Seizures | 1 |
| Developmental delay | 4 |
| Hypotonia | 6 |
| Connective tissue | 9 |
| Cephalhematoma | 4 |
| Generalized CTD | 3 |
| Inguinal hernia | 2 |
| Urogenital | 5 |
| Bladder diverticula | 3 |
| Urinary infections | 2 |
| Skeletal | 3 |
| Pectus deformity | 1 |
| Skeletal dysplasia | 1 |
| Joint pain | 1 |
| Other | 3 |
| Vomiting and diarrhea | 1 |
| Dysautonomia | 1 |
| Apnea | 1 |
| Segregation analysis | 1 |
| Unknown | 2 |
Occipital horn syndrome diagnosis
Occipital horn syndrome is often diagnosed upon the identification of connective tissue anomalies, including cutis laxa, joint hypermobility with hypotonia, mild intellectual impairment and bladder diverticula 1. Later on, the pathognomonic occipital horns become more evident. The initial differential diagnosis includes other forms of cutis laxa, including FBLN5- and LTBP4-related cutis laxa 12 and the dermatosparaxis type of Ehlers–Danlos syndrome 13, which may present with cutis laxa, hypotonia and bladder diverticula, but with normal mental development. However, in occipital horn syndrome there is no early onset emphysema as in FBLN5- and LTBP4- related cutis laxa, nor easy bruising or severe skin fragility as in the dermatosparaxis type of Ehlers–Danlos syndrome. ATP6V0A2-related cutis laxa may also present with mild intellectual impairment but does not commonly show bladder diverticula or exostoses. Later on the exostoses can be mistaken for hereditary multiple exostoses due to defects in EXT1 or EXT2 14.
Males with classic occipital horn syndrome have low serum copper concentration and low serum ceruloplasmin concentration and high copper levels in cultured fibroblasts (see Table 2) 6. However, genetics has nowadays the main role in this diagnostic process, with the identification of the ATP7A gene mutations enabling the definitive diagnosis 7.
Table 2. Serum copper and serum ceruloplasmin concentration in males with occipital horn syndrome
| Serum concentration | Menkes Disease 1 | Occipital horn syndrome | ATP7A-Related Distal Motor Neuropathy | Normal |
|---|---|---|---|---|
| Copper | 0-55 µg/dL | 40-80 µg/dL | 80-100 µg/dL | 70-150 µg/dL; (birth – 6 mos: 20-70 µg/dL) |
| Ceruloplasmin | 10-160 mg/L | 110-240 mg/L | 240-310 mg/L | 200-450 mg/L; (birth – 6 mos: 50-220 mg/L) |
Footnote: 1. Diagnosis of Menkes disease using these studies alone in males under age six months is problematic given the normally low serum concentration in all children at this age.
Occipital horn syndrome treatment
To establish the extent of disease and needs in a male diagnosed with occipital horn syndrome, evaluations for the following are recommended:
- Bladder diverticula
- Inguinal hernias
- Vascular tortuosity
- Dysautonomia (chronic diarrhea, orthostatic hypotension). Note: Some medical centers have clinical autonomic testing laboratories.
- Mild cognitive deficits
- Consultation with a clinical geneticist and/or genetic counselor
Supportive treatment
- Physical therapy (strength and stretching exercises)
- Occupational therapy
- Ankle foot orthotics
Although there is no evidence that copper replacement therapy for occipital horn syndrome is clinically beneficial, it would be reasonable to expect even better overall neurodevelopmental and neurocognitive outcomes if individuals with occipital horn syndrome were identified early and treated with copper during their first three years.
Follow-up
Concerning follow-up, routine evaluation of neurodevelopment and early intervention with physiotherapy is recommended, as well as regular urological evaluation, including video urodynamic studies of bladder function. Any pain, or vascular or neurological symptoms that may relate to exostoses should be promptly investigated. To date, there are no data on a possible risk for chondrosarcomas in exostoses although these have been described in hereditary multiple exostoses 15. It could be recommended to perform MR angiography and echocardiography from puberty onwards. Aortic root dilatation and dissection, and an increased risk for intracranial bleeds and ischemic complications have been described in other related connective tissue disorders such as Ehlers–Danlos, arterial tortuosity, Marfan and some cutis laxa syndromes. Furthermore, spirometry might detect asthma at an early stage as it was reported in four individuals. Prolonged vigilance for apnea is necessary following surgery.
Occipital horn syndrome prognosis
Occipital horn syndrome patients show prominent connective tissue abnormalities and have a better prognosis and survival rate than patients with Menkes disease 10, with low childhood mortality (1/34) in occipital horn syndrome. Affected individuals typically live to at least mid-adulthood. However, there is still significant mortality in (young) adults, often related to gastrointestinal, respiratory, or bleeding complications, such as an intestinal perforation due to a gastric ulcer and (post-surgery) apnea. Fertility is unknown.
References- Defining the Clinical, Molecular and Ultrastructural Characteristics in Occipital Horn Syndrome: Two New Cases and Review of the Literature. Genes 2019, 10(7), 528; https://doi.org/10.3390/genes10070528
- Yasmeen S, Lund K, De Paepe A, De Bie S, Heiberg A, Silva J, Martins M, et al. Occipital horn syndrome and classical Menkes Syndrome caused by deep intronic mutations, leading to the activation of ATP7A pseudo-exon. Eur J Hum Genet. 2014;22(4):517-521.
- Dozza ALCB, Fernandes GD, Yuen CT, Santanna BA, Souza KS, Araujo APQC. Doenca de Menkes: Relato de Caso. Rev Bras Neurol. 2009;45(4):43-47.
- Kaler SG. ATP7A-Related Copper Transport Disorders. 2003 May 9 [Updated 2016 Aug 18]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1413
- Cutis laxa. https://ghr.nlm.nih.gov/condition/cutis-laxa
- Bazzocchi A, Femia R, Feraco P, Battista G, Canini R, Guglielmi G. Occipital horn syndrome in a woman: skeletal radiological findings. Skeletal Radiol. 2011;40(11):1491-1494.
- Tumer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013;34(3):417-429.
- Desai V, Donsante A, Swoboda KJ, Martensen M, Thompson J, Kaler SG. Favorably skewed X-inactivation accounts for neurological sparing in female carriers of Menkes disease. Clin Genet. 2011;79:176–82.
- Moore CM, Howell RR. Ectodermal manifestations in Menkes disease. Clin Genet. 1985;28:532–40.
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 71, 15–29.
- Tang, J.; Robertson, S.; Lem, K.E.; Godwin, S.C.; Kaler, S.G. Functional copper transport explains neurologic sparing in occipital horn syndrome. Genet. Med. 2006, 811, 711–718.
- Callewaert, B.; Su, C.T.; Van Damme, T.; Vlummens, P.; Malfait, F.; Vanakker, O.; Schulz, B.; Mac Neal, M.; Davis, E.C.; Lee, J.G.; et al. Comprehensive clinical and molecular analysis of 12 families with type 1 recessive cutis laxa. Hum Mutat. 2013, 341, 111–121.
- Malfait, F.; De Coster, P.; Hausser, I.; van Essen, A.J.; Franck, P.; Colige, A.; Nusgens, B.; Martens, L.; De Paepe, A. The natural history, including orofacial features of three patients with Ehlers-Danlos syndrome, dermatosparaxis type (EDS type VIIC). Am. J. Med. Genet. Part A. 2004, 1311, 18–28.
- Wuyts, W.; Van Hul, W.; De Boulle, K.; Hendrickx, J.; Bakker, E.; Vanhoenacker, F.; Mollica, F.; Lüdecke, H.J.; Sayli, B.S.; Pazzaglia, U.E.; et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet. 1998, 622, 346–354.
- Jurik, A.G.; Jorgensen, P.H.; Mortensen, M.M. Whole-body MRI in assessing malignant transformation in multiple hereditary exostoses and enchondromatosis: Audit results and literature review. Skeletal Radiol. 2019.





{kind=link}