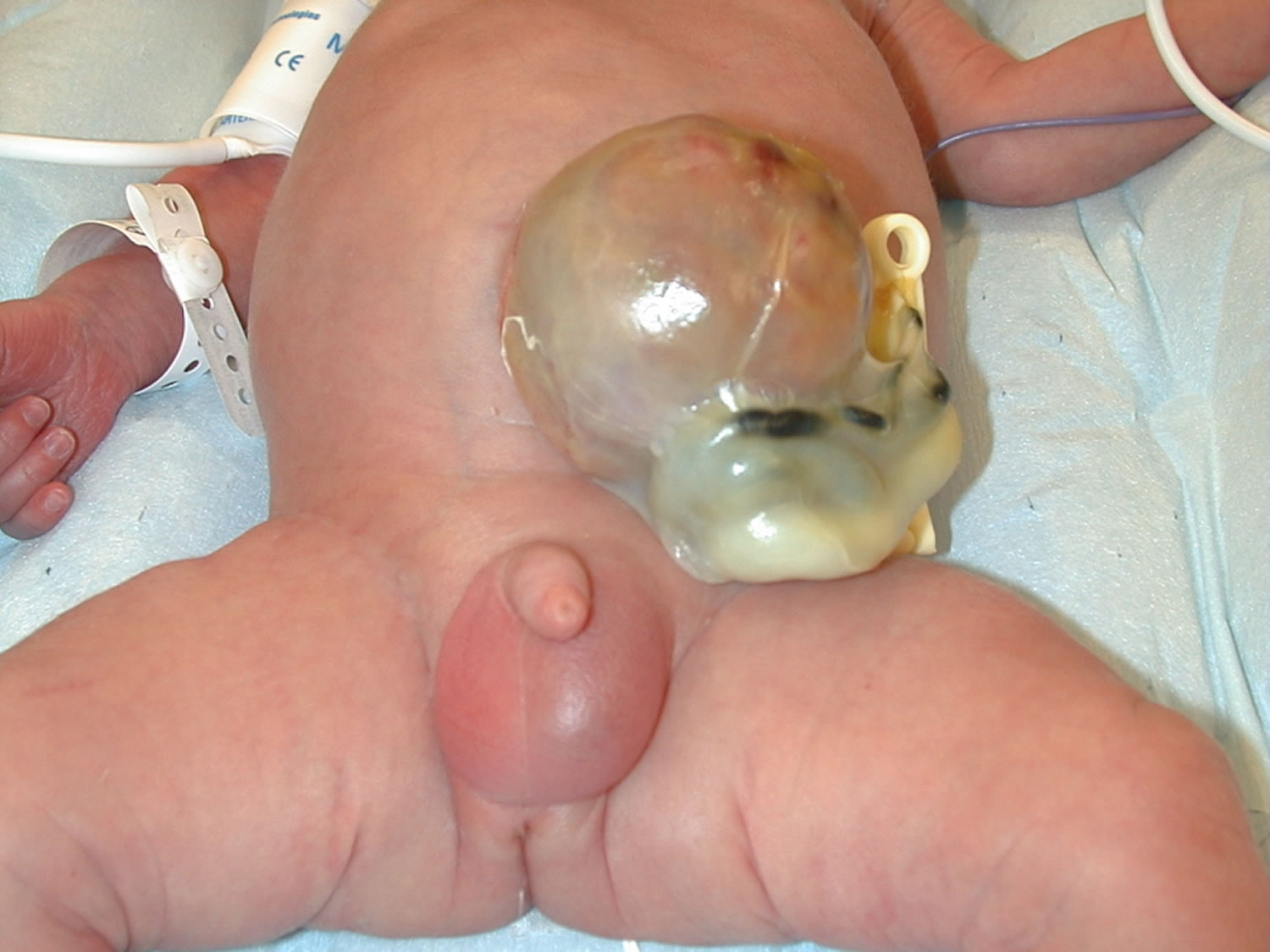
What is omphalocele
An omphalocele is also called exomphalos, is a birth defect in which an infant’s intestine, liver and other abdominal organs are outside of the body because of a hole in the belly button (navel) area at the base of the umbilical cord insertion. The intestines are covered only by a thin layer of tissue (omphalocele sac) that hardly ever is open or broken and can be easily seen. Omphalocele is a life-threatening condition. It needs to be treated soon after birth so that the baby’s organs can develop and be protected in the belly. Surgical repair is performed primarily in stages, or after a period of waiting which can last several months.
Because some or all of the abdominal (belly) organs are outside of the body, babies born with an omphalocele can have other problems. The abdominal cavity, the space in the body that holds these organs, might not grow to its normal size. Also, infection is a concern, especially if the sac around the organs is broken. Sometimes, an organ might become pinched or twisted, and loss of blood flow might damage the organ.
One-third of all babies with omphalocele have liver herniation, which is often associated with a small belly size and small lungs (known as pulmonary hypoplasia), two factors that can affect treatment and long-term outcomes. Up to one-third can also have a heart defect which can also affect long-term outcome.
Omphalocele is a rare birth defect that occurs in 1 in 4,000 — 7,000 live births. The Centers for Disease Control and Prevention (CDC) estimates that each year about 775 babies in the United States are born with an omphalocele 1. Many babies born with an omphalocele also have other birth defects, such as heart defects, neural tube defects, and chromosomal abnormalities 2. Therefore, all babies born with an omphalocele should have chromosome testing. This will help parents understand the risk for this disorder in future pregnancies.
Omphalocele occurs very early in pregnancy when the abdominal cavity fails to form normally. The abdominal cavity is normally formed at three to four weeks gestation when the disk-like embryo undergoes infolding. A large or “giant omphalocele” forms when there is a failure of lateral infolding of the embryo, resulting in an inadequate abdominal cavity with containment of the abdominal organs only by a thin clear membrane called the omphalocele sac.
Smaller omphaloceles, also referred to as “hernia of the cord,” form later (eight to 11 weeks gestation) after normal infolding of the embryo occurs (resulting in a formed abdominal cavity), when the umbilical ring fails to close around the umbilical cord resulting in a small defect that usually contains only intestine. Small omphaloceles are more likely to be associated with chromosomal defects or syndromes.
Omphalocele differs from gastroschisis in that the protruding organs are covered by the omphalocele sac. Gastroschisis has no sac and is likely caused by a rupture of a hernia of the cord, resulting in extrusion of intestine through the small umbilical defect. In contrast to gastroschisis, a ruptured giant omphalocele has all of the organs, including liver, outside the abdomen without a covering membrane.
In addition, compared to gastroschisis, giant omphaloceles are frequently associated with small lung size. Finally, whereas gastroschisis often develops in the first pregnancy of young mothers, omphaloceles typically develop in the pregnancies of older women.
If the omphalocele is identified before birth, the mother should be closely monitored to make sure the unborn baby remains healthy.
Plans should be made for careful delivery and immediate management of the problem after birth. The baby should be delivered in a medical center that is skilled at repairing abdominal wall defects. Babies are likely to do better if they do not need to be taken to another center for further treatment.
For more information about the diagnosis, delivery and treatment of babies with omphalocele, watch the educational video series about abdominal wall defects below.
Figure 1. Omphalocele
Omphalocele vs Gastroschisis
Omphalocele looks similar to gastroschisis. An omphalocele is a birth defect in which the infant’s intestine or other abdominal organs protrude through a hole in the belly button area and are covered with a membrane. In gastroschisis, there is no covering membrane (the omphalocele sac). Presence of liver tissue within the herniated sac is more common finding in omphalocele than gastroschisis. Gastroschisis has no sac and is likely caused by a rupture of a hernia of the umbilical cord, resulting in extrusion of intestine through the small umbilical defect. In contrast to gastroschisis, a ruptured giant omphalocele has all of the organs, including liver, outside the abdomen without a covering membrane.
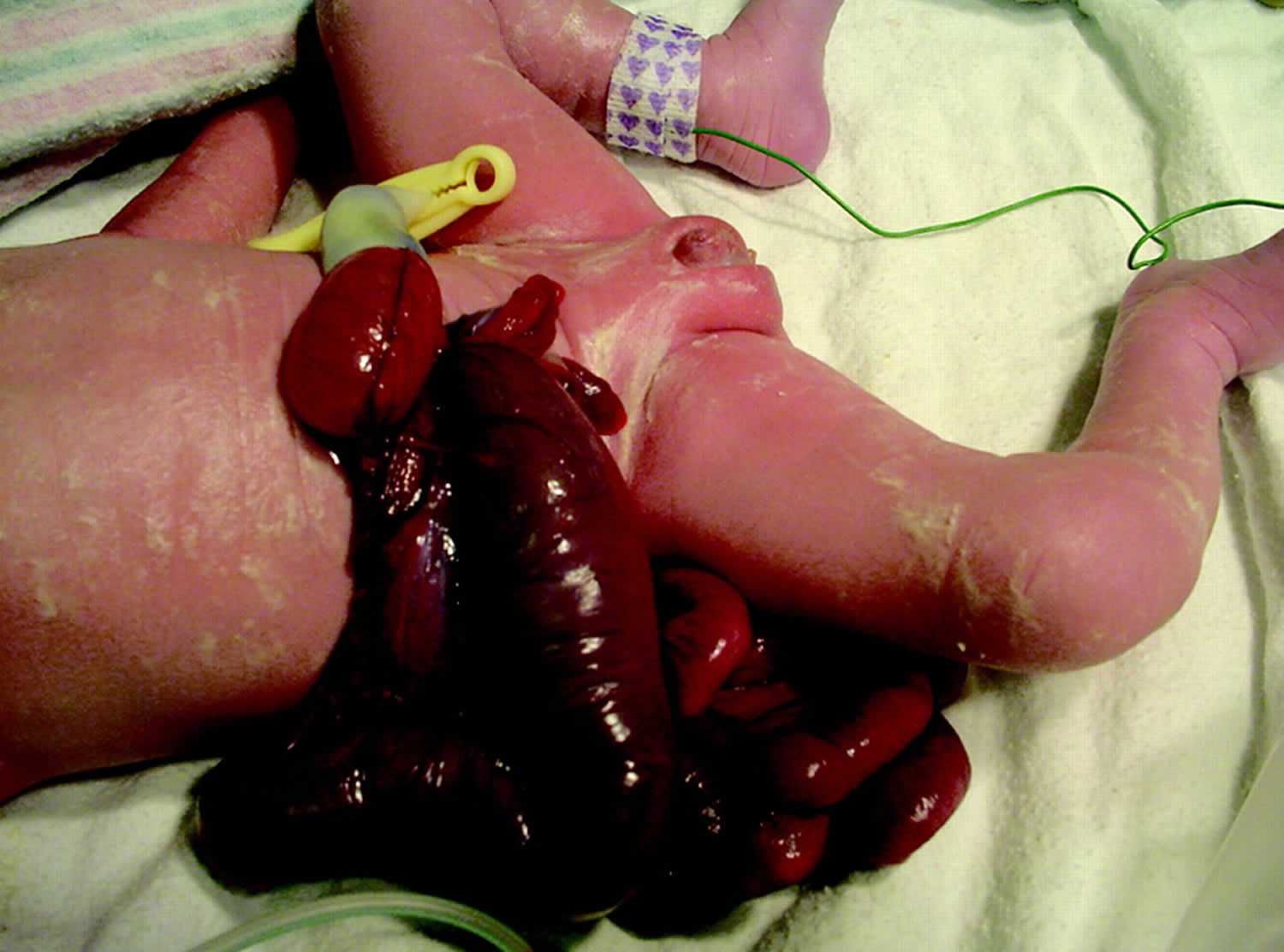
Gastroschisis is a congenital anterior abdominal wall defect, adjacent and usually to the right of the umbilical cord insertion 3. Gastroschisis occurs as a small, full-thickness periumbilical cleft either immediately adjacent to the umbilicus or separated from it by a strip of skin. This results in herniation of the abdominal contents into the amniotic sac, usually just the small intestine, but sometimes also the stomach, colon, and ovaries (Figure 2). The abdominal wall defect is relatively small compared with the size of the eviscerated bowel, which often develops walls that are matted and thickened with a fibrous peel. Gastroschisis has no covering sac and no associated syndromes. Babies with gastroschisis usually do not have other related birth defects. This differentiates it from an omphalocele, which usually is covered by a membranous sac and more frequently is associated with other structural and chromosomal anomalies (Table 1). In addition, although gastroschisis may be associated with gastrointestinal anomalies such as intestinal atresia, stenosis, and malrotation, it has a much better prognosis than omphalocele.
Mothers with the following may be at higher risk of having babies with gastrochisis:
- Younger age
- Fewer resources
- Poor nutrition during pregnancy
- Use illegal substances
If gastroschisis is found before birth, the mother will need special monitoring to make sure her unborn baby remains healthy.
Table 1. Differences Between Omphalocele and Gastroschisis
| Gastroschisis | Omphalocele | |
|---|---|---|
| Incidence | 1 in 10,000 (now increasing) | 1 in 5,000 |
| Defect Location | Right paraumbilical | Central |
| Covering Sac | Absent | Present (unless sac ruptured) |
| Description | Free intestinal loops | Firm mass including bowel, liver, etc |
| Associated With Prematurity | 50% to 60% | 10% to 20% |
| Necrotizing Enterocolitis | Common (18%) | Uncommon |
| Common Associated Anomalies | Gastrointestinal (10% to 25%)
| Trisomy syndromes (30%) |
| Cardiac defects (20%) | ||
| Beckwith-Weidemann syndrome | ||
| Cryptorchidism (31%) | Bladder extrophy | |
| Prognosis | Excellent for small defect | Varies with associated anomalies |
| Mortality | 5% to 10% | Varies with associated anomalies (80% with cardiac defect) |
Figure 2. Gastroschisis
Gastroschisis treatment
Treatment for gastroschisis is surgery to repair the defect. The surgeon will put the bowel back into the abdomen and close the defect, if possible. If the abdominal cavity is too small, a mesh sack is stitched around the borders of the defect and the edges of the defect are pulled up. The sack is called a silo. Over 5 to 7 days, the intestine returns into the abdominal cavity and the defect can be closed.
Other treatments for the baby include nutrients by IV and antibiotics to prevent infection. The baby’s temperature must be carefully controlled, because the exposed intestine allows a lot of body heat to escape.
Gastroschisis prognosis
The baby has a good chance of recovering if there are no other problems and if the abdominal cavity is large enough. A very small abdominal cavity may result in complications that require more surgeries.
Plans should be made for careful delivery and immediate management of the problem after birth. The baby should be delivered in a medical center that is skilled at repairing abdominal wall defects. Babies are likely to do better if they do not need to be taken to another center for further treatment.
Gastroschisis possible complications
A small number of babies with gastroschisis (about 10%) may have parts of the intestines that did not develop normally in the womb. With these babies, their intestines may not work normally even after the organs are put back inside the abdominal cavity.
The increased pressure from the misplaced abdominal contents can decrease blood flow to the intestines and kidneys. It can also make it difficult for the baby to expand the lungs, leading to breathing problems.
Another complication is bowel death necrosis. This occurs when intestinal tissue dies due to low blood flow or infection.
This condition is apparent at birth and will be detected in the hospital at delivery if it has not already been seen on routine fetal ultrasound exams during pregnancy. If you have given birth at home and your baby appears to have this defect, call the local emergency number (such as 911) right away.
Omphalocele causes
The causes of omphalocele among most infants are unknown. Some babies have omphalocele because of a change in their genes or chromosomes. Omphalocele might also be caused by a combination of genes and other factors, such as the things the mother comes in contact with in the environment or what the mother eats or drinks, or certain medicines she uses during pregnancy.
Omphalocele is considered an abdominal wall defect (a hole in the abdominal wall). Disturbance of organogenesis during the embryonic period results in omphalocele 4. Around weeks six through ten of pregnancy, the intestines get longer and push out from the abdominal cavity and protrude at the base of the umbilical cord. This event is known as physiologic midgut herniation and is easily identified in prenatal ultrasound between the 9 and 11 weeks of gestation 4. The liver is never present in the physiologic midgut herniation. By 11th to 12th weeks of gestation, the intestines normally go back into the belly. If this does not happen, an omphalocele occurs. Omphalocele occurs when the gut contents fail to rotate and return to the abdominal cavity. It can occasionally contain the liver in the presence of a large abdominal wall defect. The omphalocele can be small, with only some of the intestines outside of the belly, or it can be large, with many organs outside of the belly.
Recently, the Centers for Disease Control and Prevention (CDC) researchers have reported important findings about some factors that can affect the risk of having a baby with an omphalocele:
- Alcohol and tobacco: Women who consumed alcohol or were heavy smokers (more than 1 pack a day) were more likely to have a baby with omphalocele 5
- Certain medications: Women who used selective serotonin-reuptake inhibitors (SSRIs) during pregnancy were more likely to have a baby with an omphalocele 6
- Obesity: Women who were obese or overweight before pregnancy were more likely to have a baby with an omphalocele 7
Omphalocele looks similar to gastroschisis. An omphalocele is a birth defect in which the infant’s intestine or other abdominal organs protrude through a hole in the belly button area and are covered with a membrane. In gastroschisis, there is no covering membrane.
Abdominal wall defects develop as a baby grows inside the mother’s womb. During development, the intestines and other organs (liver, bladder, stomach, and ovaries or testes) develop outside the body at first and then usually return inside. In babies with omphalocele, the intestines and other organs remain outside the abdominal wall, with a membrane covering them. The exact cause for abdominal wall defects is not known.
Infants with an omphalocele often have other birth defects. Defects include genetic problems (chromosomal abnormalities), congenital diaphragmatic hernia, and heart and kidney defects. These problems also affect the overall outlook (prognosis) for the baby’s health and survival.
Omphalocele associated anomalies
Associated anomalies are high (27-91% 7) and are thought to be even commoner with smaller omphalocoele containing bowel only 8.
Omphalocele can be associated with several syndromes; the most common is Beckwith-Wiedemann syndrome 4. Beckwith-Wiedemann syndrome is an overgrowth syndrome characterized by macrosomia, enlarged tongue, neonatal hypoglycemia, ear creases, and pits, hemihypertrophy, visceromegaly, umbilical hernia, embryonal tumors, omphalocele, nephrocalcinosis, medullary sponge kidney disease, cardiomegaly, and nephromegaly. Traditionally, the macrosomia, macroglossia, and hypoglycemia are noted in the neonatal period. Hemihyperplasia is noted in segmental regions of the body or specific organs 9. Developmental and cognitive outcomes are typically normal. Patients with Beckwith-Wiedemann syndrome have an increased risk of cancer during the first eight years of life with embryonal tumors such as neuroblastoma, hepatoblastoma and Wilms tumor. These embryonal tumors have a higher cure rate when diagnosed early, making screening paramount for prevention. Screening for hepatoblastoma is performed by measuring serum alpha fetoprotein every 3 months until 4 years of age and screening for Wilms tumor is done every 3 months through 8 years of age with a complete abdominal ultrasonography 10.
Associated anomalies include:
- Chromosomal anomalies: can occur in 20-50% of cases; the risk of an associated chromosomal anomaly gets higher when the omphalocele is detected earlier in gestation
- Trisomy 18 (Edwards syndrome): considered the most common associated chromosomal anomaly. Dolichocephaly, external ear anomalies, micrognathia, short palpebral fissures, small face, clenched fist with overriding fingers, hypotonia and rocker bottom feet 11
- Trisomy 13 (Patau syndrome). Small eyes, cleft lip and palate, microcephaly, cryptorchidism, polydactyly, hypertelorism, micrognathia, cutis aplasia and external ears anomalies 12
- Trisomy 21 (Down syndrome). Hypotonia, upslanting palpebral fissures, brachycephaly, low set ears, single palmar crease, flat nasal bridge, brushfield spots around the iris, in-curved fifth digits and a gap between the first and second toes 13
- Turner syndrome
- Klinefelter syndrome
- Pallister-Killian syndrome
- Other syndromic associations
- Beckwith-Wiedemann syndrome
- Carpenter syndrome: Kleeblattschadel skull deformity (trilobed cloverleaf skull) from pancraniosynostosis, syndactyly in the hands and feet, and mental retardation 13
- Marshall-Smith syndrome: Prominent forehead, shallow orbits, blue sclerae, depressed nasal bridge, micrognathia, accelerated skeletal maturation, respiratory difficulties, mental retardation 13
- Meckel-Gruber syndrome: Occipital encephalocele, cleft lip and palate, microcephaly, microphthalmia, abnormal genitalia, polycystic kidneys, and polydactyly 13
- Pentalogy of Cantrell. Ectopia cordis, midline supraumbilical abdominal defect, sternal cleft and intracardiac defect 13
- Shprintzen-Goldberg syndrome: Craniosynostosis, dolichocephaly, hypertelorism, exophthalmos, strabismus, elongated fingers and limbs, umbilical and abdominal hernias 14
- Charge syndrome: Coloboma, heart defect, choanal atresia, growth or developmental retardation, genital abnormality, and ear anomalies 13
- OEIS complex: omphalocele, bladder/cloacal exstrophy, imperforate anus, spinal anomalies 15
- Lethal omphalocele- cleft palate syndrome
- Other fetal gastrointestinal anomalies: which confer a poor prognosis
- Fetal CNS anomalies
- Fetal cardiac anomalies: can occur in 50% of cases
- Fetal genitourinary anomalies
- Bladder exstrophy
- Cloacal exstrophy
- Fetal skeletal anomalies
- Omphalocele-radial ray (ORR) complex
Omphalocele symptoms
An omphalocele can be clearly seen. This is because the abdominal contents stick out (protrude) through the belly button area.
There are different sizes of omphaloceles. In small ones, only the intestines remain outside the body. In larger ones, the liver or other organs may be outside as well.
Omphalocele diagnosis
Omphalocele is usually seen on prenatal ultrasounds before the baby is born. The diagnosis of omphalocele is usually made by ultrasound in the middle or second trimester of pregnancy (about 20 weeks). An amniocentesis is recommended to evaluate for chromosomal abnormalities or genetic syndromes.
After an omphalocele is found, your baby will be followed very closely to make sure he or she is growing.
Your baby should be delivered at a hospital that has a neonatal intensive care unit (NICU) and a pediatric surgeon. A NICU is set up to handle emergencies that occur at birth. A pediatric surgeon has special training in surgery for babies and children. Delivery of babies with omphalocele may be vaginal or cesarean (C-section) depending on the size and contents of the omphalocele. However, most babies who have omphalocele are delivered by cesarean section in order to protect the omphalocele and prevent organs from rupturing or bleeding, which can be life-threatening.
Testing is often not necessary to diagnose omphalocele. However, babies with an omphalocele should be tested for other problems that often go with it. This includes ultrasounds of kidneys and heart (fetal echocardiogram), blood tests for genetic disorders, among other tests.
Ultrafast fetal MRI — an additional imaging technique that shows the omphalocele and the entire fetus. The MRI is used to confirm ultrasound findings and evaluate for the presence of any other anatomic abnormalities, especially central nervous system anomalies. Lung volumes are determined and compared to normal values at that gestational age (this comparison is called the observed-to-expected lung volume ratio, or O/E ratio).
Figure 3. Omphalocele ultrasound
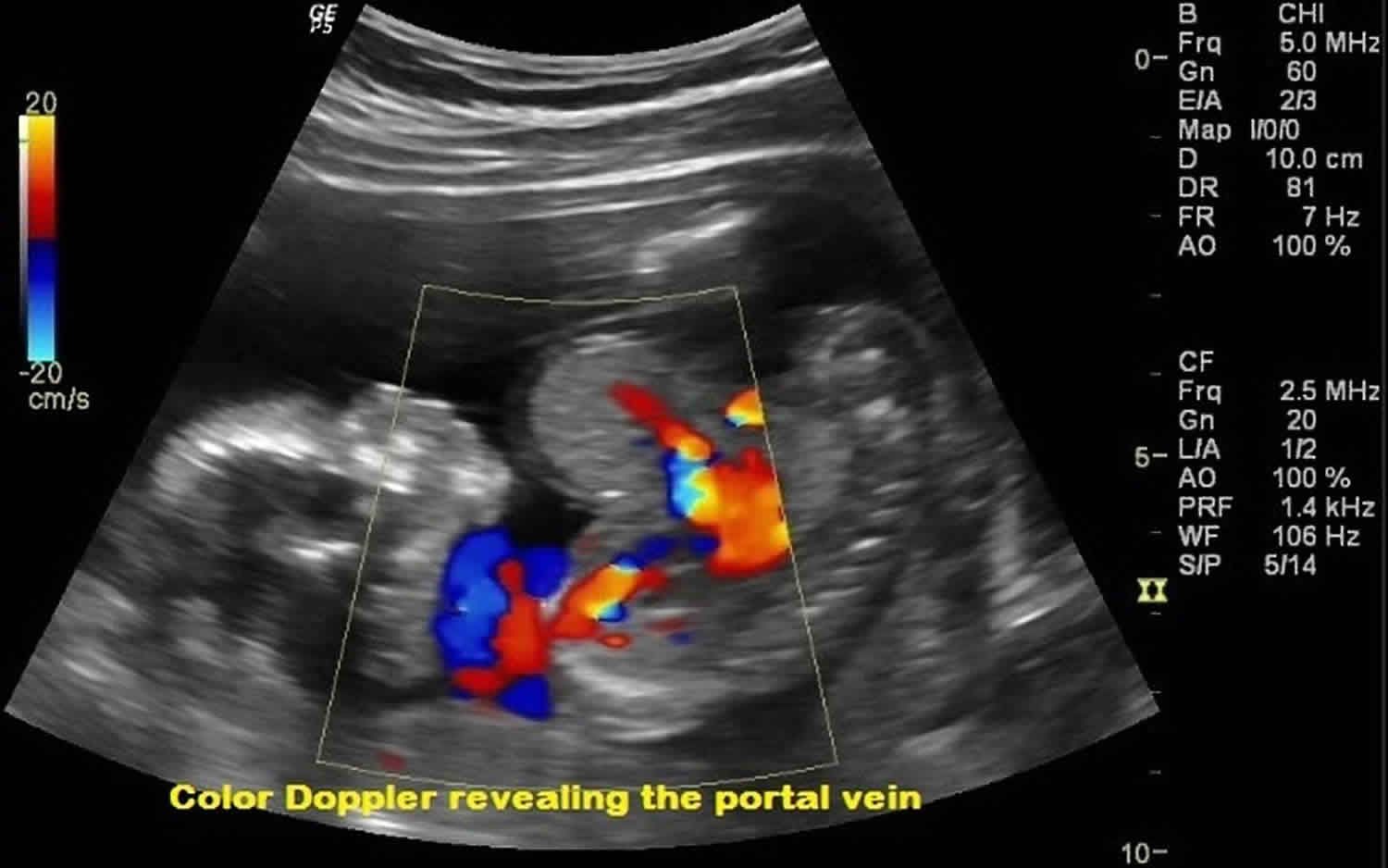
Footnote: Routine 2nd trimester antenatal care at 17 weeks of gestation. Ultrasound image and correlated doppler scanning revealed abnormal herniation of the bowel loops outside the abdominal cavity along with herniation of liver tissue. No evidence of rupture. The mean gestational age was equal to 17 weeks.
[Source 16 ]Omphalocele possible complications
Omphalocele complications can be categorized according to their time of occurrence. Prenatally and during delivery, the omphalocele may rupture, and in case of a giant omphalocele, the liver can be injured. Postnatally and after surgical repair, the complications consist of feeding difficulties, failure to thrive, inguinal hernias, gastroesophageal reflux, and occasionally esophagitis.
Another complication is bowel death (necrosis). This occurs when intestinal tissue dies due to low blood flow or infection.
The increased pressure from the misplaced abdominal contents can decrease blood flow to the intestine and kidneys. It can also make it difficult for the baby to expand the lungs, leading to breathing problems.
This condition is apparent at birth and will be detected in the hospital at delivery if it has not already been seen on routine fetal ultrasound exams during pregnancy. If you have given birth at home and your baby appears to have this defect, call the local emergency number right away.
Omphalocele survival rate
Omphalocele survival rate is close to 80%, and it is directly related to the severity of the associated anomalies as infants with isolated omphalocele have a higher survival rate (90%) 17.
Omphalocele prognosis
The prognosis for a baby with an omphalocele largely depends upon the size of the herniation and the presence or absence of other birth defects. Complete recovery is expected after surgery for an omphalocele. However, omphaloceles often occur with other birth defects. How well a child does depends on which other conditions the child has.
More than half of all babies born with omphalocele have other birth defects, including brain, spine, heart, gastrointestinal issues, genitourinary problems or Pentalogy of Cantrell. Other syndromes more commonly seen with small omphaloceles include chromosomal abnormalities such as trisomy 18 (Edwards syndrome) or genetic syndromes such as Beckwith-Wiedemann syndrome.
An omphalocele is associated with a higher morbidity and mortality than a gastroschisis, primarily due to a higher incidence of associated congenital anomalies. Smaller omphaloceles are thought to carry a worse prognosis due to increased risk of associated abnormalities.
Mortality rates can approach 80% when associated anomalies are present and increase to ~100% when chromosomal or cardiovascular anomalies exist. However, if found in isolation, then the associated mortality rate decreases to ~10% 18.
Parents should consider testing the baby, and possibly family members, for other genetic problems that are associated with this condition.
Omphalocele long-term outlook
Babies who have had small omphaloceles receive follow-up through their pediatrician and the pediatric surgeon. Those without associated defects generally have good long-term outcomes. Babies with giant omphaloceles typically need to be followed more closely by a multidisciplinary team as part of ongoing omphalocele treatment. The pulmonary hypoplasia (small lungs) associated with giant omphalocele can affect not only breathing, but also heart function, ability to feed, and overall development. This represents a significant long-term health issue.
The multidisciplinary team that follows children throughout infancy and well into school age includes:
- Pediatric surgeons
- Pediatric pulmonologists
- Pediatric cardiologists
- Developmental pediatricians
- Developmental psychologists
- Dieticians
- Audiologists
- Social workers
- Other specialties as needed, including gastroenterology, orthopedics, urology, physical therapy and occupational therapy.
Omphalocele treatment
Surgical repair of the omphalocele takes place after birth. The overall health of your baby, especially his/her respiratory status, the size of the omphalocele and the degree of liver involvement, determine the type of omphalocele treatment. Babies with small omphaloceles are monitored closely until they are ready to undergo primary repair. This means the herniated organs are placed back into the abdominal cavity and the defect is completely closed in one operation.
For babies with giant omphalocele that contain the liver and other organs, a staged repair (involving several steps, also called the Schuster procedure) is needed to gradually return the abdominal contents to the belly. This gradual process provides time for the abdominal wall to stretch to accommodate the viscera, and ensures that the lungs can continue to grow and expand without immediate pressure of surgical closure.
In a staged repair, a mesh fabric is sewn to the fascia (connective tissue) and muscle on each side of the omphalocele defect. The two pieces of fabric are then sewn together over the defect, and the omphalocele sac remains intact. Your baby returns to the NICU, where his organs are gradually returned to the abdominal cavity and the mesh is continuously tightened over the course of days or weeks. Once all of his organs are back in his belly, your child’s surgeons can remove the mesh and safely perform the final closure. Babies are monitored very closely throughout this process.
In some cases of babies with giant omphaloceles, the amount of organs protruding may be so large that there isn’t enough room in your baby’s body to fit them all inside, preventing omphalocele closure in the neonatal period. Small lung size may also delay closure. If this is the case, surgery may be postponed for months to allow the lungs and body to grow. During this time, a technique called “paint and wait” is used. The sac covering the omphalocele is painted with an antibiotic cream and covered with elastic gauze. Your baby’s skin will grow over the sac with time.
Some babies do not need to remain hospitalized during the paint and wait treatment. Your healthcare team will teach you how to do this technique so that you can bring your baby home. When all of the contents of the omphalocele are covered with skin and the lungs have had a chance to grow, your child’s surgeon will talk with you about options for surgically closing the remaining hole.
NICU stays for babies with omphalocele can range from several days to several months, depending on your baby’s lung function, the size of the defect and timing of surgical repair. Infants are monitored for common complications of omphalocele, such as feeding difficulties, bowel obstruction and gastroesophageal reflux. Babies with omphalocele also frequently have inguinal hernias, another condition that requires surgical repair.
Before your baby is ready to go home, he will need to gradually meet certain milestones, including:
- breathing on his own (may need supplemental oxygen)
- full enteral feedings (by mouth or feeding tube)
- maintaining his own temperature
- gaining weight
Another important milestone is making sure you and any other caretakers are ready to take care of your child at home. Parents are an integral part of your baby’s healthcare team and play an important role in caring for your baby from the start. During the stay in the NICU, a specialized team of surgeons, nurses, speech therapists (for feeding therapy), lactation consultants, respiratory therapists and social workers are available as needed to help educate your family about what you can do during the hospital stay, as well as caring for your baby after discharge. The nursing staff teaches you special feeding techniques and other specialized care that your child might need.
Omphalocele repair
Omphalocele repair is a procedure done on an infant to correct a birth defect in the wall of the belly (abdomen) in which all or part of the small intestine, liver, and large intestine stick out of the belly button (navel) in a thin sac.
The goal of the omphalocele repair procedure is to place the organs back into the baby’s belly and fix the defect. Repair may be done right after the baby is born. This is called primary repair. Or, the repair is done in stages. This is called staged repair.
Surgery for primary repair is most often done for a small omphalocele:
- Right after birth, the sac with the organs outside the belly is covered with a sterile dressing to protect it.
- When the doctors determine your newborn is strong enough for surgery, your baby is prepared for the operation.
- Your baby receives general anesthesia. This is medicine that allows your baby to sleep and be pain-free during the operation.
- The surgeon makes a cut (incision) to remove the sac around the organs.
- The organs are examined closely for signs of damage or other birth defects. Unhealthy parts are removed. The healthy edges are stitched together.
- The organs are placed back into the belly.
- The opening in the wall of the belly is repaired.
Staged repair is done when your baby isn’t stable enough for primary repair. Or, it is done if the omphalocele is very large and the organs can’t fit into the baby’s belly. The repair is performed the following way:
- Right after birth, a plastic pouch (called a silo) or a mesh-type of material is used to contain the omphalocele. The pouch or mesh is then attached to the baby’s belly.
- Every 2 to 3 days, the doctor gently tightens the pouch or mesh to push the intestine into the belly.
- It may take up to 2 weeks or more for all of the organs to be back inside the belly. The pouch or mesh is then removed. The opening in the belly is repaired.
Omphalocele repair risks
Risks for anesthesia and surgery in general are:
- Allergic reactions to medicines
- Breathing problems
- Bleeding
- Infection
Risks for omphalocele repair are:
- Breathing problems. The baby may need a breathing tube and breathing machine for a few days or weeks after surgery.
- Inflammation of the tissue that lines the wall of the abdomen and covers the abdominal organs.
- Organ injury.
- Problems with digestion and absorbing nutrients from food, if a baby has a lot of damage to the small bowel.
After the omphalocele repair procedure
After surgery, your baby will receive care in the neonatal intensive care unit (NICU). Your baby will be placed in a special bed to keep your baby warm.
Your baby may need to be on a breathing machine until organ swelling has decreased and the size of the belly area has increased.
Other treatments your baby will probably need after surgery are:
- Antibiotics
- Fluids and nutrients given through a vein
- Oxygen
- Pain medicines
- A nasogastric (NG) tube placed through the nose into the stomach to drain the stomach and keep it empty
Feedings are started through the nasogastric tube as soon as your baby’s bowel starts working after surgery. Feedings by mouth will start very slowly. Your baby may eat slowly and may need feeding therapy, lots of encouragement, and time to recover after a feeding.
How long your baby stays in the hospital depends on whether there are other birth defects and complications. You may be able to take your baby home once he or she is taking all foods by mouth and gaining weight.
Omphalocele repair prognosis
After you go home, your child may develop a blockage in the intestines (bowel obstruction) due to a kink or scar in the intestines. The doctor can tell you how this will be treated.
Most of the time, surgery can correct omphalocele. How well your baby does depends on how much damage or loss of intestine there was, and whether your child has other birth defects.
Some babies have gastroesophageal reflux after surgery. This condition causes food or stomach acid to come back up from the stomach into the esophagus.
Some babies with large omphaloceles may also have small lungs and may need to use a breathing machine.
References- Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, et al; for the National Birth Defects Prevention Network. Updated national birth prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res A Clin Mol Teratol. 2010;88(12):1008-16.
- Stoll C, Alembik Y, Dott B, Roth MP. Omphalocele and gastroschisis and associated malformations. Am J Med Genet A. 2008 May 15;146A(10):1280-5.
- Gastroschisis. Shilpi Chabra, Christine A. Gleason. NeoReviews Nov 2005, 6 (11) e493-e499; DOI: 10.1542/neo.6-11-e493 http://neoreviews.aappublications.org/content/6/11/e493
- Zahouani T, Mendez MD. Omphalocele. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519010
- Bird TM, Robbins JM, Druschel C, Cleves MA, Yang S, Hobbs CA, & the National Birth Defects Prevention Study . Demographic and environmental risk factors for gastroschisis and omphalocele in the National Birth Defects Prevention Study. J Pediatr Surg, 2009;44:1546-1551.
- Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM, & the National Birth Defects Prevention Study. Use of Selective Serotonin-Reuptake Inhibitors in Pregnancy and the Risk of Birth Defects. N Engl J Med, 2007;356:2684-92.
- Waller DK, Shaw GM, Rasmussen SA, Hobbs CA, Canfield MA, Siega-Riz AM, Gallaway MS, Correa A, & the National Birth Defects Prevention Study. Prepregnancy obesity as a risk factor for structural birth defects. Arch Pediatr Adolesc Med, 2007;161(8):745-50.
- Getachew MM, Goldstein RB, Edge V et-al. Correlation between omphalocele contents and karyotypic abnormalities: sonographic study in 37 cases. AJR Am J Roentgenol. 1992;158 (1): 133-6. https://www.ajronline.org/doi/pdf/10.2214/ajr.158.1.1727339
- Barisic I, Boban L, Akhmedzhanova D, Bergman JEH, Cavero-Carbonell C, Grinfelde I, Materna-Kiryluk A, Latos-Bieleńska A, Randrianaivo H, Zymak-Zakutnya N, Sansovic I, Lanzoni M, Morris JK. Beckwith Wiedemann syndrome: A population-based study on prevalence, prenatal diagnosis, associated anomalies and survival in Europe. Eur J Med Genet. 2018 Sep;61(9):499-507.
- Brioude F, Hennekam R, Bliek J, Coze C, Eggermann T, Ferrero GB, Kratz C, Bouc YL, Maas SM, Mackay DJG, Maher ER, Mussa A, Netchine I. Revisiting Wilms tumour surveillance in Beckwith-Wiedemann syndrome with IC2 methylation loss, reply. Eur. J. Hum. Genet. 2018 Apr;26(4):471-472.
- Karaman A, Aydin H, Göksu K. Concomitant omphalocele, anencephaly and arthrogryposis associated with trisomy 18. Genet. Couns. 2015;26(1):77-9.
- Hsu HF, Hou JW. Variable expressivity in Patau syndrome is not all related to trisomy 13 mosaicism. Am. J. Med. Genet. A. 2007 Aug 01;143A(15):1739-48.
- Chen CP. Syndromes and disorders associated with omphalocele (III): single gene disorders, neural tube defects, diaphragmatic defects and others. Taiwan J Obstet Gynecol. 2007 Jun;46(2):111-20.
- Zelante L, Germano M, Sacco M, Calvano S. Shprintzen-Goldberg omphalocele syndrome: a new patient with an expanded phenotype. Am. J. Med. Genet. A. 2006 Feb 15;140(4):383-4.
- Boujoual M, Madani H, Benhaddou H, Belahcen M. [Conjoined twins at common omphalocele and cloacal exstrophy with sexual ambiguity]. Pan Afr Med J. 2014;17:243.
- Omphalocele. https://radiopaedia.org/cases/omphalocele-4?lang=us
- Hijkoop A, Peters NCJ, Lechner RL, van Bever Y, van Gils-Frijters APJM, Tibboel D, Wijnen RMH, Cohen-Overbeek TE, IJsselstijn H. Omphalocele: from diagnosis to growth and development at 2 years of age. Arch. Dis. Child. Fetal Neonatal Ed. 2018 Mar 21
- Emanuel PG, Garcia GI, Angtuaco TL. Prenatal detection of anterior abdominal wall defects with US. Radiographics. 1995;15 (3): 517-30. https://pubs.rsna.org/doi/abs/10.1148/radiographics.15.3.7624560





{kind=link}