Pelizaeus Merzbacher disease
Pelizaeus-Merzbacher disease also called hypomyelinating leukodystrophy 1, is an inherited condition involving the brain and spinal cord (central nervous system) that primarily affects males 1. Pelizaeus Merzbacher disease rarely affects females. Pelizaeus-Merzbacher disease is one of a group of genetic disorders called leukodystrophy. Leukodystrophies are conditions that involve abnormalities of the nervous system’s white matter, which consists of nerve fibers covered by a fatty substance called myelin. Myelin insulates nerve fibers and promotes the rapid transmission of nerve impulses. In particular, Pelizaeus-Merzbacher disease involves hypomyelination, which means that the nervous system has a reduced ability to form myelin. As a result, overall neurological function is reduced.
Pelizaeus-Merzbacher disease is divided into classic and connatal (present from birth) types. Although these two types differ in severity, their features can overlap.
Classic Pelizaeus-Merzbacher disease is the more common type. Within the first year of life, those affected with classic Pelizaeus-Merzbacher disease typically experience weak muscle tone (hypotonia), involuntary movements of the eyes (nystagmus), and delayed development of motor skills, such as sitting or grasping objects. Some individuals are able to walk with assistance. Despite these neurological problems, intellectual and motor skills develop throughout childhood, but development usually stops around adolescence, and these skills are slowly lost (developmental regression). As the condition worsens, nystagmus usually goes away but other movement disorders develop, including muscle stiffness (spasticity), problems with movement and balance (ataxia), head and neck tremors (titubation), involuntary tensing of the muscles (dystonia), and jerking (choreiform) movements.
Connatal Pelizaeus-Merzbacher disease is the more severe of the two types. Symptoms can begin in infancy and include problems with feeding, poor weight gain and slow growth, high-pitched breathing caused by an obstructed airway (stridor), nystagmus, progressive speech difficulties (dysarthria), severe ataxia, hypotonia, and seizures. As the condition worsens, affected children develop spasticity leading to joint deformities (contractures) that restrict movement. Individuals with connatal Pelizaeus-Merzbacher disease are never able to walk, and many are not able to purposefully use their arms. They also have problems producing speech (expressive language) but can generally understand speech (receptive language).
The prevalence of Pelizaeus-Merzbacher disease is estimated to be 1 in 200,000 to 500,000 males in the United States 2.
Pelizaeus-Merzbacher disease is caused by an inability to form myelin due to mutations in the PLP1 gene. It is passed through families in an X-linked recessive pattern. The condition primarily affects males. Treatment requires a multidisciplinary team approach dictated by the presenting symptoms 3. There is no cure for Pelizaeus-Merzbacher disease, nor is there a standard course of treatment. Treatment is symptomatic and supportive and may include medication for movement disorders.
Pelizaeus Merzbacher disease causes
Mutations in the proteolipid protein-1 gene or PLP1 gene cause Pelizaeus-Merzbacher disease. The PLP1 gene provides instructions for making proteolipid protein 1 and a modified version (isoform) of that protein called DM20. Proteolipid protein 1 is found primarily in nerves in the central nervous system and DM20 is produced mainly in nerves that connect the brain and spinal cord to muscles (peripheral nervous system). These two proteins are found within the cell membrane of nerve cells, where they make up the majority of myelin and anchor it to the cells.
Most mutations that cause Pelizaeus-Merzbacher disease copy (duplicate) the PLP1 gene, which results in increased production of proteolipid protein 1 and DM20. Other mutations lead to production of abnormal proteins that are often misfolded. Excess or abnormal proteins become trapped within cell structures and cannot travel to the cell membrane. As a result, proteolipid protein 1 and DM20 are not available to form myelin. The accumulation of excess proteins leads to swelling and breakdown of nerve fibers. Still other mutations delete the PLP1 gene, which prevents proteolipid protein 1 and DM20 protein production and results in a lack of these proteins in the cell membrane, which causes any myelin that is formed to be unstable and quickly broken down. All of these PLP1 gene mutations lead to hypomyelination, nerve fiber damage, and impairment of nervous system function, resulting in the signs and symptoms of Pelizaeus-Merzbacher disease.
It is estimated that 5 to 20 percent of people with Pelizaeus-Merzbacher disease do not have identified mutations in the PLP1 gene. In these cases, the cause of the condition is unknown 2. Some of these patients have a variant of the GJC2 gene (autosomal recessive) that causes a Pelizaeus-Merzbacher-like disease, which is clinically indistinguishable from Pelizaeus Merzbacher disease. Others have variants in a growing list of other leukodystrophy genes that are being discovered.
Spastic paraplegia 2 (SPG2), hypomyelination of early myelinating structures (HEMS) and Pelizaeus Merzbacher disease result from different variants of the same gene (allelic disorders) on the X chromosome, the PLP1 gene.
Pelizaeus Merzbacher disease inheritance pattern
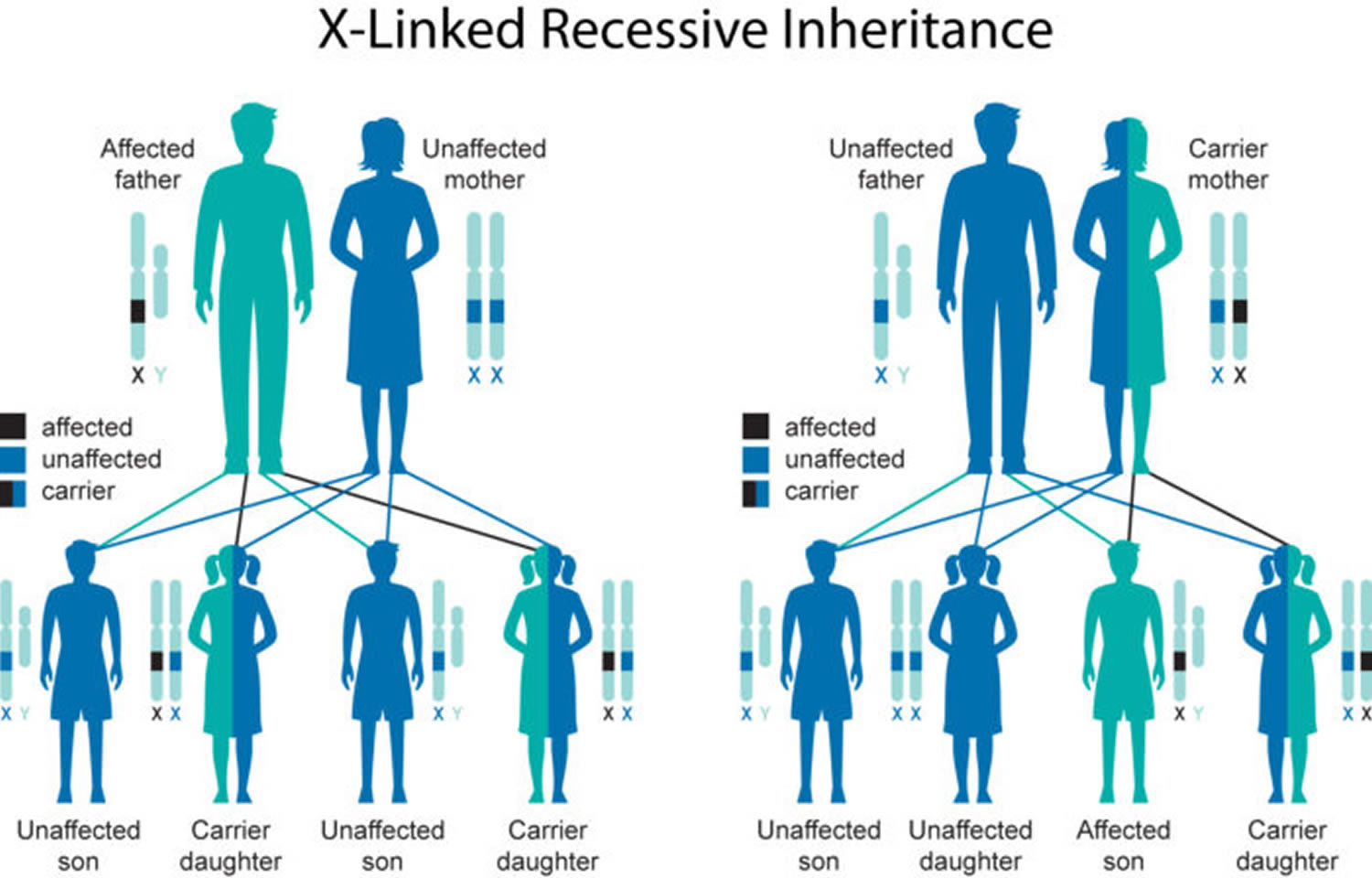
Pelizaeus-Merzbacher disease is inherited in an X-linked recessive pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males, who have only one X chromosome, a mutation in the only copy of the PLP1 gene in each cell is sufficient to cause the condition. In females, who have two copies of the X chromosome, one altered copy of the PLP1 gene in each cell can lead to less severe features of the condition, such as muscle stiffness or a decrease in intellectual function, or may cause no signs or symptoms at all. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Female carriers of Pelizaeus Merzbacher disease have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son. Females from families where males have a milder phenotype, such as SPG2 or the PLP1 null syndrome, should be more cautiously counseled. In some of these families, the disorder behaves more like an X-linked dominant disorder with reduced penetrance in which females can be affected but less severely than the affected males in the family.
Male Pelizaeus Merzbacher disease patients usually do not reproduce, but males with X-linked disorders who do reproduce pass the disease gene to all of their daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome with the PLP1 gene on it to male offspring.
Figure 1. Pelizaeus-Merzbacher disease X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Pelizaeus Merzbacher disease symptoms
The signs of Pelizaeus Merzbacher disease may vary widely from person to person. The signs of the classical form of Pelizaeus Merzbacher disease usually begin during early infancy, typically before 2 months of age. Initially, affected infants may fail to develop normal control of the head and eyes, specifically abnormal head bobbing and rapid, involuntary, jerky eye movements (nystagmus). Abnormally slow growth may also be an early sign. As affected infants and children age, additional signs may become apparent, including muscle tremors, weakness, facial grimacing, lack of muscle tone (hypotonia), impaired ability to coordinate voluntary movements (ataxia), and/or impairment in the acquisition of skills requiring the coordination of muscular and mental activities (psychomotor retardation) including delays in reaching developmental milestones such as sitting, standing, and walking. Affected individuals may also develop involuntary muscle spasms (spasticity) that result in slow, stiff movements of the legs and potentially partial paralysis of the arms and legs (spastic quadriparesis); abnormal, permanent fixation of certain joints (contractures); progressive degeneration of the nerves that lead to the eyes (optic atrophy); and/or difficulty speaking (dysarthria). As some affected children age, nystagmus may disappear. Some children may also develop skeletal deformities secondary to the severe spasticity that typically develops over time.
The signs of connatal Pelizaeus Merzbacher disease are present at birth or are observed during the first few weeks of life. This form of the disorder is characterized by weakness, spasticity, high-pitched sound when breathing (stridor), nystagmus, and seizures. Severe difficulty while swallowing (dysphagia) may also occur, necessitating gastrostomy feeding. Affected infants may also exhibit deterioration of mental functions and failure to reach developmental milestones such as speaking and walking. The progression of this form of Pelizaeus Merzbacher disease is more rapid and severe than the classic form and is often fatal during childhood.
Transitional Pelizaeus Merzbacher disease is a form of disease that is intermediate between the classical and connatal forms. The signs are similar to those of the classical and connatal forms of the disorder. However, the rate of progression is faster than the classical form but slower than the connatal form.
The PLP1 null syndrome is characterized by mild spastic quadriparesis, mild ataxia, absence of nystagmus during infancy and a mild demyelinating peripheral neuropathy. Patients with this form typically learn to walk, but deteriorate more rapidly beginning in late adolescence or early adulthood. Female carriers of Pelizaeus Merzbacher disease-related PLP1 variants may have mild to moderate signs of the disease. In some cases, these signs resolve with age.
Pelizaeus Merzbacher disease diagnosis
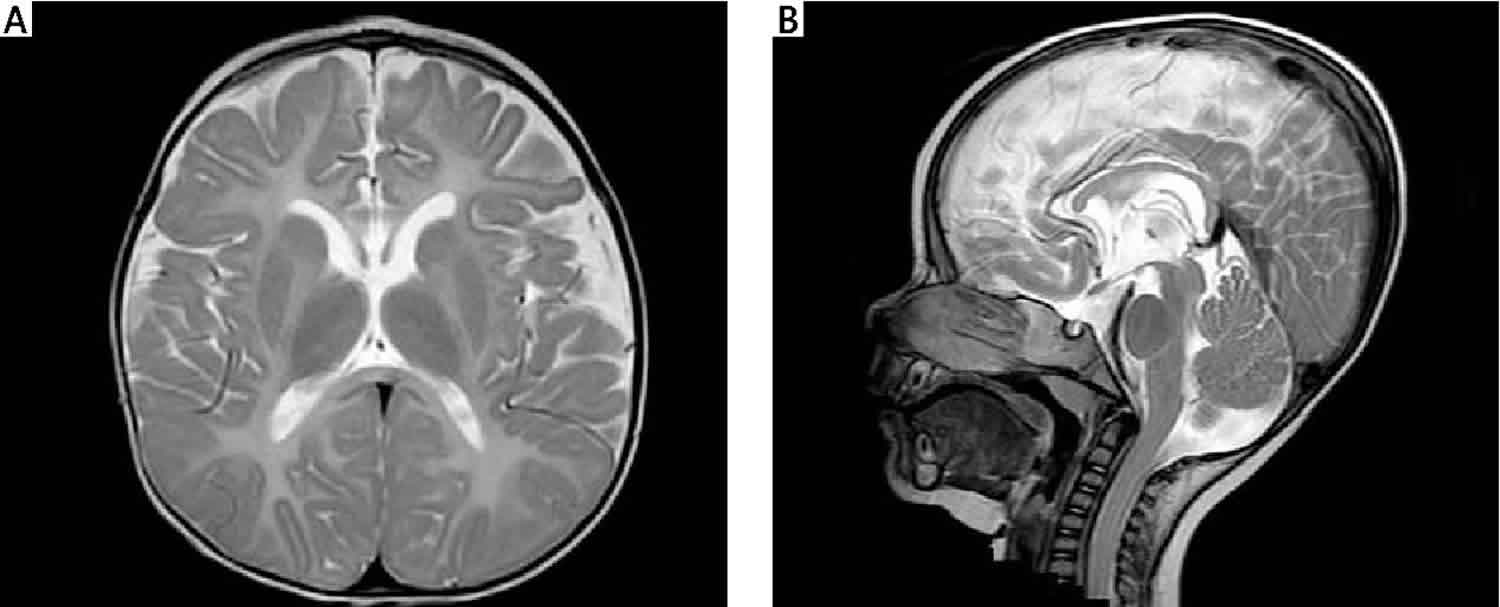
A diagnosis of Pelizaeus Merzbacher disease may be suspected based upon a thorough clinical evaluation, a detailed patient history, and a variety of specialized tests such as magnetic resonance imaging (MRI) to detect deficiency of white matter. Recognition of early myelination defects, such as lack of myelination in the cerebellum and brainstem, may aide in early diagnosis of the severe forms of Pelizaeus Merzbacher disease. Molecular genetic testing for the PLP1 gene is available to confirm the diagnosis.
Carrier testing is possible if a disease-causing variant in the PLP1 gene has been identified in an affected family member.
Prenatal diagnosis and preimplantation genetic diagnosis is available if a PLP1 gene variant is identified in an affected family member.
Pelizaeus Merzbacher disease treatment
There is no cure for Pelizaeus-Merzbacher disease, nor is there a standard treatment method or regimen 4. Management typically involves a multidisciplinary team made of specialists in neurology, physical medicine, orthopedics, pulmonary medicine, and gastroenterology. Treatment is based upon specific symptoms present and may include gastrostomy for individuals with severe dysphagia; antiepileptic drugs (AEDs) for seizures; and routine management of spasticity including physical therapy, exercise, medications (baclofen, diazepam, tizanidine, botulinum toxin), orthotics, and surgery for joint contractures. Individuals with scoliosis may benefit from proper wheelchair seating and physical therapy, with surgery reserved for the most severe cases. Specialized education and assessments are generally necessary, and assisted communication devices may be helpful 3. Supportive care, including emotional support for family members, is recommended as needed.
Genetic Counseling is recommended for individuals affected with Pelizaeus Merzbacher disease and their families.
Pelizaeus Merzbacher disease prognosis
The prognosis for those with the severe forms of Pelizaeus-Merzbacher disease is poor, with progressive deterioration until death. Individuals affected with the severe (connatal) type may die during infancy or childhood, usually of aspiration. With attentive care, these individuals may live into the third decade or longer. On the other end of the disease spectrum, individuals with the mild form, in which spastic paraplegia is the chief symptom, may have nearly normal activity and life span and may even reproduce. Survival into the sixth or seventh decade has been observed for individuals with the classic type 3.
Heterozygous females
In heterozygous females with alleles that are severe in males, the defective oligodendrocytes die and are replaced by healthy oligodendrocytes, and neurologic function is maintained or improves with maturation. Females who are heterozygous for less severe alleles of PLP1 gene that are not believed to cause oligodendrocyte cell death or apoptosis may develop a more progressive and nonremitting syndrome, which usually begins during adulthood 5.
Some females with Pelizaeus-Merzbacher disease (such as the original Pelizaeus-Merzbacher disease family) probably have a clinical course much like that of affected males, in which the symptoms do not remit and may be the result of skewed X inactivation (ie, most oligodendrocytes have inactivated the normal X chromosome, and insufficient healthy oligodendrocytes are available to effectively myelinate the central nervous system).
References- Gruenenfelder FI, McLaughlin M, Griffiths IR, et al. Neural stem cells restore myelin in a demyelinating model of Pelizaeus-Merzbacher disease. Brain. 2020;143(5):1383-1399. doi:10.1093/brain/awaa080 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7462093
- Pelizaeus Merzbacher disease. https://ghr.nlm.nih.gov/condition/pelizaeus-merzbacher-disease
- Wolf NI, van Spaendonk RML, Hobson GM, et al. PLP1 Disorders. 1999 Jun 15 [Updated 2019 Dec 19]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1182
- Pelizaeus-Merzbacher Disease Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Pelizaeus-Merzbacher-Disease-Information-Page
- Hurst S, Garbern J, Trepanier A, Gow A. Quantifying the carrier female phenotype in Pelizaeus-Merzbacher disease. Genet Med. 2006 Jun. 8(6):371-8.





{kind=link}