
What is periodic fever syndrome
Periodic fever syndromes are a group of diseases characterized by recurrent episodes of fever with healthy intervals between febrile episodes 1. Periodic fever syndromes are self-limited and improve spontaneously without specific therapy. Episodes of fever are usually associated with presence of inflammation in different parts of the body such as peritoneum, pleural spaces, testis, etc. with elevated serum acute phase reactants level 1. These diseases should be differentiated from infections, malignancies and other autoimmune disorders 2. Existence of family history of periodic fever and prolonged illnesses are diagnostic clues of periodic fever syndrome. Arthritis or arthralgia, abdominal or chest pain due to serositis, skin rash, and oral ulcers are other common presentations of periodic fever syndromes, although these symptoms can be found in infectious diseases with prolonged or recurrent fever 3. Headache, seizure, aseptic meningitis, development delay as well as adenopathy, organomegaly and cardiac involvement are uncommon findings in periodic fever syndrome. Renal involvement is uncommon but in some types, renal failure due to amyloidosis may occur.
Periodic fever is named when there are 3 episodes of fever in at least 3-6 months with 7 days asymptomatic period between each episode 3. Rarely, periodic fever syndrome without fever has been reported 4. In this situation, one of the other symptoms is repeated periodically. For this reason, experts believe that periodic fever should be considered as a diagnosis in a patient, who shows similar problem repeatedly. Some periodic fever syndromes occur in regular intervals especially periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis (PFAPA) and cyclic neutropenia, however, Familial Mediterranean Fever (FMF) and hyperimmunoglobulin D (hyper IgD) generally have regular patterns. On the other hand, auto-inflammatory syndromes in infancy such as chronic infantile neurologic cutaneous and articular (CINCA) syndrome, Muckle-Wells syndrome, familial cold urticaria and tumor necrosis factor receptor–associated periodic syndrome (TRAPS) usually have irregular patterns 5.
The first manifestation of periodic fever syndromes are present in childhood and adolescence, but infrequently it may be presented in young and middle ages. Genetic base has been known for all types of periodic fever syndromes except periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA). Common periodic fever disorders are Familial Mediterranean fever (FMF) and periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA). In each patient with periodic fever, acquired infection with chronic and periodic nature should be ruled out.
At least eight hereditary periodic fever syndromes have been known so far which are common in fever with cutaneous manifestations, and musculoskeletal involvement 6, each of them having specific characteristics. Fever and symptoms occur due to the increment of inflammation which is caused by mutation in genes. Most of these disorders result from gene mutations that cause defect in production of some proteins which have important role in control of inflammation and apoptosis.
Usually there are symptoms of the disease for a long time prior to the definite diagnosis. Mostly fever attacks start during childhood, but sometimes they may start in adolescence or in adulthood. Clinical examinations are helpful for diagnosis which is confirmed by specific genetic testing 7.
Periodic fever syndrome causes
Periodic fever syndrome disorders are categorized under the term monogenic auto-inflammatory syndromes. The characteristic of these disorders is episodic attacks of systemic inflammations without presence of infection or auto-antibodies 8. The pathogenesis of periodic fever syndrome disorders is disregulation of inflammation control due to mutations of genes coding for some proteins with regulation role 8. It has been believed that dysregulation of innate immune response and abnormalities in activity of interleukin-1 and pro-inflammatory cytokines may contribute to fever production and systemic inflammation 9. Innate immune cells such as macrophages, neutrophils, and monocytes are involved 9. Unlike adaptive immunity, innate immunity is programmed genetically 10 and most of these diseases are caused by mutation in genes which make proteins participant in inflammatory response 11.
It is thought that mutate proteins lead to increased or prolonged secretion of pro-inflammatory cytokines 12. Activation of caspase-1 and the release of IL-1β are the end point of pathophysiologic cycle of these disorders 13. Treatment with anti-IL1 drugs is mandatory 12.
Table 1. Characteristics of classic periodic fever and cryopyrin associated periodic syndromes (CAPS)
| Autoinflmatory disorder | Characteristics |
|---|---|
| Hyper IgD syndrome |
|
| TRAPS |
|
| CINCA or NOMID |
|
| Muckle-Wells Syndrome |
|
| Familial Cold Autoinflammatory Syndrome |
|
Abbreviations: IgD = Immunoglobulin D; TRAPS = Tumor Necrosis Factor Receptor–Associated Periodic Syndrome; CINCA = Chronic Infantile Neurologic Cutaneous and Articular Syndrome; NOMID = Neonatal Onset Multisystem Inflammatory Disease; CAPS = Cryopyrin Associated Periodic Syndromes
[Source 14 ]Familial Mediterranean Fever
Familial Mediterranean Fever (FMF) or recurrent hereditary polyserositis is the most common disease among hereditary periodic fever syndromes, and it is an autosomal recessive disorder 15.
Familial Mediterranean Fever is seen most frequently in people from Mediterranean area such as Jews, Arabs, Turks and Armenians, but it is sometimes reported from all over the world. Prevalence of familial mediterranean fever is 1:200 to 1:1000 5. Although familial mediterranean fever is a autosomal receive disorders, positive family history is present in less than 50% of patients 16. Familial Mediterranean Fever is slightly more common in women perhaps due to the influence of sex hormones. Sometimes fever attacks disappear in pregnancy and return after delivery. Rarely infertility may occur due to defect in ovulation and adhesion in pelvic peritoneum. Some complications of familial mediterranean fever such as renal amyloidosis are more common in males 15.
Familial Mediterranean Fever (FMF) is characterized by periodic fever that occasionally occurs at regular intervals, usually with neutrophil induced serositis. The responsible gene for the disease is MEFV that is located on short arm of chromosome 16 6. This gene produces a protein called Pyrin or Marenostrin that is found in neutrophils and plays an important role in reduction of inflammation 17. Mutation in this gene leads to defection in production of these proteins and onset of inflammatory cycle. In addition to gene mutation, environmental factors are important and in many patients there is a trigger factor such as emotional stress, infection, extreme physical exercise, fatigue, trauma and menses 15.
Familial Mediterranean Fever Treatment
The drug of choice for Familial Mediterranean Fever (FMF) is colchicine that is also used for prevention. It is recommended to use this drug for a long time. A minority group of patients do not respond to colchicine may be due to non-adherence 15. Starting dose is 1mg/day and it may be increased to 1.5 to 2 mg/day until remission is achieved 5. Based on weight or body surface area, the colchicine dose is 0.03±0.02 mg/kg/day and 1.16±0.45 mg/m2/day, respectively[8]. In children younger than 5 years, higher doses of colchicine, 0.07 mg/kg/day or 1.9 mg/m2/day, may be required 5. The most common side effects of colchicine are diarrhea and nausea which are seen rarely. Fetal malformation from colchicine has not been reported 5.
In colchicine resistance cases interferon-alpha may be helpful 18. In patients with incomplete remission with colchicine, or in whom amyloid A level is high despite treatment with colchicine, IL1 blockade (Anakinra) may be effective 13. Other drugs such as NSAIDs and corticosteroids have limited indication for treatment of FMF. Myalgia responds to treatment with corticosteroids, but not colchicines. NSAIDs are used for treatment of arthralgia 15. Febrile attacks do not respond to and are not preventable by NSAIDs or steroids.
Periodic Fever, Aphthous Stomatitis, Pharyngitis, Cervical Adenitis (PFAPA)
Periodic Fever, Aphthous Stomatitis, Pharyngitis, Cervical Adenitis (PFAPA) is a syndrome that consists of recurrent episodes of fever, sore throat, mouth sores and swelling of the glands in the neck. The frequency of PFAPA is not known, but the disease appears to be more common than originally thought, and may be the most common recurrent fever (autoinflammatory) syndrome that does not come from an infection. Both males and females and all ethnic groups can develop PFAPA. Periodic Fever, Aphthous Stomatitis, Pharyngitis, Cervical Adenitis (PFAPA) usually starts in early childhood, between the ages of two to five years. Occasionally PFAPA may develop at an older age, including rare cases in adults.
Clinical criteria of Periodic Fever, Aphthus stomatitis, Pharyngitis, Cervical Adenitis (PFAPA)
- Regulatory recurring fevers with an early age of onset (<5 years of age)
- Symptoms in the absence of upper respiratory tract infection with at least one of the following clinical signs:
- a) aphthous stomatitis
- b) cervical lymphadenitis
- c) pharyngitis
- Exclusion of cyclic neutropenia, completely asymptomatic interval between episodes, normal growth and development.
It is not yet known what causes Periodic Fever, Aphthous Stomatitis, Pharyngitis, Cervical Adenitis (PFAPA). No gene defect has yet to be found in PFAPA, although sometimes more than one family member has the disease and a history of a tonsillectomy (surgical removal of the tonsils) may be reported among family members. No infection has been found in PFAPA, and it is not a contagious disease. It is clear that the inflammatory process is active during episodes, but it is not clear why this happens.
There are no laboratory tests specific for diagnosing PFAPA. The disease is diagnosed based on symptoms and physical examination. White blood cell counts and markers of inflammation including the sedimentation rate and the C-reactive protein, all of which can be measured with a blood test, increase during episodes. It is important to exclude all other diseases that may present with similar symptoms (especially a Streptococcus infection) before confirming the diagnosis. The dramatic response to treatment with steroids also helps diagnose PFAPA. In cases without a classic presentation it may be necessary to exclude other causes of recurrent fever (see other patient sheets on this topic), especially if symptoms start in the first year of life and markers of inflammation are increased also between episodes.
Periodic Fever, Aphthous Stomatitis, Pharyngitis, Cervical Adenitis Treatment
The aim of the treatment will be to control symptoms during the episodes of fever, to shorten the duration of the episodes, and to prevent episodes from occurring. The fever usually does not respond well to acetaminophen (Tylenol) or nonsteroidal anti-inflammatory drugs (NSAIDs) like ibuprofen (Advil or Motrin). A single dose of steroids (usually prednisone or prednisolone), given when the symptoms first start, dramatically shortens, and often even ends, the episode. However, the time between episodes also may be shortened with this treatment, and the next episode may occur earlier than expected.
Medications like cimetidine and colchicine, when used regularly, may prevent future episodes in about a third (cimetidine) to half (colchicine) of the children. Several studies have found that a tonsillectomy (removing the tonsils by surgery) cures PFAPA in the majority of patients (more than 80%) but the role and timing of surgery in treating PFAPA has still not been fully clarified. Alternatives to steroids should be considered in patients needing steroids more frequently than once every three – four weeks.
Hyperimmunoglobulin D syndrome
Hyperimmunoglobulin D syndrome or mevalonate kinase deficiency is a hereditary periodic fever syndrome that occurs in unpredictable intervals[2-4]. Dutch-type periodic fever is another name for this autosomal recessive disorder 19.
First cases of hyperimmunoglobulin D syndrome were reported in Dutch population in 1984 but later several patients were reported from other countries and in other races 20. Hyperimmunoglobulin D syndrome usually starts in childhood and 70% of patients are younger than 1 year old at the beginning of the disease 15. Sometimes fever attacks are stimu-lated with immunization, trauma, and stress 20.
Hyperimmunoglobulin D syndrome results from a mutation in a gene which is located on chromosome 12q24 and encodes mevalonate kinase (MVK) enzyme 21. Mevalonate kinase (MVK) enzyme contributes to cholesterol synthesis and it has an anti-inflammatory effect 20. Mutated enzyme has a decreased function with a pro-inflammatory response 20.
The typical symptoms of hyperimmunoglobulin D syndrome are fever, malaise, chills, cervical lymphadenopathy, and diarrhea. Abdominal pain, arthritis or arthralgia, skin rash, and hepatosplenomegaly are other signs 20. Fever and other symptoms resolve after 3 to 7 days with an afebrile interval lasting 6-8 weeks, but skin rash and arthritis may last longer 22. Arthritis in hyperimmunoglobulin D syndrome is transient and non-destructive 23.
Hyperimmunoglobulin D syndrome treatment
There are no absolutely effective drugs for treatment of hyperimmunoglobulin D syndrome, thalidomide, etanercept (anti-tumor necrosis factor), and corticosteroids are used successfully in some patients, but efficacy of these drugs is doubt-ful 24. Anti interleukine-1 (IL-1) (anakinra or canakinumab) has been effective in severe cases unresponsive to other treatments 23.
Prognosis of hyperimmunoglobulin D syndrome is excellent even without treatment 25 and amyloidosis is very rare in these patients 15. Episodes of fever decreases by aging 26.
Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS)
Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) or familial Hibernian fever 15, is an autosomal dominant disease that occurs in irregular intervals and is characterized by episodes of fever, pain in muscle groups and painful skin lesions on trunk or extremi-ties 27.
For the first time, TRAPS was described in a large Irish family, but then it was reported in all countries 27. The male to female ratio is 3:2 15 or 1:1 27 and Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) is more common in Caucasians and Arabs but less common in Indians[20]. This syndrome has been reported in patients 2 weeks to 53 years of age 28.
The cause of Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) is mutation in the gene that encodes the type 1 TNF receptor 27. The gene is TNFRSF1A and is located on chromosome 12p13.2 27. TNFR1 is a trans-membrane receptor and the main mediator of signaling by TNF-α 29. As binding of TNF-α to mutate TNFR1 lessens, as a result, TNF induced apoptosis decreases 30. Consequently, following increase the activity of TNF, inflammatory response enhances 28.
Similar to other periodic syndromes, fever is the main symptom in children, in adults attack may be without fever 30. Fever duration in Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) usually is longer than in other periodic fever syndromes such as Familial Mediterranean Fever (FMF) or in hyperimmunoglobulin D syndrome and it may last up to 3 weeks 15.
Interval between fever episodes has variable frequency but in average it occurs every 5 to 6 weeks 28. The onset of the disease occurs along with fever and muscular spasm 28. No known triggers for the onset of the illness are identified 28. The muscle pain is migratory and affected muscles are tender in palpation and warm 15. This finding is usually associated with erythematous patches that are warm and tender and are resolved with pressure 20. Other symptoms of Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) are abdominal pain (due to inflammation in peritoneal cavity or muscular cramp in abdominal wall) that is seen in 92% of patients, conjunctivitis or periorbital edema, which is a characteristic for TRAPS and is seen in over 80% of patients, chest pain (in 57% of patients), and arthralgia. Other less common symptoms are arthritis, scrotal pain and lymphadenopathy 15.
Tumor Necrosis Factor Receptor–Associated Periodic Syndrome Treatment
Non steroidal anti inflammatory drugs (NSAIDs) are used for treatment of fever but musculoskeletal and abdominal pain do not respond to NSAIDs, so they should be treated with corticosteroids; neither of these drugs can prevent episodes 30. Etanercept is an anti-TNF agent that decreases severity, duration, and frequency of the attacks 31. Treatment with anakinra, which is an IL-1 blocker, may be also effective 29. Treatment with infliximab, an anti-TNF antibody, may lead to paradoxical inflammatory reactions, so it should not be used 31.
The most severe complication of Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) is amyloidosis that occurs in less than 25% of untreated patients and manifests with dysfunction of involved organs 28. So, prognosis of Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) is related to subsequent amyloidosis 15. Risk of amyloidosis increases in mutation that affects cysteine amino acids 28.
Cryopyrin Associated Periodic Syndromes (CAPS)
Cryopyrin Associated Periodic Syndromes (CAPS) refer to a group of diseases which have similar phenotypes all of which result from mutation in the CIAS1 (cold-induced autoinflammatory syndrome) gene, which encodes cryopyrin 32. This protein regulates generation of pro-inflammatory cytokines[2,29]. Chronic infantile neurologic cutaneous and articular syndrome (CINCA), Muckle-Wells syndrome (MWS), and familial cold autoinflammatory syndrome (FCAS), which will be explained respectively, belong to cryopyrin associated periodic syndromes (CAPS). These syndromes usually have overlapping symptoms the severity of which are affected by other genetic mutations and environmental factors 32.
After ruling out infection and malignancies, auto-inflammatory disorders should be considered. However, physician needs to reevaluate each patient with periodic disorder for non-inflammatory syndromes.
Chronic infantile neurologic cutaneous and articular syndrome
Chronic infantile neurologic cutaneous and articular (CINCA) syndrome is the most severe one among the cryopyrin associated periodic syndromes[30]. These syndromes occur in all ethnic groups without any superiority 33. Chronic infantile neurologic cutaneous and articular (CINCA) syndrome that is also named neonatal onset multisystem inflammatory disease (NOMID) is a congenital inflammatory disorder that starts in neonatal period and it has various symptoms such as recurrent fever, central nervous system (brain and spinal cord) involvement, cutaneous manifestations, chronic arthropathy, and morphologic feature 20. Skin manifestations which are the first presentation at birth in 75% of patients, are similar to urticaria and vary during the day 34. Urticaria is stimulated with cold. Central nervous system involvement exists in almost all patients and hydrocephalus and ventriculomegaly may be present prenatally 20. Neurologic manifestations result from chronic meningitis and brain atrophy, and include seizure, sensory organ involvement such as ocular manifestations, progressive sensory neural hearing loss, and headache. Mental retardation exists in some patients 35 but it is not an initial manifestation 34. Relapsing joint involvement is also seen in CINCA patients 36 and severity of arthropathy is related to the age of onset, i.e. patients with a later onset have milder manifestations without destruction 20. Knees are the most common involved joints and then ankles, feet, and elbows 20. Patients with CINCA have a typical morphology. They have a short stature, macrocephalus, hoarseness, saddle nose, short extremities, and clubbing 20.
Diagnosis of CINCA is based on the above mentioned specific features 37. Triad of continuous rash, CNS involvement, and relapsing joint involvement is helpful for diagnosis but this syndrome should be differentiated from other hereditary periodic fever syndromes such as Familial Mediterranean Fever (FMF), Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS), and hyperimmunoglobulin D syndrome.
The majority of these syndromes occur sporadically 36 but there is a mutation in the CIAS1 (now named NLRP3) gene on chromosome 1q44 20 that encodes the cryopyrin protein in 60% of patients 38. However, this mutation can be found in two other syndromes including Muckle-Wells Syndrome (MWS) and Familial Cold Autoinflammatory Syndrome (FCAS) 37.
Treatment with anti IL-1 drugs especially with anakinra is effective in CINCA and results in significant improvement in symptoms of the disease 39. Nonsteroidal anti-inflammatory drugs and glucocorticosteroids are effective on fever and pain but not on skin lesions 30.
Muckle-Wells Syndrome
Muckle-Wells Syndrome (MWS) also called urticarial deafness amyloidosis syndrome, is a rare autosomal dominant syndrome 40. It is of middle severity between Cryopyrin Associated Periodic Syndromes (CAPS). Episodes of this syndrome are presented with fever, limb pain, recurrent or sub-chronic urticaria-like lesions and conjunctivitis. Episcleritis and optic disc edema are other eye related findings 41. Fever is usually mild and disappears in 12 to 36 hours 29. Two-thirds of patients have sensory neural hearing loss that usually starts in the second decade of life 20. Amyloidosis occurs in one-fourth of patients which can lead to renal failure[36] and peripheral neuropathy that occurs later than renal amyloidosis 20. Unlike other Cryopyrin Associated Periodic Syndromes (CAPS), urticarial rash is not always stimulated with cold 15. The time of onset is during infancy and sometimes in adolescence. Lifelong arthralgia and non-erosive arthritis may exist[34]. There is a mutation in NLRP3 gene in MWS as in other CAPS, but some mutations are typically found in Muckle-Wells syndrome 15. Incidence of CAPS syndromes is not subject to sex 15.
Three anti-IL-1 drugs including anakinra (an IL-1 receptor antagonist), rilonacept (an IL-1 soluble receptor), and canakinumab (a human monoclonal antibody against IL-1β) are effective on CAPS. Canakinumab and rilonacept have FDA approval for treatment of Familial Cold Autoinflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS) 15. Treatment with canakinumab was very effective and in 96% of patients complete remission was achieved 42. Studies show that treatment cannot eradicate existing damages but hearing loss may return 20.
Familial Cold Autoinflammatory Syndrome
Familial Cold Autoinflammatory Syndrome (FCAS), formerly called familial cold urticaria, is the mildest CAPS disease 20. As other Cryopyrin Associated Periodic Syndromes (CAPS), FCAS is inherited in an autosomal dominant fashion 43. Familial Cold Autoinflammatory Syndrome (FCAS) usually begins before 6 months of age 35 and in 60% of patients symptoms are seen during the first days of life 20. Clinical symptoms occur within 8 hours 43 after generalized exposure to cold 35 and last for 12 to 48 hours 43. Fever, arthralgia, conjunctivitis, non-itching urticarial like rash, and headache are indicators of familial cold autoinflammatory syndrome (FCAS) 43. There are no signs of CNS involvement 32. Audiology symptoms and deafness is absent 44. Localized exposure to cold is not a trigger in this disease and it distinguishes familial cold autoinflammatory syndrome from acquired cold urticaria 35.
Treatment with anakinra before exposure to cold may be effective in prevention of inflammatory symptoms 45. Moving to warm places is recommended to these patients 46. In all three diseases of CAPS, treatment with anakinra is effective 47. Rilonacept and canakinumab as long acting IL1 blockers have been approved by FDA for treatment of familial cold autoinflammatory syndrome and Muckle-Wells syndrome 45. Long time prognosis of familial cold autoinflammatory syndrome is good and amyloidosis is uncommon 43.
Autoinflammatory Bone Disorders
Autoinflammatory Bone Disorders are a group of autoinflammatory disorders with sterile bone inflammation as a hallmark finding. Some of the disorders of this group are monogenic such as pyogenic arthritis, pyoderma gangraenosum and acne (PAPA) syndrome, the deficiency of IL-1 receptor antagonist (DIRA) and Majeed syndrome (or familial chronic multifocal osteomyelitis). Some other sterile osteitises have polygenic background or are without specific genetic basis, so these disorders are classified as sporadic group,. In which chronic recurrent multifocal osteomyelitis (CRMO) and synovitis, acne, pustulosis, hyperostosis and osteitis (SAPHO) syndrome are also classified 48.
Pyogenic arthritis, Pyoderma gangaenosum and Acne (PAPA) syndrome
Pyogenic arthritis, Pyoderma gangaenosum and Acne (PAPA) syndrome is a rare autosomal-dominant disease that characterizes with sterile inflammation in joints and cutaneous manifestation. The cause of the disease is mutation in PSTPIP1 (proline/serine/threonine phosphatase–interacting protein 1) located on chromosome 15q24-25 44. This gene produces a protein that mostly exists in hematopoietic cells and regulates T cell activation, cytoskeletal organization and releases of interleukin-1B (IL-1B). Mutation in this gene leads to dysregulation of IL-1B production 77 and macrophages. Pyogenic arthritis, Pyoderma gangaenosum and Acne (PAPA) syndrome patients have impaired invasive motility 77 and might have dysregulated apoptosis 44.
Two major manifestations of Pyogenic arthritis, Pyoderma gangaenosum and Acne (PAPA) syndrome are erosive arthritis and cutaneous syndromes both of which start in childhood. Joints involvement is recurrent, sterile and destructive and may develop spontaneously or after minor trauma. Skin symptoms of the disease are severe acne, recurrent non healing sterile ulcer called pyoderma gangraenosum and pathergy.
For treatment, the disease responds well to TNFα blockers such as Infliximab, adalimumab and anakinra. Some new immunosuppressive drugs may also be used to treat PAPA syndrome.
Majeed Syndrome
Majeed Syndrome is an autosomal recessive disease caused from a mutation in LPIN243. Majeed Syndrome was firstly reported in 1989 43. Majeed syndrome is a chronic disease which affects the whole life and is characterized by early onset of multifocal osteomyelitis. Other features of Majeed syndrome are congenital anemia and transient inflammatory dermatosis which result from infiltration of neutrophils into dermis and is called Sweet syndrome. Diagnosis is suspected based on clinical manifestations and is confirmed with genetical studies 44. Treatment of the disease includes non-steroidal anti-inflammatory drugs eventually in combination with corticosteroids or interferon-α.
Deficiency of the Interlukin-1 Receptor Antagonist
Deficiency of the IL-1 receptor antagonist (DIRA) is a new TNF-receptor-associated periodic syndrome which was primarily described in 2009 35. Deficiency of the IL-1 receptor antagonist (DIRA) results from mutation in the IL1RN (2q14) 31. It is an autosomal recessive disease. Signs of the disease usually start in prenatal period and include systemic inflammation, multifocal osteolytic lesions, periostitis, and a pustular rash. Other manifestations are heterotopic bone formation around the proximal femur, thrombosis, and rarely vasculitis. Some manifestations which are seen in Neonatal Onset Multisystem Inflammatory Disease (NOMID) but not in deficiency of the Interlukin-1 Receptor Antagonist (DIRA) are meningitis, cochlear inflammation, hearing loss, and conjunctivitis or uveitis. Deficiency of the interlukin-1 receptor antagonist (DIRA) patients have a good response to treatment with the recombinant interlukin-1 receptor antagonist anakinra 38.
Synovitis, Acne, Pustulosis, Hyperostosis and Osteitis (SAPHO) syndrome or Chronic Recurrent Multifocal Osteomyelitis (CRMO)
Synovitis, Acne, Pustulosis, Hyperostosis and Osteitis (SAPHO) syndrome and Chronic Recurrent Multifocal Osteomyelitis (CRMO) are chronic nonbacterial or sterile osteomyelitises due to an auto-inflammatory process. Some authorities believe this disorders are different diseases with similar pathway and they have common clinical, radiological and maybe histological findings 49. Innate immune system is involved and any auto-antibodies or autoreactive T-cells have not been fouind in these disorders. SAPHO is usually described in adult patients with similar symptoms of spondylarthropathies 50. Vertebral bone is the most common site of involvement and it may lead to vertebral collapse. In SAPHO syndrome, skin involvement such as acne and pustulosis is prominent. CRMO is suggested to be a variant of SAPHO syndrome in children 51, but in CRMO skin manifestations are rare findings 52. Exacerbations and remissions of the inflammation is a hallmark for differentiation of these disorders from similar disorders. Increasing number of lesions over the time and self-limiting course without major sequelae are other characteristics. The patients present symptoms of CRMO at school age that include bone lesions. One fourth of the patients had spinal involvement and half of them developed vertebral deformities 38.
Treatment is based on NSAIDs and/or corticosteroids. In cases with resistance to these drugs methotrexate, sulfasalazine, and TNF inhibitors are used. CRMO is usually self-limited but a large number of patients experience a prolonged disease 38. In patients with bone lesions, treatment with bisphosphonates and pamidronate may be helpful and pain resolves 3 months after treatment 53. MRI is the most sensitive way of diagnosing bone lesions and finding those lesions that are not observed in a simple radiography.
SWEET’s Syndrome
In 1964, Sweet described a syndrome with fever and neutrophilic dermatitis 54. Three types of disease were described by Cohen and Kurzrock 55. Malignancy-associated form can be associated with hematologic malignancy (such as acute leukemia) and solid tumors. It can be first manifestation of a malignant disorder or first presentation of recurrent malignancy. Second form is classical form which usually is seen following a respiratory or gastrointestinal infection, pregnancy or rheumatologic disorders (such as irritable bowel disease, systemic lupus erythematosus, Behcet’s disease, FMF and Sarcoidosis) 56. Third form of Sweet’s syndrome is drug induced following antibiotics (trimethoprim-sulfamethoxazole, nitrofurantoin), NSAIDs (celecoxib and diclofenac), antiepileptics (diazepam and carbamazepine), antihypertensives (hydralazine), oral contraceptives, propylthiouracil and retinoids 56.
Pathogenesis of this syndrome is unknown. Some Cytokines (such as Interleukin 1, TNF-alpha, IL-8, IL-17), human leukocyte antigen serotypes have a possible role in the pathogenesis of this syndrome 57. The innate immune is involved in this syndrome, so recently it is classified as a autoinflmmatory disorder 57.
Clinical features of this syndrome are a recurrent and acute illness with high fever, peripheral neutrophillia, and skin involvement such as erythematous plaques, erythema nodosum and ulcers. Other manifestations are general malaise, headache, and myalgia, arthralgia, and conjunctivitis 58. Central nervous system (aseptic meningitis), kidneys, intestines, liver, heart, and lungs may be involved 58. Diagnostic criteria for classic and drug-induced Sweet syndrome is shown in Table 2. Criteria for malignancy-associated Sweet’s syndrome are similar to classic Sweet’s syndrome.
In histopathology, diffuse and neutrophil-rich infiltrate is seen in the dermis 59. There is usually no evidence of vasculitis (fibrin deposition or neutrophils) in the vessel wall 60.
There are some reports on successful treatment of Sweet’s syndrome with antibiotics, oral potassium iodide, NSAIDs and colchicines; oral corticosteroids or metylprednolone pulse is very useful in patients with refractory disease 61. Other treatments are infusion of intravenous immunoglobulin and biologic agents such as anakinra and anti TNF-α agants 61.
Table 2. Diagnostic criteria for classic and drug-induced Sweet’s syndrome
| Classical Sweet’s syndrome |
|---|
Major Criteria (both are mandatory)
|
Minor Criteria
ESR>20mm/hr / positive CRP / WBC>8000 /Neutrophilia > 70%. |
| Drug-induced |
|
Footnote: Diagnosis of classic Sweet’s syndrome is confirmed by 2 major criteria and two of the four minor criteria but for diagnosis of drug-induced Sweet’s syndrome all five criteria are mandatory.
Blau Syndrome
Blau syndrome or “juvenile systemic granulomatosis” 44 or early onset sarcoidosis 43 was reported for the first time in 1985 43. It is a rare disease related to mutation in the CARD15/NOD2 gene localized on chromosome 16q12 44. The disease is autosomal dominant and begins in early childhood usually before 4 years of age 43.
The first episode of Blau syndrome is in the first 2 years of age with continuous attack 35. The most common sites of this syndrome are skin, joints and eyes but not lungs or hilar lymph nodes. Skin lesions which are rarely seen in sarcoidosis in adults include brown colored scaly maculopapules with tapioca-like appearance which are lichenoid-like. Another skin manifestation is erythema nodosum 43. Destructive arthritis and tendinitis are other manifestations of musculoskeletal involvement 35. Pneumonitis, eye involvement (uveitis and iritis) and involvement of other organs such as liver and kidney is seen in this syndrome 35. Characteristic feature of the disease is non-caseating granulomatous lesions involving joints, skin and eyes that lead to symmetric polyarthritis and cataract in 50% of patients. Differentiation of Blau syndrome from sarcoidosis based on histological findings is difficult but clinical symptoms are clearly different 43.
Treatment options for Blau syndrome are corticosteroids, immunosuppressant drugs (such as methotrexate and cyclosporine), infliximab or anakinra 44.
Periodic fever syndrome symptoms
Periodic fever syndromes are a group of diseases characterized by episodes of fever with healthy intervals between febrile episodes. These disorders are self-limited and improve spontaneously without specific therapy. Episodes of fever are usually associated with presence of inflammation in different parts of the body such as peritoneum, pleural spaces, testis, etc. with elevated serum acute phase reactants level. Periodic fever syndromes should be differentiated from infections, cancers and other autoimmune disorders 1.
Periodic fever syndrome diagnosis
In a patient with periodic fever, acquired infection with chronic and periodic nature should be ruled out. It depends on epidemiology of infectious diseases. Malaria and borrelliosis can be ruled out by evaluation of peripheral blood smear and brucellosis and Lyme disease can be excluded by serological studies or blood culture. In children, urinary tract infection may present as a periodic disorder, so urine analysis and culture are mandatory in a child with periodic symptoms 62. Some malignancies such as leukemia and tumoral lesions should be excluded in patients with periodic syndromes and weight loss in any age and weight gain disorder or malaise in children 63. For this reason, abdominal ultrasound and chest X-ray are necessary and bone marrow aspiration in selected patients is recommended. Table 2 shows paraclinical evaluation in suspected patients with periodic fever syndromes.
After ruling out infection and malignancies, auto-inflammatory disorders should be considered. However, physician needs to reevaluate each patient with periodic disorder for non-inflammatory syndromes. Hyper IgD, Chronic Infantile Neurologic Cutaneous and Articular Syndrome (CINCA syndrome), Tumor Necrosis Factor Receptor–Associated Periodic Syndrome (TRAPS) and other auto-inflammatory syndromes are other causes of periodic fever disorders. Figure 1 and Figure 2 show clinical approach to periodic fever patients, based on the epidemiology of periodic fever in the country.
To establish diagnosis in autoinflammatory disorders is very difficult because there are no definite diagnostic criteria or a gold standard, so it depends on both clinical findings and genetic study to get the diagnosis confirmed (Table 1) 14. All patients usually have a history of repeated hospitalizations and antibiotic therapy. It is due to similar symptoms and signs in autoinflammatory disorders and infectious diseases, immuno-deficiency and/or hematologic disorders. So in each patient, these disorders, especially infectious diseases, should be ruled out 1.
Table 3. Laboratory tests for patients with periodic fever
| Study | Indication | Time |
|---|---|---|
| CBC, ESR, CRP | All patients | At least 2 times in febrile and afebrile period |
| Chest X-ray | All patients | In febrile period |
| Abdominal ultrasound | All patients | In febrile period |
| Serum Igs | All patients | No difference |
| Wright, Coombs-Wright | In endemic region | At least once in febrile period |
| EBV serology | All patients with sore throat and lymphadenopathy | In febrile period |
| Peripheral blood smear | In endemic region | At least once in febrile period |
| Blood culture | All patients | At least once in febrile period |
| Throat culture | All patients with sore throat | In febrile period |
| Urine analysis and culture | All children | At least once in febrile period |
| Stool exam | All children | At least once, no difference |
| Bone marrow aspiration | In selected patients with cytopenia, bone pain, weight loss, abdominal and/or mediastinal adenopathy | No difference |
| Liver function tests | All patients | At least once in febrile period |
| FANA | All children | At least once, no difference |
Abbreviations: CBC = Complete Blood Cells; ESR = Erythrocyte Sedimentation Rate; CRP: C = Reactive Protein; Ig: Immunoglobulin; EBV = Epstein Barr Virus; FANA = Fluorescent Antinuclear Antibody Test
[Source 1 ]Figure 1. First step to diagnosing Periodic Fever
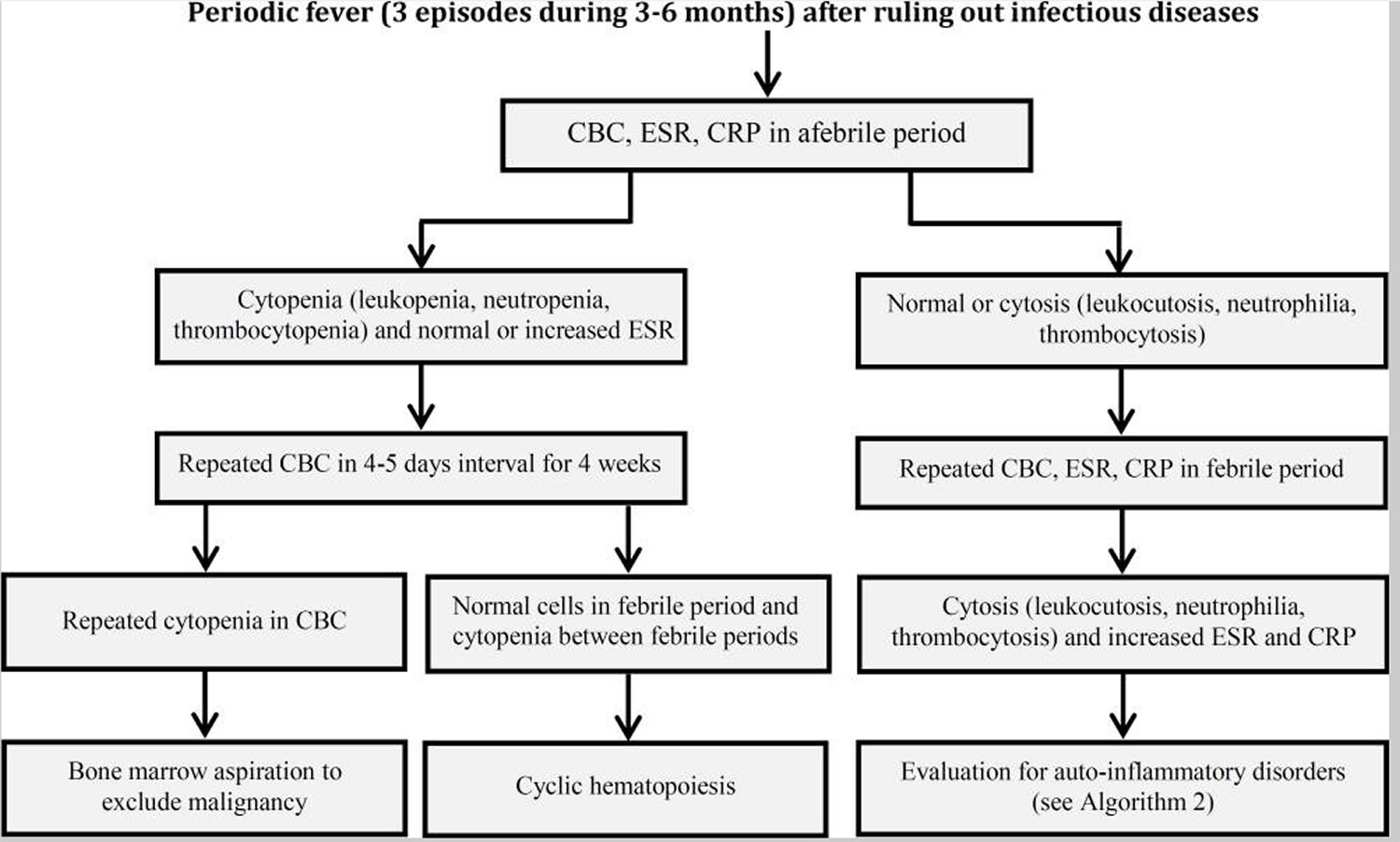
Abbreviations: CBC = Complete Blood Cells; ESR = Erythrocyte Sedimentation Rate; CRP = C-Reactive Protein
[Source 1 ]Figure 2. Second step to diagnosing Periodic Fever
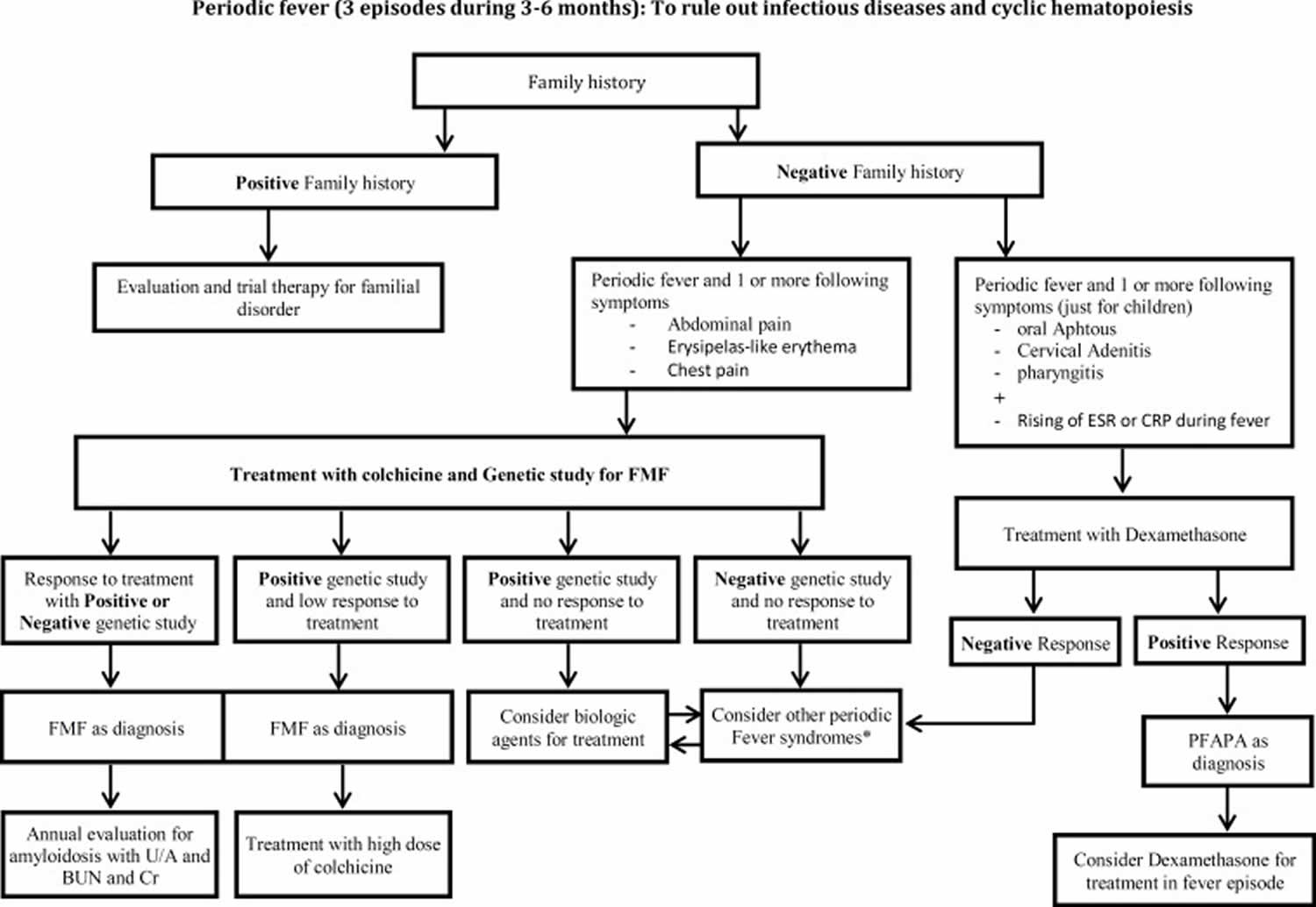
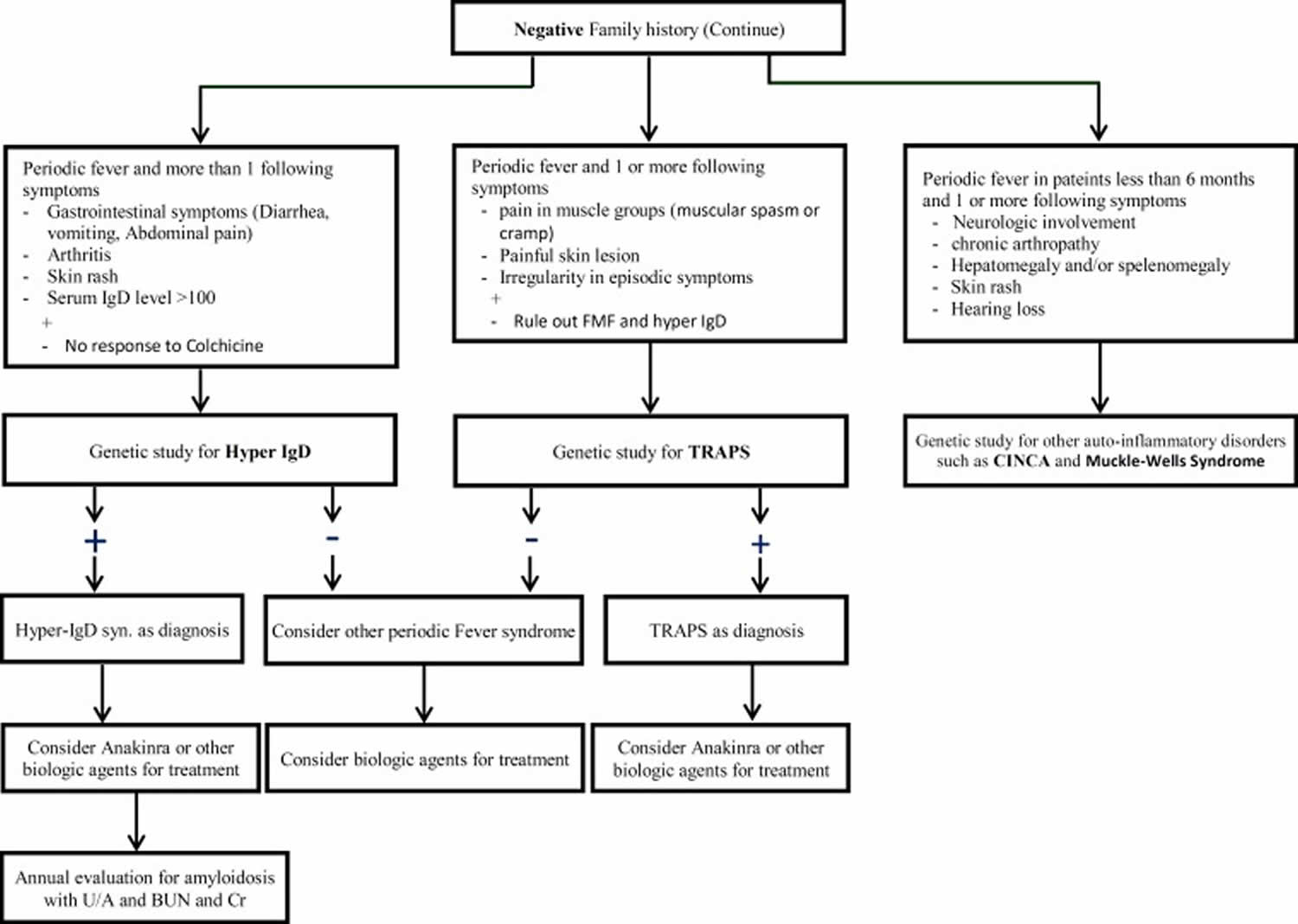
Abbreviations: FMF = Familial Mediterranean Fever; PFAPA = Periodic Fever, Aphthus stomatitis, Pharyngitis, Cervical Adenitis; U/A = Urinaly sis; BUN = Blood Urea Nitrogen; Cr = Creatinine; ESR = Erythrocyte Sedimentation Rate; CRP = C – Reactive Protein; IgD = Hyperimmunoglobulin D; TRAPS = Tumor Necrosis Factor Receptor–Associated Periodic Syndrome; CINCA = Chronic Infantile Neurologic Cutaneous and Articular Syndrome
[Source 1 ]Periodic fever syndrome treatment
Treatment options depend on the underlying cause of the periodic fever.
References- Ahmadinejad Z, Mansori S, Ziaee V, et al. Periodic Fever: a review on clinical, management and guideline for Iranian patients – part I. Iran J Pediatr. 2013;24(1):1–13. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4359590
- Autoinflammatory conditions: when to suspect? How to treat? Grateau G, Duruöz MT. Best Pract Res Clin Rheumatol. 2010 Jun; 24(3):401-11.
- Barron K, Athreya B, Kastner D. Periodic fever syndromes and other inherited autoinflammatory diseases. In: Cassidy JT, Petty RE, Laxer RM, et al., editors. Textbook of Pediatric Rheumatology. 6th ed. Philadelphia: Sanders; 2011. pp. 642–60
- Ziaee V, Aghighi Y. Familial Mediterranean Fever: a 10-year follow-up in Iranian patients. Clin Experiment Rheumatol. 2011;29(2):400
- Periodic fever syndromes. Padeh S. Pediatr Clin North Am. 2005 Apr; 52(2):577-609, vii.
- Neurological manifestations of the Mendelian-inherited autoinflammatory syndromes. Montealegre Sanchez GA, Hashkes PJ. Dev Med Child Neurol. 2009 Jun; 51(6):420-8.
- Pathogenesis of familial periodic fever syndromes or hereditary autoinflammatory syndromes. Simon A, van der Meer JW. Am J Physiol Regul Integr Comp Physiol. 2007 Jan; 292(1):R86-98.
- The autoinflammatory diseases. Federici S, Caorsi R, Gattorno M. Swiss Med Wkly. 2012; 142():w13602.
- Recurrent febrile syndromes: what a rheumatologist needs to know. Hoffman HM, Simon A. Nat Rev Rheumatol. 2009 May; 5(5):249-56.
- Genetics of monogenic autoinflammatory diseases: past successes, future challenges. Aksentijevich I, Kastner DL. Nat Rev Rheumatol. 2011 Jul 5; 7(8):469-78.
- Autoinflammatory syndromes with a dermatological perspective. Kanazawa N, Furukawa F. J Dermatol. 2007 Sep; 34(9):601-18.
- A preliminary score for the assessment of disease activity in hereditary recurrent fevers: results from the AIDAI (Auto-Inflammatory Diseases Activity Index) Consensus Conference. Piram M, Frenkel J, Gattorno M, Ozen S, Lachmann HJ, Goldbach-Mansky R, Hentgen V, Neven B, Stojanovic KS, Simon A, Kuemmerle-Deschner J, Hoffman H, Stojanov S, Duquesne A, Pillet P, Martini A, Pouchot J, Koné-Paut I, EUROFEVER and EUROTRAPS networks. Ann Rheum Dis. 2011 Feb; 70(2):309-14.
- Familial Mediterranean fever and related periodic fever syndromes/autoinflammatory diseases. Savic S, Dickie LJ, Battellino M, McDermott MF. Curr Opin Rheumatol. 2012 Jan; 24(1):103-12.
- Ahmadinejad Z, Mansouri S, Ziaee V, Aghighi Y, Moradinejad MH, Fereshteh-Mehregan F. Periodic Fever: A Review on Clinical, Management and Guideline for Iranian Patients – Part II. Iran J Pediatr. 2014;24(3):229–240. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4276575
- Hereditary Periodic Fever Syndromes. https://emedicine.medscape.com/article/952254-overview
- A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Yalçinkaya F, Ozen S, Ozçakar ZB, Aktay N, Cakar N, Düzova A, Kasapçopur O, Elhan AH, Doganay B, Ekim M, Kara N, Uncu N, Bakkaloglu A. Rheumatology (Oxford). 2009 Apr; 48(4):395-8.
- Translational research network and patient registry for auto-inflammatory diseases. Lainka E, Bielak M, Hilger V, Basu O, Neudorf U, Wittkowski H, Holzinger D, Roth J, Niehues T, Foell D. Rheumatology (Oxford). 2011 Jan; 50(1):237-42.
- Adult-onset familial mediterranean Fever in northwestern iran; clinical feature and treatment outcome. Nobakht H, Zamani F, Ajdarkosh H, Mohamadzadeh Z, Fereshtehnejad S, Nassaji M. Middle East J Dig Dis. 2011 Mar; 3(1):50-5.
- Hyperimmunoglobulinemia D with periodic fever. https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=3276&Disease_Disease_Search_diseaseGroup=Hyperimmunoglobulin-D-syndrome&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Hyperimmunoglobulinemia-D-with-periodic-fever&title=Hyperimmunoglobulinemia%20D%20with%20periodic%20fever&search=Disease_Search_Simple
- Montealegre Sanchez G, Hashkez PJ. Neurological manifestations of the Mendelian-inherited autoinflammatory syndromes. Dev Med Child Neurol. 2009;51(6):420–8
- Federici S, Caorsi R, Gattorno M. The autoinflammatory diseases. Swiss Med Wkly. 2012;142:w13602
- Jacobs Z, Ciaccio CE. Periodic fever syndromes. Curr Allergy Asthma Rep. 2010;10(6):398–404.
- Galeotti C, Meinzer U, Quartier P, et al. Efficacy of interleukin-1-targeting drugs in mevalonate kinase deficiency. Rheumatology (Oxford) 2012;51(10):1855–9
- Bodar EJ1, van der Hilst JC, Drenth JP, et al. Effect of etanercept and anakinra on inflammatory attacks in the hyper-IgD syndrome: introducing a vaccination provocation model. Neth J Med. 2005;63(7):260–4
- John CC, Gilsdorf JR. Recurrent fever in children. Pediatr Infect Dis J. 2002;21(11):1071–80
- Drenth JP, Haagsma CJ, van der Meer JW. Hyperimmuno-globulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients: International Hyper-IgD Study Group. Medicine (Baltimore) 1994;73(3):133–44
- Aguado-Gil L, Irarrazaval-Armendáriz I, Pretel-Irazabal M. Advances in the diagnosis and treatment of tumor necrosis factor receptor-associated periodic syndrome. Actas Dermosifiliogr. 2013;104(7):617–22
- Jacobs Z, Ciaccio CE. Periodic fever syndromes. Curr Allergy Asthma Rep. 2010;10(6):398–404
- De Sanctis S, Nozzi M, Scardapane A, et al. Autoinflammatory syndromes: diagnosis and management. Ital J Pediatr. 2010;36:57
- Padeh S. Periodic fever syndromes. Pediatr Clin North Am. 2005;52(2):577–609
- Savic S, Dickie LJ, Battellino M, McDermott MF. Familial Mediterranean fever and related periodic fever syndromes/autoinflammatory diseases. Curr Opin Rheumatol . 2012;24(1):103–12
- Aksentijevich I, Putnam CD, Remmers EF, et al. The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American Patients and a new cryopyrin Model. Arthritis Rheum. 2007;56(4):1273–85
- Obici L, Manno C, Muda AO, et al. First report of systemic reactive (AA) amyloidosis in a patient with the hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheum. 2004;50(9):2966–9
- Leys C, Eschard C, Motte J, et al. Chronic infantile neurological, cutaneous, and articular syndrome with severe early articular manifestations. Pediatr Dermatol. 2005;22(3):222–6.
- Bodar EJ, Drenth JP, van der Meer JW, et al. Dysregulation of innate immunity: hereditary periodic fever syndromes. Br J Haematol. 2008;144(3):279–302
- Leys C, Eschard C, Motte J, et al. Chronic infantile neurological, cutaneous, and articular syndrome with severe early articular manifestations. Pediatr Dermatol. 2005;22(3):222–6
- Caroli F, Pontillo A, D’Osualdo A, et al. Clinical and genetic characterization of Italian patients affected by CINCA syndrome. Rheumatology. 2007;46(3):473–8
- Henderson C, Goldbach-Mansky R. Monogenic autoinflammatory diseases: new insights into clinical aspects and pathogenesis. Curr Opin Rheumatol. 2010;22(5):567–78
- Grateau G, Duruoz MT. Autoinflammatory conditions: when to suspect? How to treat? . Best Practice Res Clin Rheumatol. 2010;24:401–11
- Simon A, van der Meer JW, Vesely R, et al. Approach to genetic analysis in the diagnosis of hereditary autoinflammatory syndromes. Rheumatology. 2006;45(3):269–73
- Ozen S, Frenkel J, Ruperto N, et al. The Eurofever Project: towards better care for autoinflammatory diseases. Eur J Pediatr. 2011;170(4):445–52
- Curran MP. Canakinumab: in patients with cryopyrin-associated periodic syndromes. Bio Drugs. 2012;26(1):53–9.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601–8
- Rigante D. The protean visage of systemic autoinflammatory syndromes: a challenge for inter-professional collaboration. Eur Rev Med Pharmacol Sci. 2010;14(1):1–18
- Hoffman HM. Therapy of autoinflammatory syndromes. J Allergy Clin Immunol . 2009;124:1129–38.
- Lierl M. Periodic fever syndromes: a diagnostic challenge for the allergist. Allergy. 2007;62(12):1349–58
- Hoffman HM, Simon A. Recurrent febrile syndromes – what a rheumatologist needs to know. Rheumatology. 2009;5:249–55
- Hedrich CM, Hofmann SR, Pablik J, et al. Autoinflammatory bone disorders with special focus on chronic recurrent multifocal osteomyelitis (CRMO) Pediatr Rheumatol Online J. 2013;11(1):47
- Baltensperger M, Gratz K, Bruder E, et al. Is primary chronic osteomyelitis a uniform disease? Proposal of a classification based on a retrospective analysis of patients treated in the past 30 years. J Craniomaxillofac Surg. 2004;32(1):43–50.
- Vittecoq O, Said LA, Michot C, et al. Evolution of chronic recurrent multifocal osteitis toward spondylarthropathy over the long term. Arthritis Rheum. 2000;43(1):109–19
- Job-Deslandre C, Krebs S, Kahan A. Chronic recurrent multifocal osteomyelitis: five-year outcomes in 14 pediatric cases. Joint Bone Spine. 2001;68(3):245–51
- Sayili A, Tosun O, Cobanoglu N, et al. Synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome in childhood: A case report and review of literature. Iran J Pediatr. In Press.
- Miettunen PM1, Wei X, Kaura D, et al. Dramatic pain relief and resolution of bone inflammation following pamidronate in 9 pediatric patients with persistent chronic recurrent multifocal osteomyelitis (CRMO) Pediatr Rheumatol Online J. 2009;7:2
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349–56
- Raza S, Kirkland RS, Patel AA, et al. Insight into Sweet’s syndrome and associated-malignancy: a review of the current literature. Int J Oncol. 2013;42(5):1516–22
- Barton JL, Pincus L, Yazdany J, et al. Association of sweet’s syndrome and systemic lupus erythematosus. Case Rep Rheumatol. 2011;2011:242681
- Marzano AV1, Ishak RS, Saibeni S, et al. Autoinflammatory skin disorders in inflammatory bowel diseases, pyoderma ;gangraenosum and Sweet’s syndrome: a comprehensive review and disease classification criteria. Clin Rev Allergy Immunol. 2013;45(2):202–10
- Cohen PR1, Kurzrock R. Sweet’s syndrome: a neutrophilic dermatosis classically associated with acute onset and fever. Clin Dermatol. 2000;18(3):265–82
- Tabanlıoglu D, Boztepe G, Erkin G, et al. Sweet’s syndrome and erythema nodosum: a companionship or a spectrum? – a case report with review of the literature. Int J Dermatol. 2010;49(1):62–6.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol . 2003;42(10):761–78.
- Anzalone CL, Cohen PR. Acute febrile neutrophilic dermatosis (Sweet’s syndrome) Curr Opin Hematol . 2013;20(1):26–35
- Distinguishing among prolonged, recurrent, and periodic fever syndromes: approach of a pediatric infectious diseases subspecialist. Long SS. Pediatr Clin North Am. 2005 Jun; 52(3):811-35, vii
- Periodic fever as a presenting sign of childhood acute lymphoblastic leukaemia. Koffeman EC, Wulffraat NM, Bruin M, Hogeman PH, Frenkel J. Rheumatology (Oxford). 2005 Dec; 44(12):1583-4.





{kind=link}