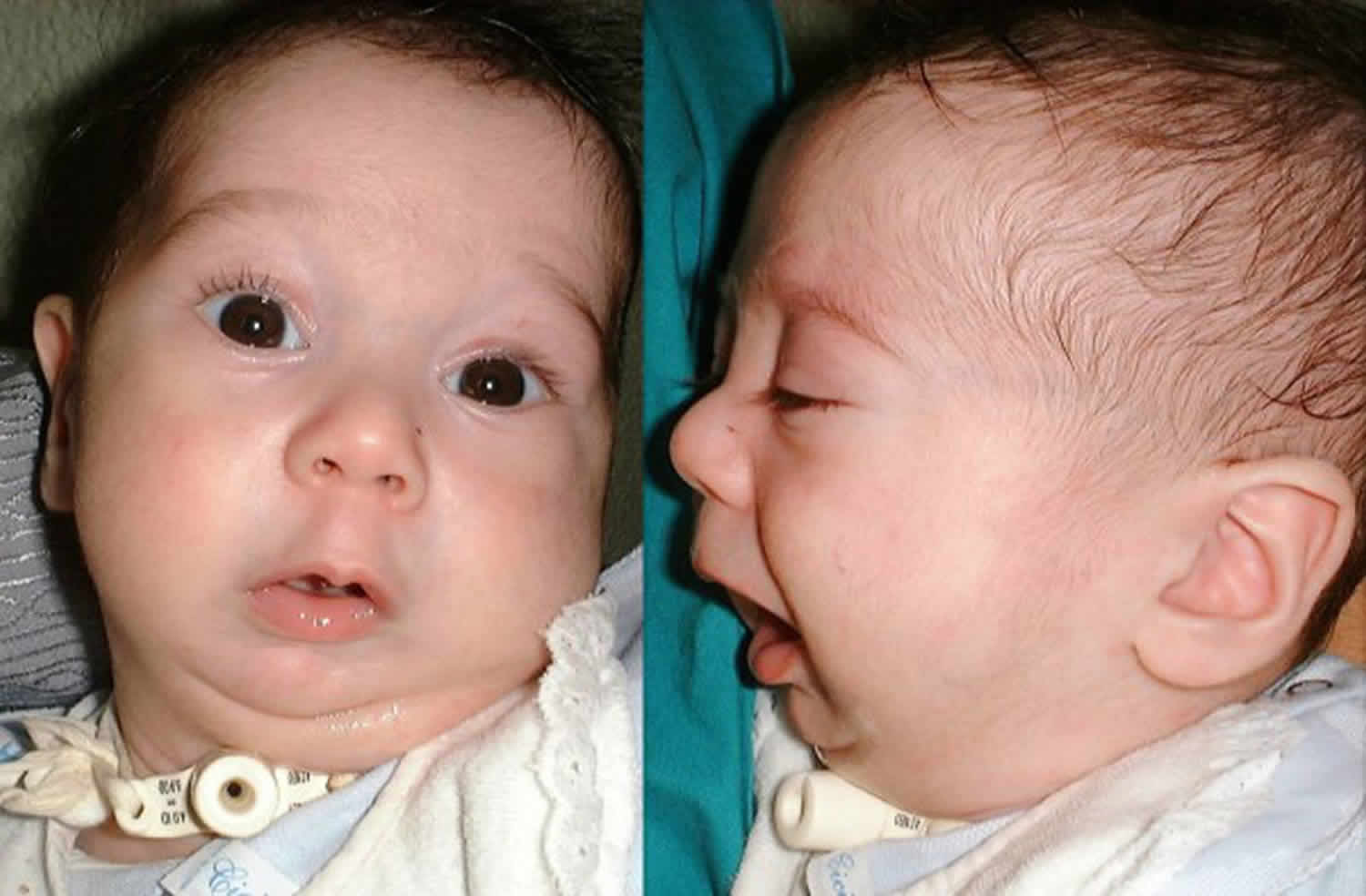
What is Pierre Robin sequence
Pierre Robin sequence also known as Robin sequence, is a set of abnormalities affecting the head and face, consisting of a small lower jaw (micrognathia), a tongue that is placed further back than normal (glossoptosis), and breathing difficulty due to airway obstruction. This combination of features can lead to breathing and feeding problems early in life. As a result, some affected babies have difficulty growing and gaining weight at the expected rate. If the airway obstruction is not identified and managed, it may lead to hypoxia, right heart failure, failure to thrive, and feeding difficulties, in addition to cerebral impairment. In addition, many children with Pierre Robin sequence have an opening in the roof of the mouth (cleft palate). This also affects a child’s ability to feed. This feature is not generally considered necessary for diagnosis of the condition, although there is some disagreement among doctors.
Pierre Robin sequence affects an estimated 1 in 8,500 to 14,000 births, making it one of the most common facial differences.
Experts describe Pierre Robin sequence as a “sequence” because they believe that as the embryo forms in early pregnancy, the underdeveloped lower jaw (mandible) sets off a sequence of events before birth that causes the other signs and symptoms. When the lower jaw does not grow properly, the tongue can prevent the palate (roof of the mouth) from closing, resulting in a cleft palate. The underdeveloped lower jaw also causes the tongue to be positioned at the back of the mouth can block the airways, making breathing difficult.
The combination of features characteristic of Pierre Robin sequence can lead to difficulty breathing and problems eating early in life. As a result, some affected babies have an inability to grow and gain weight at the expected rate (failure to thrive). In some children with Pierre Robin sequence, growth of the mandible catches up, and as adults these individuals have normal-sized chins.
Pierre Robin syndrome can occur as an isolated anomaly or part of a syndrome. Some people have the features of Pierre Robin sequence as part of a syndrome that affects other organs and tissues in the body, such as Stickler syndrome (20-25% of these cases), campomelic dysplasia, trisomy 11q syndrome, deletion 4q syndrome, CHARGE association, velocardiofacial syndrome, and Treacher-Collins syndrome 1. These instances are described as syndromic. When Pierre Robin sequence occurs by itself, it is described as nonsyndromic or isolated. Approximately 20 to 40 percent of cases of Pierre Robin sequence are isolated.
Treatment is focused on the specific needs of each patient, but may include surgery to assist with breathing and feeding modifications to prevent choking 2. Children with Pierre Robin syndrome may require a series of surgeries over years.
In more mild cases, management may consist of positioning your child in a side or face-down position to allow the tongue to fall forward and relieve the obstruction. Care may also include the addition of a nasopharyngeal airway until your child matures neurologically and has grown enough to bring the tongue into a more favorable position. In patients who are more severely affected or have abnormal sleep study results, surgical alternatives can be considered.
Surgical intervention includes tongue-lip adhesion, mandibular distraction or tracheostomy. Tongue-lip adhesion and mandibular distraction osteogenesis have both been performed with favorable outcomes.
In tongue-lip adhesion, the undersurface of the tip of the tongue is sutured to the inside of the lower lip to hold it in a more forward position. If successful, this is released in three to six months.
In distraction osteogenesis, an internal or external device is applied to the mandible and a cut is made posteriorly. By activating the device, the jaw is gradually brought forward carrying the tongue with it. This procedure improves the airway as new bone forms in the gap.
If neither of these procedures is successful or your child has disease below the base of the tongue, she may need a tracheostomy.
Pierre Robin sequence causes
Experts do not know the exact causes of Pierre Robin sequence, but there may be a genetic link. Changes in the DNA near the SOX9 gene (located in chromosome 17 [17q24]) are the most common genetic cause of isolated Pierre Robin sequence. It is likely that changes in other genes, some of which have not been identified, are also involved in Pierre Robin sequence. Doctors speculate that nongenetic factors, for example conditions during pregnancy that restrict growth of the jaw, may cause some cases of isolated Pierre Robin sequence. There is a higher incidence of Pierre Robin sequence of defects in twins. This may be due to crowding in the uterus, which restricts the growth of the lower jaw.
The SOX9 gene provides instructions for making a protein (protein SOX9) that plays a critical role in the formation of many different tissues and organs during embryonic development. The SOX9 protein regulates the activity of other genes, especially those that are important for development of the skeleton, including the mandible.
The genetic changes near the SOX9 gene that are associated with isolated Pierre Robin sequence are thought to disrupt regions of DNA called enhancers, which normally regulate the activity of the SOX9 gene. These changes reduce SOX9 gene activity. As a result, the SOX9 protein cannot properly control the genes essential for normal development of the lower jaw, causing micrognathia, and consequently, glossoptosis, airway obstruction, and, often, cleft palate.
Pierre Robin sequence facial differences can be seen in other syndromes as well, including:
- Stickler syndrome: This is a genetic malfunction in the tissue that connects bones, heart, eyes and ears. A child with Stickler syndrome may have problems with vision, hearing, bones and joints, the heart and facial formation.
- Velocardiofacial syndrome: The most common features of this syndrome are cleft palate, heart defects and characteristic facial appearance, as well as minor learning problems and speech and feeding challenges.
Pierre Robin sequence inheritance pattern
Isolated Pierre Robin sequence is usually not inherited. Pierre Robin sequence typically results from new (de novo) genetic changes and occurs in people with no history of the disorder in their family. When the condition is inherited, it follows an autosomal dominant pattern, which means one copy of the DNA alteration in each cell is sufficient to cause the disorder.
Syndromic Pierre Robin sequence is inherited in the same pattern as the condition it is associated with.
Pierre Robin sequence signs and symptoms
Pierre Robin sequence is a combination of facial differences that occurs in a developing fetus and that is apparent when your baby is born. These differences include:
- Micrognathia: An underdeveloped or abnormally small lower jaw (mandible) that is much shorter than the rest of the face. The growth of the lower jaw may speed up during the first year of life.
- Glossoptosis: A tongue that is positioned further back in the mouth than normal. Because of the small mandible, the tongue is too large for the airway. This can interfere with breathing.
- Cleft palate: A cleft palate is a separation in the tissue that makes up the roof of the mouth (palate). For some babies, both the front and back parts of the palate are open. For other babies, only part of the palate is open. The cleft palate in Robin sequence is usually a rounded, or U-shaped cleft palate, in comparison to the more common V-shaped cleft palate seen in other conditions.
These differences result in a variety of symptoms, including:
- Feeding difficulties: The smaller size of the lower jaw, position of the tongue and the cleft of the palate can make it difficult for your baby to feed normally. Your baby might be able to learn to feed with specially adapted nipples and bottles.
More severe Pierre Robin sequence may require a temporary feeding tube.
- Trouble breathing: Your child’s tongue may fall backwards, especially when lying on his or her back. This can block the throat and obstruct breathing and is of special concern when your baby sleeps. According to literature, some babies respond well to positioning on the stomach, which helps pull the tongue forward during sleep. Other infants may require nasal tubes or surgery to pull or push the tongue forward.
As a result of these symptoms, some babies struggle with growth and weight gain until treated.
Pierre Robin sequence possible complications
These complications can occur:
- Breathing difficulties, especially when the child sleeps
- Choking episodes
- Congestive heart failure
- Death
- Feeding difficulties
- Low blood oxygen and brain damage (due to difficulty breathing)
- Type of high blood pressure called pulmonary hypertension
Pierre Robin sequence diagnosis
A doctor can diagnose Pierre Robin sequence during an exam immediately after your baby is born. The visible symptoms of Pierre Robin sequence are:
- A small lower jaw (mandible)
- An opening in the roof of the mouth (cleft palate)
- A tongue that is positioned further back in the mouth than normal (glossoptosis)
Infants with these facial differences are commonly taken to the neonatal intensive care unit (NICU) in the first day of life to be monitored for breathing problems (oxygen desaturations) and to provide assistance with feeding. If there is an associated laryngotracheal anomaly present, a tracheostomy may be required.
Observation in a hospital and a sleep study (polysomnogram) are helpful in assessing the degree of airway obstruction.
If your child has been diagnosed with Pierre Robin sequence, a geneticist can provide a full genetic evaluation to determine if the sequence is part of a syndrome that affects other organs and tissues.
Pierre Robin sequence treatment
Treatment for Pierre Robin sequence ensures your baby can eat and breathe safely and comfortably. Consultation with a feeding specialist is advised. In many cases, when carefully instructed, a mother is able to manage bottle feeding while her baby is in a semisitting position. Special cleft palate nipples and squeezing bottles are helpful. Feeding must be done very carefully to avoid choking and breathing liquids into the airways. In patients with severe problems, tube feeding may be necessary in the beginning of the baby’s life to prevent choking.
Do NOT put infants with this condition on their back. This is to prevent their tongue from falling back into their airway.
In moderate cases, the child will need to have a tube placed through the nose and into the airway to avoid airway blockage. In severe cases, surgery is needed to prevent a blockage in the upper airway. Some children need surgery to make a hole in their airway or to move their jaw forward.
Airway and breathing treatments
Children with Pierre Robin sequence may have trouble getting enough oxygen. This is due to glossoptosis — a tongue that is positioned further back in the mouth than normal.
Nonsurgical treatments to assist with breathing include:
- Positioning. Laying your baby on his or her side or belly will help to pull the tongue forward and unblock the airway. Your child will need to be continuously monitored with an oxygen saturation monitor.
- Nasopharyngeal airway. A craniofacial doctor can insert a small, flexible tube into your child’s nose and down the throat (ending just above the vocal cords). This tube keeps the airway open by preventing the tongue from falling back and blocking it. This tube will need to be changed every few days. If your baby needs to use this tube over the long term, the size and length may need adjusting to accommodate your baby’s growth.
Some children may also require surgery, potential surgical interventions include:
- Tongue-lip adhesion:
- This procedure:
- Corrects the position of your baby’s tongue by pulling the base of the tongue forward and stitching it to the lower lip
- Holds the tongue forward until your child develops a more stable airway with growth
- Is reversed when your baby’s cleft palate is repaired, at approximately 9 to 11 months old
- This procedure:
- Mandibular distraction osteogenesis:
- This process:
- Lengthens the jaw in a forward direction
- Pulls the tongue base forward
- Relieves the obstruction of the airway
- Involves generating new bone by progressively stretching divided segments of the jaw
- This process:
Feeding treatments
A team of craniofacial nurses will weigh your child and provide frequent follow-ups. As they monitor your child’s growth and weight gain, a team of doctors and nurses may suggest the following:
- Specialty feeders: The Haberman feeder is the most commonly used bottle for infants with Robin sequence, a cleft palate or both. In this specially designed bottle, the nipple is separated from the bottle by a one-way valve. It helps your baby feed by rewarding even the weakest suck with formula or breast milk. It also:
- Never floods
- Reduces wind, vomiting and colic
- Has variable flow settings
- Allows to you apply compression to help your baby receive milk
- Nasogastric feeding tube (NG tube): This is a flexible tube made of rubber that is passed through your baby’s nose and into the stomach. Nasogastric tubes are utilized as a temporary solution (weeks to months) to ensure weight gain.
- Gastrostomy feeding tube (G-tube): A feeding tube that is inserted directly into the stomach to provide nutrition. This tube is surgically placed and is used as a long-term solution (six months to years).
Cleft palate repair
Your child’s cleft palate will be treated when he or she is 9 to 11 months old. The type of treatment depends on the extent of the cleft and may include:
- Dentofacial orthopedics: Repairs of the face and jaw that may require appliances in upper jaw to widen the palate or headgear to correct an underbite.
- Corrective surgery and follow-up surgery to make additional corrections.
Pierre Robin sequence prognosis
Choking and feeding problems may go away on their own over the first few years as the lower jaw grows to a more normal size. There is a high risk for problems if the child’s airways are not kept from getting blocked.
All neonates with significant Pierre Robin sequence are at risk for sudden death 1. The sudden infant death syndrome (SIDS) data show that the risk of SIDS is increased when infants sleep in the prone position. Neonates with Pierre Robin sequence already have a compromised airway, and they also typically require prone positioning. Accordingly, monitoring of these neonates should be strongly considered.
Infants with Pierre Robin sequence deserve to be treated with a multidisciplinary approach that involves a knowledgeable and experienced team capable of providing a comprehensive assessment, a realistic plan of treatment, and appropriate follow-up. Engaging the family in the early stages of the evaluation, the ongoing medical investigations, the issues regarding the child’s care, and future planning generally leads to satisfaction, even in the most difficult of medical scenarios.
In a study of 103 patients followed for a median of 8.6 years (range, 0.1-21.9 years), Logjes et al 3 documented a 10% mortality (n = 10) at a median patient age of 0.8 years (range, 0.1-5.9 years). Of the 10 infants who died, nine had syndromic Pierre Robin sequence; seven of the nine died of respiratory insufficiency due to various causes, and the other two died of arrhythmia due to hypernatremia and of West syndrome with status epilepticus. The infant with nonsyndromic Pierre Robin sequence died of brain ischemia after mandibular distraction osteogenesis.
References- Pierre Robin Sequence. https://emedicine.medscape.com/article/995706-overview
- Pierre Robin sequence. https://medlineplus.gov/ency/article/001607.htm
- Logjes RJH, Haasnoot M, Lemmers PMA, Nicolaije MFA, van den Boogaard MH, Mink van der Molen AB, et al. Mortality in Robin sequence: identification of risk factors. Eur J Pediatr. 2018 May. 177 (5):781-789.





{kind=link}