What is Pompe disease
Pompe disease also called acid maltase deficiency or glycogen storage disease type II 1, is a rare (estimated at 1 in every 40,000 births in the United States) inherited and often fatal disorder due to buildup of a complex sugar called glycogen in the body’s cells that disables the heart and skeletal muscles 2. Pompe disease is caused by mutations in a gene that makes an enzyme called acid alpha-glucosidase (also known as acid maltase). Normally, the body uses acid alpha-glucosidase to break down glycogen, a stored form of sugar used for energy. The enzyme performs its function in intracellular compartments called lysosomes. Lysosomes are known to function as cellular clearinghouses; they ingest multiple substances including glycogen, which is converted by the acid alpha-glucosidase into glucose, a sugar that fuels muscles. In Pompe disease, mutations in the GAA gene reduce or completely eliminate this essential enzyme. Excessive amounts of lysosomal glycogen accumulate everywhere in the body, but the cells of the heart and skeletal muscles are the most seriously affected. Researchers have identified up to 300 different mutations in the GAA gene that cause the symptoms of Pompe disease, which can vary widely in terms of age of onset and severity. The severity of the Pompe disease and the age of onset are related to the degree of enzyme deficiency. The incidence of Pompe disease varies among different ethnic groups.
Researchers have described three types of Pompe disease, which differ in severity and the age at which they appear. These types are known as classic infantile-onset, non-classic infantile-onset, and late-onset.
- Classic form of infantile-onset Pompe disease begins within a few months of birth. Infants with this disorder typically experience muscle weakness (myopathy), poor muscle tone or floppiness (hypotonia), an enlarged liver (hepatomegaly), feeding problems, poor weight gain, and head lag and heart defects (grossly enlarged). Many infants with Pompe disease also have enlarged tongues. Affected infants may also fail to gain weight and grow at the expected rate (failure to thrive) and have breathing problems often complicated by lung infections. If untreated, most babies die from heart failure or respiratory complications before their first birthday.
- Non-classic form of infantile-onset Pompe disease usually appears by age 1. It is characterized by delayed motor skills (such as rolling over and sitting) and progressive muscle weakness. The heart may be abnormally large (cardiomegaly), but affected individuals usually do not experience heart failure. The muscle weakness in this disorder leads to serious breathing problems, and most children with non-classic infantile-onset Pompe disease live only into early childhood.
- Late onset (or juvenile/adult) Pompe disease may not become apparent until later in childhood, adolescence, or adulthood. Late-onset Pompe disease is usually milder than the infantile-onset forms of this disorder and is less likely to involve the heart. Most individuals with late-onset Pompe disease experience progressive muscle weakness, especially in the legs and the trunk, including the muscles that control breathing. As the disorder progresses, breathing problems can lead to respiratory failure. The primary symptom is muscle weakness progressing to respiratory weakness and death from respiratory failure after a course lasting several years. The heart is usually not involved.
A diagnosis of Pompe disease can be confirmed by screening for the common genetic mutations or measuring the level of acid alpha-glucosidase enzyme activity in a blood sample. Once Pompe disease is diagnosed, testing of all family members and a consultation with a professional geneticist are recommended. Carriers are most reliably identified via genetic mutation analysis.
Individuals with Pompe disease are best treated by a team of specialists (such as cardiologist, neurologist, and respiratory therapist) knowledgeable about the disease, who can offer supportive and symptomatic care. The discovery of the GAA gene has led to rapid progress in understanding the biological mechanisms and properties of the GAA enzyme. As a result, an enzyme replacement therapy has been developed that has shown, in clinical trials with infantile-onset patients, to decrease heart size, maintain normal heart function, improve muscle function, tone, and strength, and reduce glycogen accumulation. A recombinant human acid alpha-glucosidase called alglucosidase alfa (Myozyme©), has received FDA approval for the treatment of infants and children with Pompe disease. Another algluosidase alfa drug, Lumizyme©, has been approved for late-onset (non-infantile) Pompe disease.
Enzyme replacement therapy has been shown to dramatically improve the prognosis and quality of life of patients with infantile-onset Pompe disease. Early initiation of enzyme replacement therapy in infantile-onset Pompe disease reduces the risk of death and invasive ventilation in the first year of life. To achieve the optimal outcome, enzyme replacement therapy should be started before symptoms are apparent and irreversible damage has occurred 3. Newborn screening is the best way to diagnose patients and enable initiation of treatment in a timely manner in the pre-symptomatic period 4. Newborn screening for Pompe disease has already started in several US states, and it has been successful in improving the prognosis of patients with infantile-onset Pompe disease 5.
Pompe disease life expectancy
Without enzyme replacement therapy, the hearts of babies with infantile onset Pompe disease progressively thicken and enlarge. These babies die before the age of one year from either cardiorespiratory failure or respiratory infection. For individuals with late onset Pompe disease, the prognosis is dependent upon the age of onset. In general, the later the age of onset, the slower the progression of the disease. Ultimately, the prognosis is dependent upon the extent of respiratory muscle involvement.
Pompe disease symptoms
The clinical spectrum of Pompe disease is continuous and broad. In the severe, infantile onset cases, signs and symptoms usually present within the first months of life. In many late-onset patients, symptoms may not develop (or be brought to clinical attention) for several years or decades. Initial signs and symptoms in children and adults may be subtle (such as having difficulty rising from a chair or climbing stairs, or morning headaches and fatigue), but the severity of symptoms and the rate of progression will vary from person to person. Pompe disease expression is modified by the age of onset, underlying genetic mutations, and as-of-yet unknown genetic and environmental interactions.
Patients with the classic infantile form of Pompe disease are the most severely affected. Although hardly any symptoms may be apparent at birth, the disease usually presents within the first three months of life with rapidly progressive muscle weakness (floppy infants), diminished muscle tone (hypotonia), respiratory insufficiency, and a type of heart disease known as hypertrophic cardiomyopathy, a condition characterized by abnormal thickening of the walls of the heart (mainly the left chamber and the wall between the left and right chamber) resulting in diminished cardiac function. These problems together culminate in cardio-respiratory failure within the first 2 years of life.
Many infants have a large, protruding tongue and a moderate enlargement of the liver. The legs often rest in a frog position and feel firm on palpation (pseudo-hypertrophy).
Feeding and swallowing problems as well as respiratory difficulties, which are often combined with respiratory tract infections, are common. Major developmental milestones such as rolling over, sitting up, and standing are delayed or not achieved. Mental development is usually normal. Virtually all infants experience hearing loss. The classic infantile form of Pompe disease is characterized by a total lack of acid alpha-glucosidase (GAA) activity and by a rapid buildup of glycogen in skeletal muscle and heart.
Childhood Pompe disease typically presents during childhood and adult Pompe disease during adulthood. Both these forms of Pompe disease are often grouped together as late-onset Pompe disease despite the fact that the time of presentation can vary from the first year to the eighth decade. Patients who develop symptoms early in life tend to be the more severely affected and to have a faster rate of disease progression than those who develop symptoms later in life. Both children and adults usually have more acid alpha-glucosidase activity present than those who show symptoms as infants, and the glycogen buildup is not usually as rapid. However, symptoms do progress, can greatly affect the quality of life, and diminish the lifespan of affected individuals.
Childhood and adult Pompe disease are associated with progressive weakness of mainly the proximal muscles (limb girdle, upper arms and upper legs), and varying degrees of respiratory weakness due to dysfunction of the diaphragm and intercostal muscles (muscle between ribs). The lower limbs are more affected than the upper limbs. The extent of muscle involvement is highly variable. The muscles adjacent to the spinal column (para-spinal muscles) and neck are usually also involved. Weakness of the para-spinal muscles around puberty can cause abnormal curvature of the spine (scoliosis). As a result of the combination of these serious symptoms, affected individuals may become wheelchair and/or ventilator dependent.
Other symptoms can include chewing and swallowing difficulties and drooping of the upper eyelids (ptosis). Additionally, blood vessel abnormalities due to smooth muscle weakness and problems of the urinary and digestive systems have been reported.
What causes Pompe disease
Mutations in the GAA gene cause Pompe disease. The GAA gene provides instructions for producing an enzyme called acid alpha-glucosidase (also known as acid maltase). This enzyme is active in lysosomes, which are structures that serve as recycling centers within cells. The enzyme normally breaks down glycogen into a simpler sugar called glucose, which is the main energy source for most cells.
Mutations in the GAA gene prevent acid alpha-glucosidase from breaking down glycogen effectively, which allows this sugar to build up to toxic levels in lysosomes. This buildup damages organs and tissues throughout the body, particularly the muscles, leading to the progressive signs and symptoms of Pompe disease.
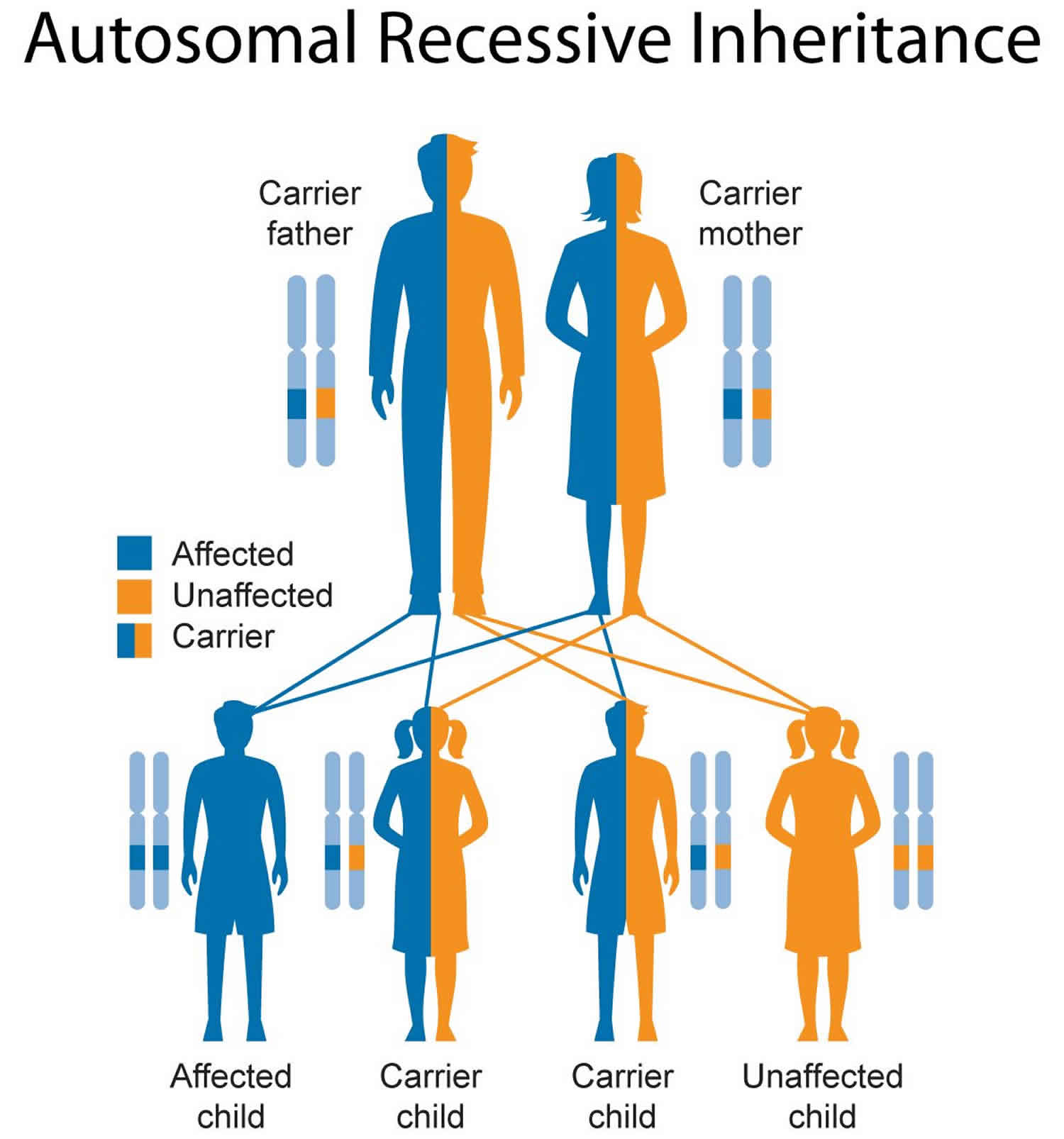
Inheritance Pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Figure 1. Pompe disease autosomal recessive inheritance pattern
Pompe disease diagnosis
A diagnosis of Pompe disease is based upon a thorough clinical evaluation, a detailed patient and family history, and a variety of tests. Prenatal diagnosis is possible when a pregnancy is believed to be at risk for Pompe disease.
Pompe disease can be diagnosed by determining the activity of the enzyme acid alpha glucosidase. This deficiency can be shown with a specific enzyme testing that can be performed on blood samples, muscle biopsy or cultured cells from a skin biopsy. This type of testing is very specific and available at only a few specialized laboratories in the USA. Genetic testing for mutation finding and gene sequencing for pompe disease is also available clinically from a limited number of laboratories in the USA. For many families, genetic testing is most helpful in providing additional information for the person with pompe disease and other family members after the diagnosis is made and confirmed using enzyme (acid alpha glucosidase) testing.
If someone with muscle weakness has a muscle biopsy examined at their local hospital by pathology, testing called histopathology will show a great increase of glycogen of normal structure, and microscopic studies will show increased glycogen enclosed within the lysosomes. This type of testing, while helpful in determining that pompe disease is a possible diagnosis, is not diagnostic for pompe disease. In some people with pompe disease, depending on where the piece of muscle was obtained, the muscle histopathy in the piece of muscle from the the muscle biopsy can be normal. If pompe disease is suspected, then diagnostic testing is needed. These diagnostic tests can either test the function of specific enzyme acid alpha glucosidase or genetic testing looking for changes in the GAA gene can be performed.
Clinical Testing and Work Up
In individuals suspected of having Pompe disease, blood can be drawn and the function/activity of the acid alpha glucosidase enzyme can be measured in white blood cells (leukocytes), but only if the proper assay conditions are being used and acarbose is added to the reaction mixture to inhibit the activity of glucoamylase. The isolation of lymphocytes to prevent the interference of glucoamylase is not advised, as the successful isolation of lymphocytes is not only time consuming, but also error prone when the blood sample is not sufficiently fresh.
Alternatively, the acid alpha glucosidase enzyme activity/functional assay can also be performed on a dried blood spot, but the assay is not any quicker or more sensitive than the leukocyte assay and also requires the use of acarbose to inhibit the glucoamylase activity. The advantage of the bloodspot test is that it allows convenient shipment of samples if a certified diagnostic laboratory test is not locally available. Additionally, dried blood spot testing allows for mass screening. The blood spot test is without any argument the most convenient methodology for the screening of large populations of newborns and, for instance, large numbers of patients with undiagnosed limb-girdle muscular dystrophies and creatine kinase-non-ketotic hypoglycemia.
When a diagnosis of Pompe disease is based on a leukocyte or blood spot assay, it must be confirmed through molecular genetic testing (DNA analysis) or by another enzyme assay, preferably using cultured skin fibroblasts obtained by a skin biopsy. More invasive muscle biopsies are not needed and not optimal for obtaining material for acid alpha glucosidase enzyme activity/function assays. The advantage of DNA analysis over a acid alpha glucosidase enzyme activity assay is that the DNA test discriminates unequivocally between carriers and affected children/adults, whereas the enzyme assay does not in all cases.
The taking of a skin biopsy and the growing of a skin fibroblasts culture may not be feasible in every diagnostic setting, but should always be considered as there are important advantages to this procedure. The acid alpha glucosidase enzyme activity test on this material is superior over others as it is the most sensitive test and discriminates best between classic-infantile, childhood and adult Pompe disease. Cultured fibroblasts can be stored forever and used as eternal source of acid alpha glucosidase enzyme, DNA and mRNA for all kinds of present day and future sophisticated analyses.
A variety of additional tests may be performed to detect or assess symptoms potentially associated with Pompe disease such as sleep studies, breathing tests to measure lung capacity, and electromyography, a test to measure muscle function. Muscle MRI’s are also used to measure the degree of damage that has occurred to the muscles.
Specific tests may also performed to assess the heart including chest x-rays, electrocardiogram, and echocardiogram. Chest x-rays allows physicians to assess the size of the heart, which can be enlarged in some infants with Pompe disease. An electrocardiogram measures the electrical activity of the heart and can detect abnormal heart rhythms. An echocardiogram uses reflected sound waves to create a picture of the heart and can reveal abnormal thickening of the heart muscle tissue.
If the specific GAA gene mutations in both parents are known, prenatal diagnosis is possible through chorionic villi sampling (CVS) or amniocentesis. Pre-implantation genetic diagnosis (testing an embryo to determine whether it has the same genetic abnormalities as the parents) may also be an option. Pre-implantation genetic diagnosis can be performed on embryos created through in vitro fertilization. Families interested in pre-implantation genetic diagnosis should seek the counsel of a certified genetics professional.
Pompe disease treatment
The treatment of Pompe disease is disease-specific, symptomatic, and supportive. Treatment requires the coordinated efforts of a team of specialists with expertise in treating neuromuscular disorders. Pediatricians or internists, neurologists, orthopedists, cardiologists, dieticians, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment. Genetic counseling is of utmost importance for affected individuals and their families.
Enzyme Replacement Therapy
Enzyme replacement therapy is an approved treatment for all patients with Pompe disease. It involves the intravenous administration of recombinant human acid α-glucosidase. In 2006, enzyme replacement therapy (ERT) for Pompe disease (Myozyme®) was approved in the USA, Europe, and Canada, with subsequent approval in countries around the world. (In 2010, the Food and Drug Administration (FDA) approved Lumizyme® for patients older than 8 years of age. In 2014, the FDA, expanded their approval of Lumizyme to include patients of any age. Genzyme Corp., Cambridge, MA). In all patients, enzyme replacement therapy is recommended every two weeks via intravenous administration of recombinant human acid a-glucosidase at a dose of 20 mg/kg body weight.
Supportive Therapies
Additional treatment of Pompe disease is symptomatic and supportive. Respiratory support may be required, as most patients have some degree of respiratory compromise and/or respiratory failure. Physical therapy may be helpful to strengthen respiratory muscles. Some patients may need respiratory assistance through mechanical ventilation (i.e. bipap or volume ventilators) during the night and/or periods of the day. In addition, it may be necessary for additional support during respiratory tract infections. Mechanical ventilation support can be through noninvasive or invasive techniques. The decision about the duration of respiratory support is best made by the family in careful consultation with the patient’s physicians and other members of the healthcare team based upon the specifics of the patient.
Physiotherapy is recommended to improve strength and physical ability. Occupational therapy, including the use of canes or walkers, may be necessary. Eventually, some individuals may require the use of a wheelchair. Speech therapy can be beneficial to improve articulation and speech for some patients.
Orthopedic devices including braces may be recommended for some patients. Surgery may be required for certain orthopedic symptoms such as contractures or spinal deformity.
Since Pompe disease can weaken muscles used for chewing and swallowing, adequate measures may be required to ensure proper nutrition and weight gain. Some patients may need specialized, high-calorie diets and may need to learn techniques to change the size and texture of food to lower the risk of aspiration. Some infants may require the insertion of a feeding tube that is run through the nose, down the esophagus and into the stomach (nasogastric tube). In some children, a feeding tube may need to be inserted directly into the stomach through a small surgical opening in the abdominal wall. Some individuals with late onset Pompe disease may require a soft diet, but few require feeding tubes.
Respiratory support
This is one of the most important aspects of Pompe management, as most patients have some degree of respiratory compromise and respiratory failure is the most common cause of death in children and adults with the disease 6. Physical therapy may help to strengthen respiratory muscles and airway secretion clearance can be can be optimized through assistive cough, chest percussion and suctioning. Close monitoring for the early signs of infection is critical. Further interventions include non-invasive or invasive ventilatory support for weakened diaphragm and intercostal muscles.
Physiotherapy
Pompe disease causes progressive muscular degeneration resulting in weakness and physical disability. Therefore, stabilizing or improving physical ability it is one of the aims of Pompe management. Physiotherapy, in conjunction with enzyme replacement therapy (ERT), can help to alleviate or prevent secondary complications, such as reduced bone mineral density and contractures.
Occupational therapy
Patients may eventually need the assistance of mobility devices to remain independent. In the earlier stages of the disease, canes and walkers may suffice, but in advanced disease, patients may become non-ambulatory and require the use of wheelchairs.
Nutrition
Pompe disease can weaken muscles used for chewing and swallowing food. As a result, many people with Pompe disease have trouble gaining and/or maintaining weight. Patients are routinely given guidance on balanced, high-calorie diets, as well as on strategies to change the size and texture of food to make it easier to eat and to reduce the risk of aspiration. Nasogastric or nasoenteric tube feeding may be called for if patients are at high risk for aspiration. Permanent tube placement in the abdomen may also be required. However, in general, tube feeding is more common in infantile-onset patients due to severe muscle weakness, ventilator dependency, and macroglossia.
Orthopedic support and interventions
As in a number of neuromuscular disorders, patients with Pompe disease may experience contractures, scapular winging, and truncal weakness due to muscle wasting and reduced muscle use. Spinal deformity, particularly scoliosis, can also affect pulmonary function and may cause discomfort while sitting. If severe, orthopedic braces or surgical intervention may be necessary to alleviate pain and/or disability 7.
Speech therapy
Speech may be affected due to facial and tongue muscle weakness and macroglossia (particularly in infants). Bulbar muscle weakness affects speech intelligibility and therefore social communication. Early referral to a speech therapist may help to improve articulation and speech.
Additional assessments
Audiology assessments (particularly for children) and sleep studies are also often warranted in the management of Pompe disease.
References- Vissing J, Lukacs Z, Straub V. Diagnosis of Pompe DiseaseMuscle Biopsy vs Blood-Based Assays. JAMA Neurol. 2013;70(7):923–927. doi:10.1001/2013.jamaneurol.486
- Pompe Disease Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Pompe-Disease-Information-Page
- The emerging phenotype of long-term survivors with infantile Pompe disease. Genet. Med., 14 (2012), pp. 800-810.
- Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics, 124 (2009), pp. 1116-1125
- A large-scale nationwide newborn screening program for Pompe disease in Taiwan: towards effective diagnosis and treatment. Am. J. Med. Genet. A, 164A (2014), pp. 54-61
- Winkel, L.P., et al., The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol, 2005. 252(8): p. 875-84.
- Roberts, M., et al., The prevalence and impact of scoliosis in Pompe disease: lessons learned from the Pompe Registry. Mol Genet Metab, 2011. 104(4): p. 574-82.





{kind=link}