Sickle cell disease
Sickle cell disease is a group of inherited red blood cell disorders that affects hemoglobin (Hb), the protein that carries oxygen to cells throughout the body. Within the umbrella of sickle cell disease, many subgroups exist, namely sickle cell anemia (HbSS), hemoglobin SC disease (HbSC), and hemoglobin sickle-beta-thalassemia (HbSB or HbSBetaThal). Sickle cell beta-thalassemia (HbSB) can present in 2 forms. The first form is one in which there is a small amount of normal hemoglobin present called sickle cell beta-thalassemia+ (Sickle Beta-Plus Thalassemia or HbSB+). The sickle cell disease combined with beta-thalassemia (Sickle Beta-Plus Thalassemia) is generally a “mild” form of sickle cell disease 1. The second form is the absence of normal hemoglobin called sickle cell beta-thalassemia zero (Sickle Beta-Zero Thalassemia or HbSB0), and these patients present similarly to those with sickle cell disease 2. Several other minor variants within the group of sickle cell diseases also, albeit not as common as the aforementioned varieties. Lastly, it is important to mention the sickle cell trait (HbAS), which carries a heterozygous mutation and seldom presents with any clinical signs or symptoms. Normal hemoglobin is called hemoglobin A (α2β2) and consists of four protein subunits: two subunits of alpha (α) globin chains, which is produced by hemoglobin alpha (HBA1 or HBA2) gene and two subunits of beta (β) globin chains, which is produced by HBB gene (hemoglobin beta gene). Each of these protein subunits is attached (bound) to an iron-containing molecule called heme; each heme contains an iron molecule in its center that can bind to one oxygen molecule. Hemoglobin within red blood cells binds to oxygen molecules in the lungs. A complete hemoglobin protein is capable of carrying four oxygen molecules at a time (one attached to each heme molecule). Oxygen attached to hemoglobin gives blood its bright red color. These red blood cells then travel through the bloodstream and deliver oxygen to tissues throughout the body (see Figure 4). Adult red blood cells normally contain the following hemoglobin chain combinations: hemoglobin A (α2β2 or alpha2-beta 2) >95%; hemoglobin A2 (α2δ2 or alpha2-delta2) 2% to 3.4%; fetal hemoglobin F (α2γ2 or alpha2-gamma2) <1%. In sickle-cell disease, the sickle-shaped red blood cells do not carry as much oxygen and therefore deliver less oxygen to the body’s tissues. These cells are also fragile and can break, causing painful “crises” because they disrupt blood flow.
Sickle cell disease is the most common genetic disease in the United States, affecting 1 in 500 African Americans and 1 in 1,000 to 1,400 Hispanic Americans 3, 4. About 1 in 12 African Americans carry the autosomal recessive mutation, and approximately 300,000 infants are born with sickle cell anemia annually. Sickle cell disease affects more than 100,000 people in the United States and 20 million people worldwide.
Normally, red blood cells are disc shaped and flexible to move easily through the blood vessels. If you have sickle cell disease, your red blood cells are crescent or “sickle” shaped. The sickle-shaped red blood cells are not flexible and cannot change shape easily. Many of them burst apart as they move through your blood vessels. The sickle red blood cells usually only last 10 to 20 days, instead of the normal 90 to 120 days. Your body may have trouble making enough new red blood cells to replace the ones that you lost. Because of this, you may not have enough red blood cells. This is a condition called anemia, and it can make you feel tired.
The sickle-shaped red blood cells can also stick to blood vessel walls, causing a blockage that slows or stops the flow of blood. When this happens, oxygen can’t reach nearby tissues. The lack of oxygen can cause attacks of sudden, severe pain, called pain crises. These attacks can occur without warning. If you get one, you might need to go to the hospital for treatment.
The blocked blood flow through the body can also lead to serious problems, including stroke, eye problems and infections.
Sickle cell disease is inherited, meaning that it runs in families. People who have sickle cell disease inherit two abnormal hemoglobin genes, called hemoglobin-Beta gene (HBB gene), one from each parent. The HBB gene provides instructions for making beta-globin, which is one part of hemoglobin (see Figure 4 below). Sickle cell disease is caused by a point mutation in the hemoglobin-Beta gene (HBB gene) found on chromosome 11. Various versions of beta-globin result from different mutations in the HBB gene. A point mutation of HBB gene replaces A with T at codon 6 of beta hemoglobin chain 5. This causes the switch from glutamic acid to valine amino acid at position 6 in beta-globin, written as Glu6Val or E6V. Replacing glutamic acid with valine causes the abnormal version of beta-globin known as hemoglobin S (HbS) subunits to stick together and form long, rigid molecules that bend red blood cells into a sickle (crescent) shape when exposed to a low oxygen threshold. Other mutations in the HBB gene lead to additional abnormal versions of beta-globin such as hemoglobin C (HbC) and hemoglobin E (HbE). HBB gene mutations can also result in an unusually low level of beta-globin; this abnormality is called beta thalassemia.
If you’re a carrier of sickle cell or have sickle cell trait, it means you carry one of the hemoglobin-Beta (HBB) gene that causes sickle cell disease, but you do not have the condition yourself. In contrast, people with sickle cell disease carry two copies of the altered hemoglobin gene. With two copies of the altered gene, the red blood cells are destroyed rapidly and patients have chronic, severe anemia, or low hemoglobin levels. Red blood cells become misshapen, many of which take the “C” or sickle shape that gives the disease its name. Without proper treatment, a person with sickle cell disease can develop recurrent episodes of pain and may have life-threatening complications, including damage to organs such as brain, bones, lungs, kidneys, liver and heart. The disease affects between 70,000 and 100,000 Americans and is most common in people of African, Middle Eastern, Mediterranean, Central and South American and Asian Indian origin or descent.
A person has sickle cell disease trait (HbAS) also called sickle cell carrier when the hemoglobin S (Hb S) gene is inherited from only one parent, and a normal hemoglobin gene — hemoglobin A (Hb A) — is inherited from the other parent 6. People who have sickle cell trait (sickle cell carrier) are generally healthy. However, there have been reports of adverse conditions such as anaesthetics can cause problems due to the patient’s sickle cell trait status. If you have sickle cell trait always notify your dentist or doctor before treatment commences to be on the safe side. There is also a small chance that you may experience pain at high altitudes (generally above 10,000 feet), including long-haul flying in unpressurized planes and mountain climbing. It is important you say you have sickle cell trait before undertaking such activities as you may need to breathe oxygen. While sickle cell trait is not a barrier to playing competitive sports, athletes with sickle cell trait have experienced significant physical distress, including collapse and death during intense exercise. Heat, dehydration, inadequate acclimatization, altitude and asthma can increase the risk for medical complications in athletes with sickle cell trait 7. Therefore, sickle cell trait may not be completely benign and these patients should be managed aggressively whenever they develop some of these complications 6. The National Collegiate Athletic Association (NCAA) has some great resources for athletes and their coaches with sickle cell trait (https://www.ncaa.org/sports/2016/7/27/sickle-cell-trait.aspx).
If you have sickle cell trait, you are a carrier of the hemoglobin S (Hb S) gene, so you can pass it on when you have a child. If the child’s other parent also has sickle cell trait or another abnormal hemoglobin gene, such as beta-thalassemia, hemoglobin C, hemoglobin D, or hemoglobin E, that child has a chance of having sickle cell disease.
If both parents are sickle cell carriers (also known as having the sickle cell trait), there’s a:
- 1 in 4 chance each child they have will not inherit any sickle cell genes and will not have sickle cell disease or be able to pass it on
- 1 in 2 chance each child they have will just inherit a copy of the sickle cell gene from 1 parent and be a carrier
- 1 in 4 chance each child they have will inherit copies of the sickle cell gene from both parents and will be born with sickle cell disease
Worldwide, it is estimated that there are 300 million people with sickle cell trait and one-third of this number are in sub-Saharan Africa 8. This protection against Plasmodium explains why the prevalence of sickle cell trait is higher in areas where malaria is endemic. Gibson and colleague mentions that the prevalence is as high as 25% in some part of Africa and 60% in Saudi Arabia 9. Because of the high migration of people from areas of high prevalence like Africa, Middle East, the prevalence of both sickle cell trait and disease will increase in the western part of the world.
Many states routinely screen newborns for sickle cell so that treatment can begin as soon as possible. Early diagnosis and treatment can reduce the risk of complications.
Hemoglobin electrophoresis is a blood test that can determine if a person is a carrier of sickle cell, or has any of the diseases associated with the sickle cell gene.
Sickle cell disease is a lifelong illness. A blood and bone marrow transplant is currently the only cure for sickle cell disease, but there are effective treatments that can reduce symptoms and prolong life. Treatments for sickle cell include antibiotics, pain management and blood transfusions. A new drug treatment, hydroxyurea, which is an anti-tumor drug, appears to stimulate the production of fetal hemoglobin F (α2γ2), a type of hemoglobin usually found only in newborns. Fetal hemoglobin (α2γ2) helps prevent the “sickling” of red blood cells. Patients treated with hydroxyurea also have fewer attacks of acute chest syndrome and need fewer blood transfusions.
Figure 1. Blood composition
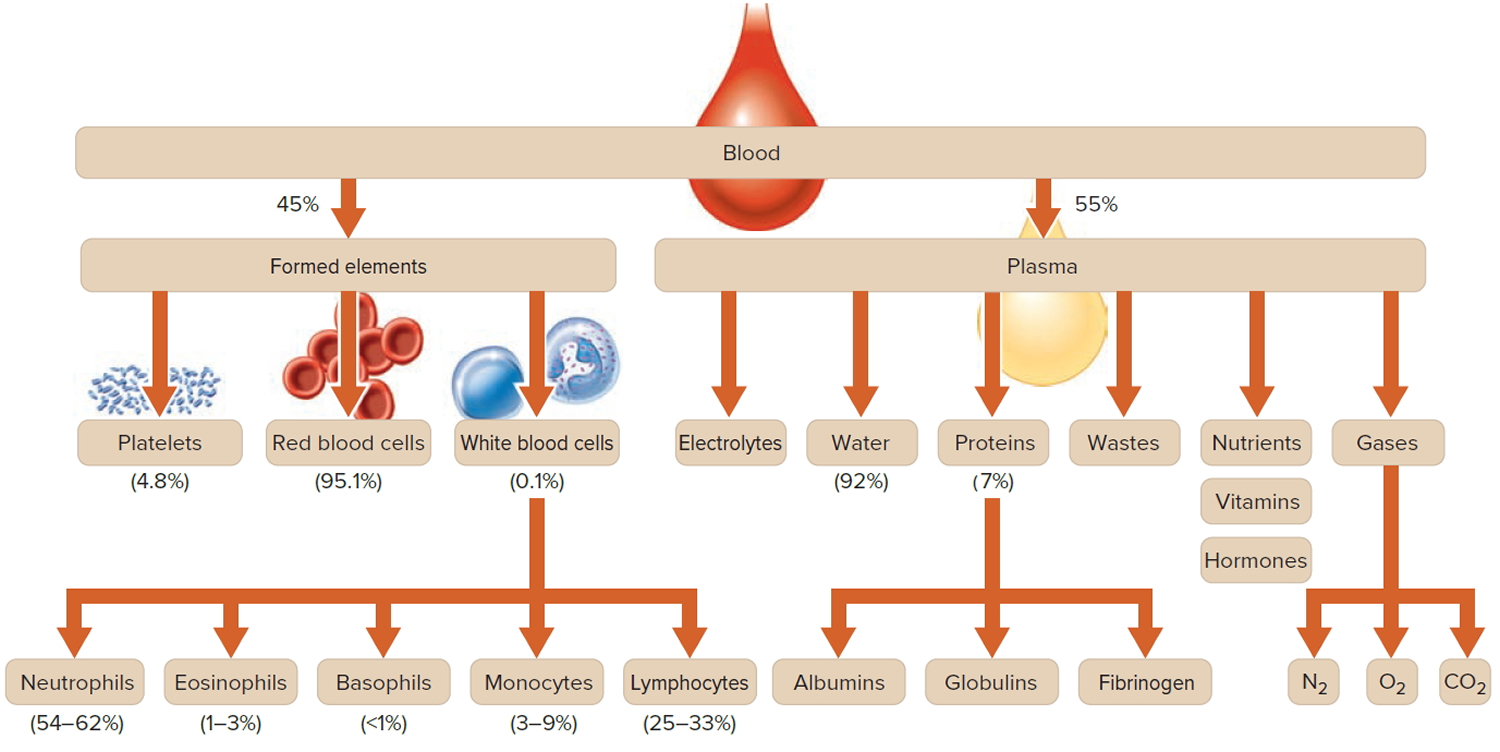
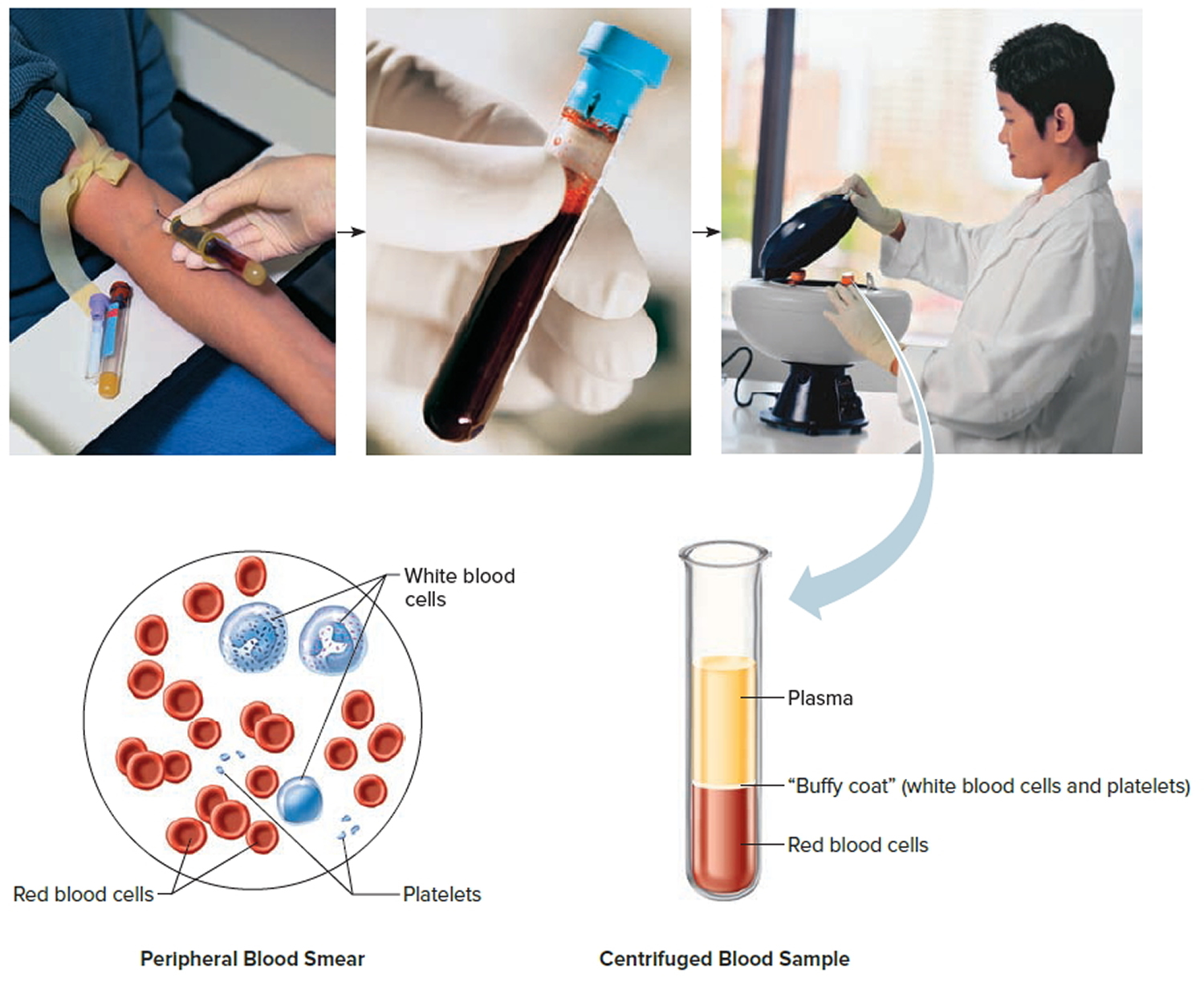
Footnote: Blood consists of a liquid portion called plasma and a solid portion (the formed elements) that includes red blood cells, white blood cells, and platelets. When blood components are separated by centrifugation, the white blood cells and platelets form a thin layer, called the “buffy coat,” between the plasma and the red blood cells, which accounts for about 1% of the total blood volume. Blood cells and platelets can be seen under a light microscope when a blood sample is smeared onto a glass slide.
Figure 2. Red blood cells (normal red blood cells)
Figure 3. Sickle cell disease
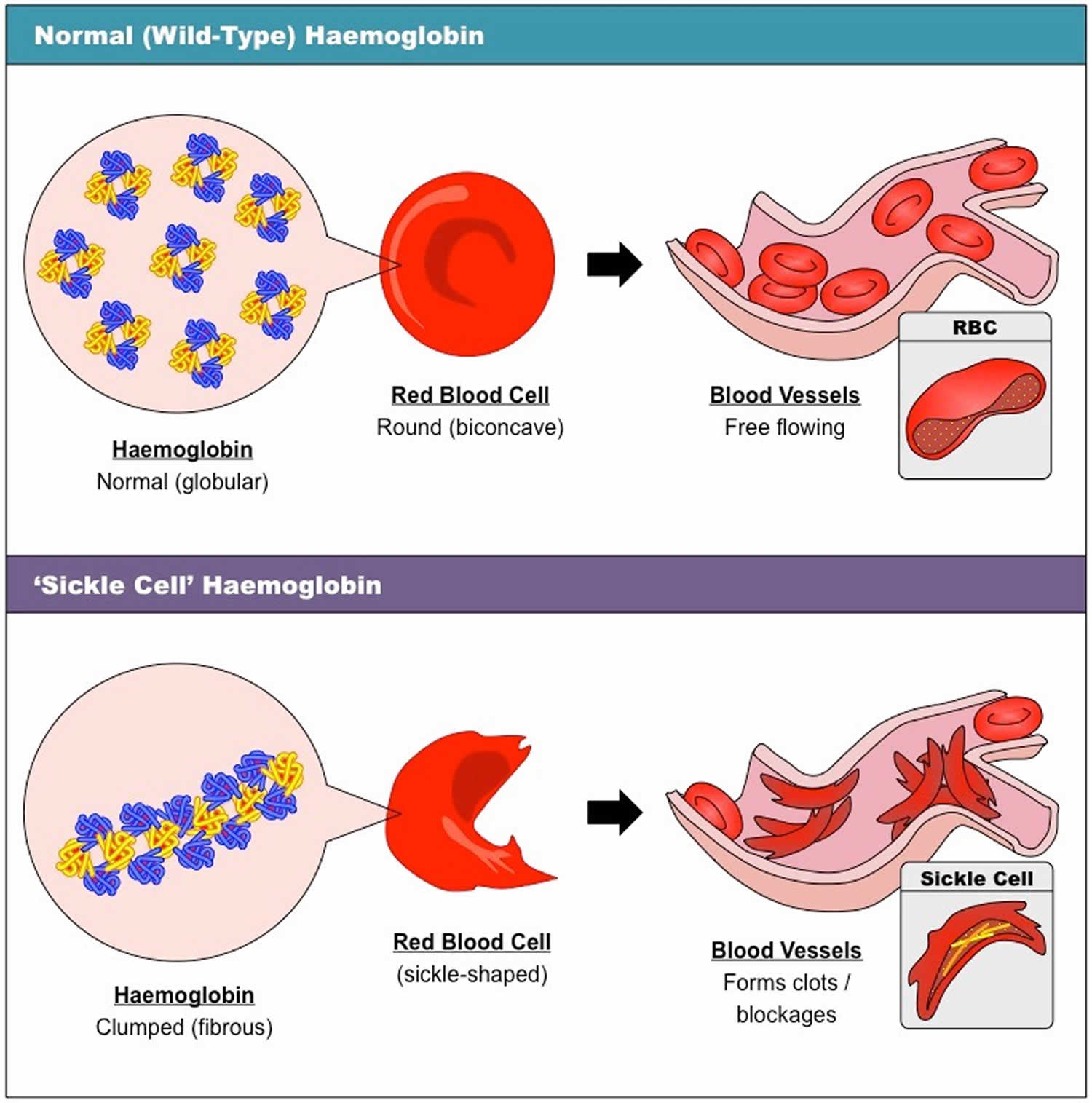
Figure 4. Normal hemoglobin structure (normal hemoglobin is called hemoglobin A [HbA] and consists of 2 alpha (α) globin chains and 2 beta (β) globin chains)
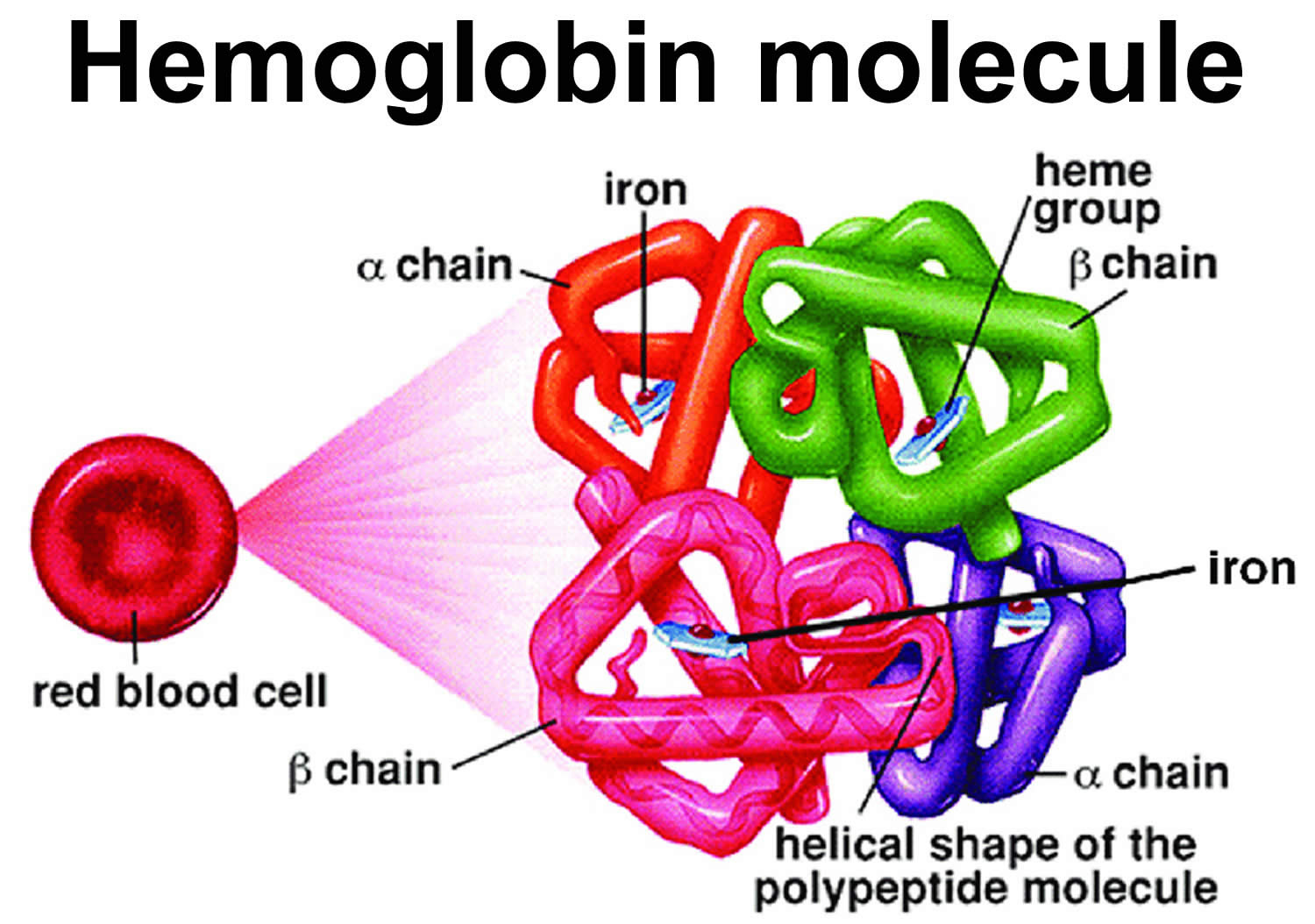
Figure 5. Sickle cell disease hemoglobin structure
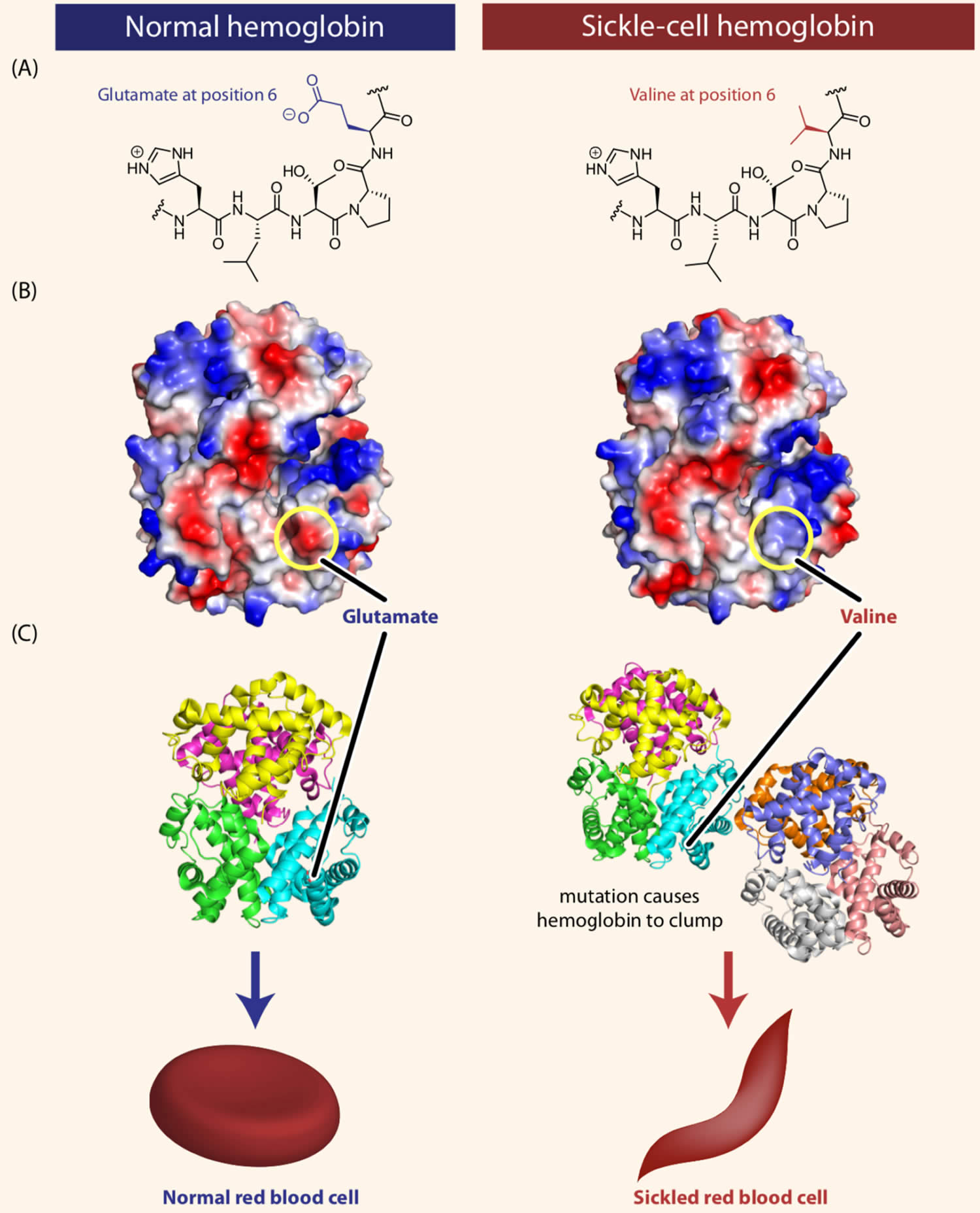
Footnotes: Sickle-cell disease is caused by a single amino acid change in the hemoglobin protein. (A) Line drawings of a portion of the hemoglobin (left) and sickle-cell hemoglobin (right) proteins. Normal hemoglobin contains the amino acid glutamate at position 6 in the primary sequence. In individuals with sickle-cell disease, this glutamate is replaced with the amino acid valine. (B) Computer-generated structure showing the charges present on the surfaces of hemoglobin (left) and sickle-cell hemoglobin (right). As we will see shortly in this chapter, some amino acids can be negatively or positively charged, while many others are neutral. In the figure, blue represents positive charge, red represents negative charge, and white represents neutral atoms. The substitution of the negatively charged amino acid glutamate at position 6 with the neutral valine removes a negative charge that is normally present in hemoglobin. (C) Computer-generated models showing the structures of the hemoglobin (left) and sickle-cell hemoglobin (right) proteins. The substitution of glutamate with valine causes hemoglobin tetramers to clump together.
[Source 10 ]Pregnancy and Sickle Cell Disease
Sickle cell disease can raise your risk of problems during pregnancy. Compared to women without sickle cell disease, women with sickle cell disease are more likely to experience preeclampsia 11, venous thromboembolism (blood clots in the veins), infections, and maternal mortality during pregnancy 12. Preeclampsia is a complication of pregnancy in which affected women develop high blood pressure (hypertension); they can also have abnormally high levels of protein in their urine (proteinuria).
During pregnancy, 40–50 percent of women with sickle cell disease require at least one hospital admission13. Although there are no data specifically for women with sickle cell disease, the presence of pulmonary hypertension increases the cardiopulmonary demands of gestation. Non-sickle cell disease maternal mortality has been reported to be as high as 30–50 percent in women with pulmonary hypertension 14. Even with current multidisciplinary care, maternal mortality in women with pulmonary hypertension is still reported to be 10 percent 15.
If you are pregnant or planning for pregnancy, meet with a healthcare provider who specializes in high-risk pregnancies and has experience with patients who have sickle cell disease. Your healthcare provider may prescribe certain vitamins and will be careful to prescribe pain medicines that are safe for you and your baby. You should not use hydroxyurea during pregnancy.
You may need to have one or more blood transfusions during pregnancy to treat problems, such as anemia symptoms that get worse. You may also experience more pain crises or be at higher risk of having acute chest syndrome. Your provider will talk to you about how to help prevent these complications.
Sickle cell trait in pregnancy
There are limited number of adequately designed and powered studies on pregnancy-related complications in sickle cell trait 16. An analysis of pregnancy-related deaths in the United States between 2007 and 2016 by the Centers for Disease Control (CDC) reported a mortality ratio for black women that was 2.8–3.3 times that of non-Hispanic white women 17. A systematic review of pregnancy complications in sickle cell disease demonstrated an increased risk for maternal mortality, as well as preeclampsia, stillbirth, preterm delivery, and small for gestational age infants 18. The effect of sickle cell trait on mortality related to pregnancy has not been studied, likely due to the relatively small number of perinatal maternal deaths (approximately 700 per year in the United States) 16. Overall, the weight of evidence currently suggests that there are no compelling data that adverse perinatal outcomes are associated with the presence of sickle cell trait 16.
Pregnancy-related bacteriuria
A possible association between pregnancy-related bacteriuria (the presence of bacteria in the urine) and sickle cell trait was proposed years ago and has subsequently been studied by several groups. Early studies reported an increased prevalence of asymptomatic bacteriuria and pyelonephritis (kidney infection) among individuals with sickle cell trait 19. A more recent study was unable to detect an association between sickle cell trait and asymptomatic bacteriuria during pregnancy, but it reported more than a three-fold increase (2.4% vs 0.7%) in the prevalence for pyelonephritis in sickle cell trait 20. The most recent study on this topic failed to find an association between sickle cell trait and asymptomatic bacteriuria or pyelonephritis 21. Mechanistically, it has been proposed that chronic renal papillary necrosis due to microinfarction could lead to an increased susceptibility to genitourinary tract infection in sickle cell trait 22. In the past, the American College of Obstetrics and Gynecology recommended routine screening for asymptomatic bacteriuria during each trimester in pregnant sickle cell trait patients 23, but the most recent guidelines do not address this recommendation 24. However, an informal survey of several large academic obstetric units suggests that the practice of screening for bacteriuria in pregnant women with sickle cell trait continues (unpublished data).
Pregnancy-related hypertensive disorders
A prospective study of pregnant black women with sickle cell trait was the first to demonstrate a significantly increased incidence of preeclampsia (24.7% vs 10.1%) compared to black women noncarriers 25. Since then, two large retrospective cohort studies and one retrospective case-control study failed to show an association between sickle cell trait and an increased risk of preeclampsia 26, 21. In a broader scope retrospective cohort study of over 25,000 women in the military from 1993–2013, the incidence of pregnancy-related hypertensive disorder (which included gestational hypertension, pre-eclampsia, and eclampsia) was compared in 5,004 individuals with sickle cell trait with 20,016 matched controls 27. The authors concluded that sickle cell trait is associated with an increased risk of pregnancy-related hypertensive disorder with a hazard ratio of 1.43, corresponding to an attributable risk of 30.6% for pregnancy-related hypertensive disorder from sickle cell trait 27. However, with pre-eclampsia and eclampsia comprising only a small portion (2%) of the pregnancy-related hypertensive disorder outcomes, no association between sickle cell trait and pre-eclampsia or eclampsia was observed 27. These findings suggest that additional studies are needed to clarify the strength of the association between sickle cell trait and pregnancy-related hypertensive disorder, as well as determine whether this association extends to pre-eclampsia and eclampsia. Given the association between sickle cell trait and acute kidney injury, as well as chronic kidney disease (CKD) 28 and possibly also end-stage renal failure (also known as end-stage renal disease) 29, it is conceivable that pregnancy-related hypertensive disorders in sickle cell trait may contribute to an increased the risk of chronic renal disease, although no studies have directly addressed this possibility.
Pregnancy loss
The most worrisome association between sickle cell trait and pregnancy outcomes is the risk for pregnancy loss. An older retrospective cohort study of 500 individuals with sickle cell trait did not find an increased risk for perinatal mortality 30. A more recent small retrospective case series of 131 pregnant patients with sickle cell trait showed an intrauterine fetal death rate of 8% (compared to 5% baseline at institution) and identified one neonatal death 31. However, the significance of this finding is unclear. The same group, in a retrospective case-control study of 180 patients at a single institution, found an almost three-fold rate (9.7% vs 3.5%) of fetal death after first-trimester viability amongst pregnant patients with sickle cell trait compared to ethnicity-matched noncarrier controls 26. Histologic examination of the placentas of pregnant mothers (regardless of pregnancy outcome) showed evidence of more amniotic fluid infection (50% vs 18%) in mothers with sickle cell trait compared to noncarriers. In addition, red cell sickling in the intervillous and decidual vessels of the placentas was observed, with evidence of placental infarctions and retroplacental haemorrhages. In contrast, a more recent and larger retrospective cohort study of over 1,800 pregnant individuals with sickle cell trait did not find an association between maternal sickle cell trait and perinatal morality 21.
Preterm delivery
Studies evaluating an association between sickle cell trait and other perinatal outcomes have generally been less controversial. Several have evaluated but failed to identify an association between sickle cell trait and preterm delivery 32, 21. In fact, a possible protective effect of sickle cell trait on risk for preterm delivery has been reported 32. The case-control study by Taylor et al did find a significantly shorter mean duration of pregnancy in individuals with sickle cell trait compared to noncarriers (233 vs 255 days; p < .001), suggesting increased risk of preterm delivery, although this was not measured directly and thus does not provide conclusive evidence for such an association (Taylor et al, 2006).
Low birthweight
A few studies have evaluated risk for low birthweight and sickle cell trait. Taylor et al 26 demonstrated that sickle cell trait was associated with decreased mean birth weight (2,114 g vs 2,672 g). However, subsequent studies failed to confirm this association 33.
Types of sickle cell disease
There are several different types of sickle cell disease that differ in symptoms and severity. The specific type of sickle cell disease a person has depends on the genes they inherited from their parents. People with sickle cell disease inherit genes that contain instructions, or code, for abnormal hemoglobin.
Below are the most common types of sickle cell disease.
Sickle cell anemia
Sickle cell anemia is also called homozygous sickle cell disease or HbSS disease, is the most common and most severe type of sickle cell disease. People who have sickle cell anemia inherit two HBB genes, one from each parent, that code for hemoglobin “S.” Hemoglobin S is an abnormal form of hemoglobin that causes the red blood cells to become rigid, and sickle shaped. These irregular blood cells die prematurely, resulting in a chronic shortage of red blood cells or anemia (low number of red blood cells).
Red blood cells are usually round and flexible, so they move easily through blood vessels. In sickle cell anemia, some red blood cells are shaped like sickles or crescent moons. These sickle cells also become rigid and sticky, which can slow or block blood flow. Signs and symptoms of sickle cell disease usually begin in early childhood and may include anemia, repeated infections, and periodic episodes of pain (called crises). Children with sickle cell anemia are prone to infections, which often start with a fever and can be life-threatening, seek prompt medical attention for a fever greater than 101.5 °F (38.5 °C).
Sickle cell anemia is caused by genetic changes (mutations) in the HBB gene and is inherited in an autosomal recessive pattern. For a baby to be born with sickle cell anemia, both parents must carry a sickle cell gene. In the United States, sickle cell anemia most commonly affects people of African, Mediterranean and Middle Eastern descent.
There’s no cure for most people with sickle cell anemia. A stem cell transplant is the only known cure for sickle cell anemia, but it is not for everyone. Most patients who have sickle cell disease either are too old for a transplant or do not have a relative who is a good enough genetic match to be a donor. A well-matched donor is needed for a patient to have the best chance for a successful transplant. Treatments for sickle cell include antibiotics, pain management and blood transfusions. A new drug treatment, hydroxyurea, which is an anti-tumor drug, appears to stimulate the production of fetal hemoglobin F (α2γ2), a type of hemoglobin usually found only in newborns. Fetal hemoglobin (α2γ2) helps prevent the “sickling” of red blood cells. Patients treated with hydroxyurea also have fewer attacks of acute chest syndrome and need fewer blood transfusions.
Causes of sickle cell anemia
Sickle cell anemia is caused by genetic changes (mutations) in the HBB gene and is inherited in an autosomal recessive pattern. The HBB gene provides instructions for making a protein called beta-globin. Beta-globin is a component (subunit) of a larger protein called hemoglobin (Hb), which is located inside red blood cells. In adults, hemoglobin normally consists of four protein subunits (also called hemoglobin A [α2β2]): two subunits of beta (β) globin chains which is produced by HBB gene (hemoglobin beta gene) and two subunits of a protein called alpha (α) globin chains, which is produced from another gene called hemoglobin alpha (HBA1 or HBA2). Each of these protein subunits is attached (bound) to an iron-containing molecule called heme; each heme contains an iron molecule in its center that can bind to one oxygen molecule. Hemoglobin within red blood cells binds to oxygen molecules in the lungs. These cells then travel through the bloodstream and deliver oxygen to tissues throughout the body.
Mutation in the HBB gene (hemoglobin beta gene) results in the production of an abnormal version of beta (β) globin called hemoglobin S or HbS. In sickle cell anemia, hemoglobin S replaces both beta-globin subunits in hemoglobin. The mutation that causes hemoglobin S changes a single protein building block (amino acid) in beta-globin. Specifically, the amino acid glutamic acid is replaced with the amino acid valine at position 6 in beta-globin, written as Glu6Val or E6V. Replacing glutamic acid with valine causes the abnormal hemoglobin S subunits to stick together and form long, rigid molecules that bend red blood cells into a sickle (crescent) shape. The sickle-shaped cells die prematurely, which can lead to a shortage of red blood cells (anemia). The sickle-shaped cells are rigid and can block small blood vessels, causing severe pain and organ damage.
Autosomal means the gene is located on any chromosome except the X or Y chromosomes (sex chromosomes). Genes, like chromosomes, usually come in pairs. Recessive means that both copies of the responsible gene must have a disease-causing change (pathogenic variant) in order for a person to have the disease. Mutation is an older term that is still sometimes used to mean pathogenic variant. A person who has an autosomal recessive disease receives a gene with a pathogenic variant from each of their parents. Each parent is a carrier which means they have a pathogenic variant in only one copy of the gene. Carriers of an autosomal recessive disease usually do not have any symptoms of the disease. When two carriers of an autosomal recessive disease have children, there is a 25% (1 in 4) chance to have a child who has the disease.
For a baby to be born with sickle cell anemia, both parents must carry a sickle cell gene. In the United States, sickle cell anemia most commonly affects people of African, Mediterranean and Middle Eastern descent.
Sickle cell anemia signs and symptoms
Signs and symptoms of sickle cell anemia usually appear around 6 months of age. They vary from person to person and may change over time. Signs and symptoms can include:
- Anemia. Sickle cells break apart easily and die. Red blood cells usually live for about 120 days before they need to be replaced. But sickle cells typically die in 10 to 20 days, leaving a shortage of red blood cells (anemia). Without enough red blood cells, the body can’t get enough oxygen and this causes fatigue.
- Episodes of pain. Periodic episodes of extreme pain, called pain crises, are a major symptom of sickle cell anemia. Pain develops when sickle-shaped red blood cells block blood flow through tiny blood vessels to your chest, abdomen and joints. The pain varies in intensity and can last for a few hours to a few days. Some people have only a few pain crises a year. Others have a dozen or more a year. A severe pain crisis requires a hospital stay. Some adolescents and adults with sickle cell anemia also have chronic pain, which can result from bone and joint damage, ulcers, and other causes.
- Swelling of hands and feet. The swelling is caused by sickle-shaped red blood cells blocking blood circulation in the hands and feet.
- Frequent infections. Sickle cells can damage the spleen, increasing vulnerability to infections. Infants and children with sickle cell anemia commonly receive vaccinations and antibiotics to prevent potentially life-threatening infections, such as pneumonia.
- Delayed growth or puberty. Red blood cells provide the body with the oxygen and nutrients needed for growth. A shortage of healthy red blood cells can slow growth in infants and children and delay puberty in teenagers.
- Vision problems. Tiny blood vessels that supply the eyes can become plugged with sickle cells. This can damage the retina — the portion of the eye that processes visual images — and lead to vision problems.
Sickle cell anemia prevention
If you carry the sickle cell trait, seeing a genetic counselor before trying to conceive can help you understand your risk of having a child with sickle cell anemia. A genetic counselor can also explain possible treatments, preventive measures and reproductive options.
Sickle cell anemia complications
Sickle cell anemia can lead to a host of complications, including:
- Stroke. Sickle cells can block blood flow to an area of the brain. Signs of stroke include seizures, weakness or numbness of the arms and legs, sudden speech difficulties, and loss of consciousness. If your child has any of these signs and symptoms, seek medical treatment immediately. A stroke can be fatal.
- Acute chest syndrome. A lung infection or sickle cells blocking blood vessels in the lungs can cause this life-threatening complication, resulting in chest pain, fever and difficulty breathing. It might require emergency medical treatment.
- Pulmonary hypertension. People with sickle cell anemia can develop high blood pressure in their lungs. This complication usually affects adults. Shortness of breath and fatigue are common symptoms of this condition, which can be fatal.
- Organ damage. Sickle cells that block blood flow to organs deprive the affected organs of blood and oxygen. In sickle cell anemia, blood is also chronically low in oxygen. This lack of oxygen-rich blood can damage nerves and organs, including kidneys, liver and spleen, and can be fatal.
- Splenic sequestration. A large number of sickle cells can get trapped in the spleen, causing it to enlarge and possibly causing belly pain on the left side of the body. This can be life-threatening. Parents of children with sickle cell anemia should learn to regularly feel their child’s spleen for enlargement.
- Blindness. Sickle cells can block tiny blood vessels that supply the eyes. Over time, this can lead to blindness.
- Leg ulcers. Sickle cell anemia can cause painful open sores on the legs.
- Gallstones. The breakdown of red blood cells produces a substance called bilirubin. A high level of bilirubin in the body can lead to gallstones.
- Priapism. In this condition, men with sickle cell anemia can have painful, long-lasting erections. Sickle cells can block the blood vessels in the penis, which can lead to impotence over time.
- Deep vein thrombosis (DVT). Sickling of red cells can cause blood clots, increasing the risk of a clot lodging in a deep vein (deep vein thrombosis) or a lung (pulmonary embolism). Either can cause serious illness or even death.
- Pregnancy complications. Sickle cell anemia can increase the risk of high blood pressure and blood clots during pregnancy. It can also increase the risk of miscarriage, premature birth and having low birth weight babies.
Sickle cell anemia diagnosis
A blood test can check for the form of hemoglobin that underlies sickle cell anemia. In the United States, this blood test is part of routine newborn screening. But older children and adults can be tested, too.
In adults, a blood sample is drawn from a vein in the arm. In young children and babies, the blood sample is usually collected from a finger or heel. The sample is then sent to a laboratory, where it’s screened for the sickle cell form of hemoglobin.
If you or your child has sickle cell anemia, your doctor might suggest other tests to check for possible complications of the disease.
If you or your child carries the sickle cell gene, you’ll likely be referred to a genetic counselor.
Sickle cell anemia assessing stroke risk
A special ultrasound machine can reveal which children have a higher risk of stroke. This painless test, which uses sound waves to measure blood flow in the brain, can be used in children as young as 2 years. Regular blood transfusions can decrease stroke risk.
Tests to detect sickle cell genes before birth
Sickle cell disease can be diagnosed in an unborn baby by sampling some of the fluid surrounding the baby in the mother’s womb (amniotic fluid). If you or your partner has sickle cell anemia or the sickle cell trait, ask your doctor about this screening.
Sickle cell anemia treatment
Management of sickle cell anemia is usually aimed at avoiding pain episodes, relieving symptoms and preventing complications. Treatments might include medications and blood transfusions. For some children and teenagers, a stem cell transplant might cure the disease.
Sickle cell anemia medications
- Hydroxyurea (Droxia, Hydrea, Siklos). Daily hydroxyurea reduces the frequency of painful crises and might reduce the need for blood transfusions and hospitalizations. But it can increase the risk of infections. Don’t take the drug if you’re pregnant.
- L-glutamine oral powder (Endari). The FDA recently approved this drug for treatment of sickle cell anemia. It helps in reducing the frequency of pain crises.
- Crizanlizumab (Adakveo). This drug, given by injection, can help reduce the frequency of pain crises in adults and children older than 16. Side effects can include nausea, joint pain, back pain and fever.
- Voxelotor (Oxbryta). This drug is used to treat sickle cell disease in adults and children older than 12. Taken orally, this drug can lower the risk of anemia and improve blood flow throughout the body. Side effects can include headache, nausea, diarrhea, fatigue, rash and fever.
- Pain-relieving medications. Your doctor might prescribe narcotics to help relieve pain during sickle cell pain crises.
Sickle cell anemia infections prevention
Children with sickle cell anemia might receive penicillin between the ages of about 2 months old until at least age 5 years. Doing so helps prevent infections, such as pneumonia, which can be life-threatening to children with sickle cell anemia.
Adults who have sickle cell anemia might need to take penicillin throughout their lives if they’ve had pneumonia or surgery to remove the spleen.
Childhood vaccinations are important for preventing disease in all children. They’re even more important for children with sickle cell anemia because their infections can be severe.
Your child’s doctor should ensure that your child receives all the recommended childhood vaccinations, as well as vaccines against pneumonia, meningitis, hepatitis B and an annual flu shot. Vaccines are also important for adults with sickle cell anemia.
Sickle cell anemia home remedies
Taking the following steps to stay healthy might help you avoid complications of sickle cell anemia:
- Take folic acid supplements daily and choose a healthy diet. Bone marrow needs folic acid and other vitamins to make new red blood cells. Ask your doctor about a folic acid supplement and other vitamins. Eat a variety of colorful fruits and vegetables, as well as whole grains.
- Drink plenty of water. Dehydration can increase your risk of a sickle cell crisis. Drink water throughout your day, aiming for about eight glasses a day. Increase the amount of water you drink if you exercise or spend time in a hot, dry climate.
- Avoid temperature extremes. Exposure to extreme heat or cold can increase your risk of a sickle cell crisis.
- Exercise regularly, but don’t overdo it. Talk with your doctor about how much exercise is right for you.
- Use nonprescription medications with caution. Use pain medications, such as ibuprofen (Advil, Motrin IB, Children’s Motrin, others) or naproxen sodium (Aleve), sparingly, if at all, because of the possible effect on your kidneys. Ask your doctor before taking nonprescription drugs.
- Don’t smoke. Smoking increases your risk of pain crises.
Sickle cell anemia during surgical and other procedures
- Blood transfusions. These are used to treat and prevent complications, such as stroke, in people with sickle cell disease. In a red blood cell transfusion, red blood cells are removed from a supply of donated blood, then given through a vein to a person with sickle cell anemia. This increases the number of normal red blood cells, which helps reduce symptoms and complications. Risks include an immune response to the donor blood, which can make it hard to find future donors; infection; and excess iron buildup in your body. Because excess iron can damage your heart, liver and other organs, you might need treatment to reduce iron levels if you undergo regular transfusions.
- Stem cell transplant also known as bone marrow transplant, this procedure involves replacing bone marrow affected by sickle cell anemia with healthy bone marrow from a donor. The procedure usually uses a matched donor, such as a sibling, who doesn’t have sickle cell anemia. Because of the risks associated with a bone marrow transplant, including death, the procedure is recommended only for people, usually children, who have significant symptoms and complications of sickle cell anemia. A stem cell transplant is the only known cure for sickle cell anemia. Clinical trials are ongoing to address stem cell transplantation in adults and gene therapies.
Sickle cell with hemoglobin C disease (HbSC)
In sickle cell with hemoglobin C disease (HbSC), the child inherits a hemoglobin “S” (HbS) gene from one parent and a gene for a different type of abnormal hemoglobin called “C” (HbC) from the other parent. This is usually a milder form of sickle cell disease.
Sickle cell beta thalassemia
People who have sickle cell beta-thalassemia (HbSβ) inherit a hemoglobin “S” gene (HbS) from one parent and a gene for beta thalassemia, another type of hemoglobin abnormality, from the other parent. Sickle cell beta-thalassemia (HbSβ) is a rarer type of sickle cell disease that can present in 2 forms: “beta-zero” (HbS beta0) and “beta-plus” (HbS beta+). The first form is one in which there is a small amount of normal hemoglobin A (HbA) and abnormal sickle hemoglobin (HbS) present is called sickle cell beta plus thalassemia (HbSbeta+ or HbS β+thal). Many babies with sickle beta plus thalassemia (HbSβ+thal) are born healthy and do not show symptoms until later in childhood. Some problems can include low red blood cell count, pain, and risk of infection. The second form is the absence of normal hemoglobin called sickle cell beta-thalassemia zero (HbSB0 or HbS beta0), and these patients usually have a severe form of sickle cell disease and present similarly to those with sickle cell anemia (HbSS).
Rarer types of sickle cell disease
Other forms of sickle cell disease (compound heterozygotes), including HbSE, HbSO, and HbSD inherit one hemoglobin “S” gene (HbS) and one gene that codes for another abnormal type of hemoglobin (“D”, “E”, or “O”). The severity of these rarer types of sickle cell disease varies depending on the specific genetic defect.
Sickle cell trait
A person has sickle cell disease trait is also called sickle cell carrier when the hemoglobin S (Hb S) gene is inherited from only one parent, and a normal hemoglobin gene — hemoglobin A (Hb A) — is inherited from the other parent 6. If you have sickle cell trait, you are a carrier of the hemoglobin S (Hb S) gene, so you can pass it on when you have a child. If the child’s other parent also has sickle cell trait or another abnormal hemoglobin gene, such as beta-thalassemia, hemoglobin C, hemoglobin D, or hemoglobin E, that child has a chance of having sickle cell disease. People with sickle cell trait should be aware of their condition for family planning purposes because they can pass the gene onto their children. If both parents have sickle cell trait, there is a greater chance that one or more of their children will be born with sickle cell disease.
Sickle cell trait (HbAS) is not considered a disease, it is an inherited red blood cell condition and in most cases, people living with sickle cell trait are generally healthy and lead normal lives as it does not typically cause the multi-organ complications associated with sickle cell disease 34. However, following certain extreme triggers, individuals with sickle cell trait may experience medical problems, including an increased risk for prevalent and incident chronic renal disease, pulmonary embolism, and rhabdomyolysis 35. There have been reports of adverse conditions such as to anaesthetics that can cause problems due to the patient’s sickle cell trait status. If you have sickle cell trait always notify your dentist or doctor before treatment commences to be on the safe side. There is also a small chance that you may experience pain at high altitudes (generally above 10,000 feet), including long-haul flying in unpressurized planes and mountain climbing. It is important you say you have sickle cell trait before undertaking such activities as you may need to breathe oxygen. Extreme exercise may also precipitate problems and if you are a professional athlete you should have a training programme that takes account of this. Therefore, sickle cell trait may not be completely benign and these patients should be managed aggressively whenever they develop some of these complications 6. Because some persons with sickle cell trait have complications from the condition, research is needed to better understand when and how sickle cell trait might affect a person’s health.
Sickle cell trait has evolutionarily persisted throughout the world because of its strong protective effect against severe and cerebral malaria 36. In the United States, about 2.5 million to 3 million persons live with sickle cell trait, including an estimated 6% to 10% of the African American population and 0.01% to 0.07% of the remaining racial/ethnic groups, primarily those of Arabs, Southeast Asians, Hispanics, or Mediterranean descent 37. Worldwide, it is estimated that there are 300 million people with sickle cell trait and one-third of this number are in sub-Saharan Africa 8. The prevalence of sickle cell trait is higher in areas where malaria is endemic. Gibson and colleague mentions that the prevalence is as high as 25% in some part of Africa and 60% in Saudi Arabia 9. Because of the high migration of people from areas of high prevalence like Africa, Middle East, the prevalence of both sickle cell trait and disease will increase in the western part of the world.
Sickle cell trait does not cause often vaso-occlusive crisis, unlike that of sickle cell disease 6. However, patients with sickle cell trait could have the same presentation as sickle cell anemia if they are exposed to conditions that favor sickling. Conditions include severe hypoxia, dehydration, increase in sympathetic outflow, hypothermia/hyperthermia, high 2,3-DPG levels, and release of inflammatory cells. The HbS will result in the clogging of tiny capillary vessels most especially in the bones by sickled red blood cells. Apart from the sickling of the cells, other cells interact to cause more adhesion of the red blood cells including inflammatory cells, and platelets. This could occur in multiple organs in the body including the chest, heart, lungs, abdomen, kidneys, and extremities. Due to the repeated attacks, organ damage may happen due to constant ischemia.
Recent epidemiological studies have identified three primary areas that require further research to understand the clinical implications of sickle cell trait. The first is exercise-related complications, which include exertional rhabdomyolysis, heat-associated collapse, and sudden death. A retrospective review of 2.1 million military personnel from 1977 to 1981 found that 12 of 28 unexplained sudden deaths were in individuals with sickle cell trait, with a relative risk of death that was 39.8 times higher among recruits with sickle cell trait than among peers without sickle cell trait 38. A more recent retrospective review of 273 deaths in the National Collegiate Athletic Association from 2004 to 2008 found 13 deaths categorized as exertion related, 5 in athletes with sickle cell trait, with a relative risk of death of 29 39. All exercise-related deaths in individuals with sickle cell trait were associated with extreme exertion and intense exercise, and both studies failed to adjust for confounders. Thus, prospective well-designed cohort studies to better elucidate the true relative risk of exertional death in sickle cell trait are urgently needed.
Epidemiological studies have lent support to the notion that sickle cell trait may predispose one to chronic kidney disease (CKD). In a pooled analysis of 15,975 self-identified African Americans from five prospective population-based cohort studies—the Atherosclerosis Risk in Communities, Jackson Heart Study, Women’s Health Initiative, Multi-Ethnic Study of Atherosclerosis, and Coronary Artery Risk Development in Young Adults—239 of the 2,233 individuals with chronic kidney disease (CKD) were found to have sickle cell trait, with a pooled adjusted odds ratio of 1.57 for chronic kidney disease (CKD) with sickle cell trait compared with those without sickle cell trait 40.
Further studies are required to better establish the relationship between sickle cell trait and chronic kidney disease (CKD) and the effect of sickle cell trait on the development of diabetic, hypertensive, and other risk-variant renal disease.
Sickle cell trait causes
Sickle cell trait is a type of sickle cell disease in which the affected individual has only one abnormal hemoglobin S (Hb S) gene that is inherited from only one parent, and a normal hemoglobin gene — hemoglobin A (Hb A) — that is inherited from the other parent 6. If you have sickle cell trait, you are a carrier of the hemoglobin S (Hb S) gene, so you can pass it on when you have a child. If the child’s other parent also has sickle cell trait or another abnormal hemoglobin gene, such as beta-thalassemia, hemoglobin C, hemoglobin D, or hemoglobin E, that child has a chance of having sickle cell disease.
How sickle cell trait is inherited?
- If both parents have sickle cell trait, there is a 50% (or 1 in 2) chance that any child of theirs also will have sickle cell trait, if the child inherits the sickle cell gene from one of the parents. Such children will not have symptoms of sickle cell disease, but they can pass sickle cell trait on to their children.
- If both parents have sickle cell trait, there is a 25% (or 1 in 4) chance that any child of theirs will have sickle cell disease. There is the same 25% (or 1 in 4) chance that the child will not have sickle cell disease or sickle cell trait.
Sickle cell trait symptoms
Most people with sickle cell trait do not have any symptoms of sickle cell disease, although—in rare cases—people with sickle cell trait might experience complications of sickle cell disease, such as pain crises.
In their extreme form, and in rare cases, the following conditions could be harmful for people with sickle cell trait:
- Increased pressure in the atmosphere (which can be experienced, for example, while scuba diving).
- Low oxygen levels in the air (which can be experienced, for example, when mountain climbing, exercising extremely hard in military boot camp, or training for an athletic competition).
- Dehydration (for example, when one has too little water in the body).
- High altitudes (which can be experienced, for example, when flying, mountain climbing, or visiting a city at a high altitude).
Some people with sickle cell trait have been shown to be more likely than those without sickle cell trait to experience heat stroke and muscle breakdown when doing intense exercise, such as competitive sports or military training under unfavorable temperatures( very high or low) or conditions. Studies have shown that the chance of this problem can be reduced by avoiding dehydration and getting too hot during training. The National Collegiate Athletic Association (NCAA) has some great resources for athletes and their coaches with sickle cell trait (https://www.ncaa.org/sports/2016/7/27/sickle-cell-trait.aspx).
People with sickle cell trait who participate in competitive or team sports (i.e. student athletes) should be careful when doing training or conditioning activities. To prevent illness it is important to:
- Set your own pace and build your intensity slowly.
- Rest often in between repetitive sets and drills.
- Drink plenty of water before, during and after training and conditioning activities.
- Keep the body temperature cool when exercising in hot and humid temperatures by misting the body with water or going to an air conditioned area during breaks or rest periods.
- Immediately seek medical care when feeling ill.
More research is needed to find out why some people with sickle cell trait have complications and others do not.
Sickle cell trait complications
Sickle cell trait is associated with hematuria (blood in urine) due to renal papillary necrosis, splenic infarction, renal medullary carcinoma, chronic kidney disease (CKD), sudden death due to exertion, and asymptomatic bacteriuria in females 8.
Papillary necrosis is one of the complications that has been reported in several case studies. In the case study presented by Li EJ and Carroll VG 41, they mentioned that there are hematologic parameters that allow sickle cell trait patients to have this complication. Sickle cell trait patients with an average HbS level of 34% or higher are more likely to get papillary necrosis compared to those who have HbS of 20% 41. Necrosis is caused by the presence of sickling hemoglobin in small capillaries or vasa recta of the kidney which could cause microthrombi formation and then infarction. Patients with papillary necrosis usually present with gross hematuria and abdominal pain. The management is conservative including IV fluids, bed rest, and pain management. The prognosis is usually very good because only a single papillary is mostly affected and there is enough viable tissue.
The pathogenesis causing splenic infarction is similar to other complications. Like other complications, it occurs when the patient is exposed to low oxygen environment in high altitudes, dehydration, increased acidity, and viscosity 42. Unlike the other complications, it also occurs even when the patient is at rest at low altitudes. Several case reports of a young sickle cell trait patient presenting with multiple infarctions in the spleen have been reported.
Renal medullary carcinoma is also another complication associated with sickle cell trait. It is usually an aggressive tumor at the time of presentation with possible metastasis on diagnosis. A case report was published by Goenaga-Vasquez Y and his colleagues on a 9-year-old boy who presented with diffuse abdominal pain and was found to have renal medullary carcinoma with metastasis to the cervical, mediastinal and retroperitoneal lymph node 43.
Sickle cell trait has also been associated with increased chronic kidney disease in African American males. Studies have shown that the presence of sickle cell trait was associated with a decline in GFR, and the development of albuminuria compared to those without the trait 40. According to Niket and his colleagues 40, GFR decreased at a rate of 0.254 mL/min/1.73 m per year in sickle cell trait individuals compared to the noncarriers. The reason for this is chronic reversible sickling induced by hypoxia in the renal medullae, leading to constant ischemia and microinfarction of the renal tubules. Ischemia of the renal medulla and tubules causes the release of vasoactive elements. These elements contribute to hyperfiltration leading to sclerosis and proteinuria.
Sudden death due to exertion has been associated with athletes, police, and military recruits. According to Harmon KG and colleagues 39, there was a 37 times higher risk of exertional death in Division 1 football players with sickle cell trait in their database study. As a result of this complication found in athletes, a mandatory policy of the National Collegiate Athletic Association (NCAA) sickle cell screening program was proposed. Tarini BA et al 44 estimated that over 2000 athletes can be identified with this screening program. These identified individuals can be prevented from having a sudden death if proper intervention is made.
Studies have also shown that sickle cell is associated with exertional rhabdomyolysis. Rhabdomyolysis is the breakdown of skeletal muscle cells during physical exertion causing myoglobinuria. There is a 54% higher rate of rhabdomyolysis during physical exertion in the presence of sickle cell trait 45. It is said to be the cause of the sudden death of a 19 -year-old college athlete during intense football training. The death of this college freshman led to the screening policy implemented by the NCAA 34. The NCAA also has some great resources for athletes and their coaches with sickle cell trait (https://www.ncaa.org/sports/2016/7/27/sickle-cell-trait.aspx).
Sickle cell trait diagnosis
Sickle cell trait is diagnosed with a simple blood test. All babies born in the United States, regardless of ethnicity, are tested for sickle cell disease as a part of regular newborn screenings. Hemoglobin electrophoresis is a blood test that can determine if a person is a carrier of sickle cell, or has any of the diseases associated with the sickle cell gene.
People at greatest risk for sickle cell disease have ancestors from Africa, South or Central America, the Caribbean, Mediterranean countries, India, or Saudi Arabia. Boys and girls are affected equally. If your baby’s sickle cell disease screening comes back positive, medical professionals will do further testing to confirm the diagnosis.
A laboratory test, called hemoglobin electrophoresis, is used to identify the types of hemoglobin in the blood. This test will determine the specific type of sickle cell disease your baby has. Sometimes, testing parents and siblings is helpful for the diagnosis.
Sickle cell trait treatment
Sickle cell trait patients usually do not require any treatment 6. Treatment is only indicated if a patient presents with medical conditions including the ones that have been associated with the trait. It is important for clinicians to recognize the complications associated with sickle cell trait so that prompt management can be started once patients present with symptoms. For instance, if a sickle cell trait patient presents with hematuria, it is important to rule out papillary necrosis and these patients should get conservative management.
Sickle cell trait prognosis
Although sickle cell trait has been associated with many complications like papillary necrosis, asymptomatic bacteriuria, splenic infarction, and exercise-induced death, the prognosis of patients with sickle cell trait is promising. Tsaras and colleagues 22 mentioned in their article that despite the associated complications of sickle cell trait, the average life expectancy of people with sickle cell trait is the same as the general population.
Sickle cell disease causes
Sickle cell disease is inherited, meaning that it runs in families (a genetic condition you have at birth). Sickle cell disease is inherited in an autosomal recessive pattern, which means both copies of the hemoglobin beta (HBB) gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, also known as sickle cell carriers or having sickle cell trait. Sickle cell carriers (having sickle cell trait) do not have sickle cell disease themselves (do not show signs and symptoms of the condition), but there’s a chance they could have a child with sickle cell disease if their partner is also a sickle cell carrier (see Figure 6 below).
To be born with sickle cell disease, a child has to inherit a copy of the sickle cell gene (HBB gene) from both their parents. This usually happens when both parents are “carriers” of the sickle cell gene, also known as having the sickle cell trait.
As this sickle cell disease inheritance patterns flowchart illustrates, each parent has one hemoglobin A gene and one hemoglobin S gene, meaning each child of these parents has:
- a 25% chance of inheriting two normal genes (the child does not have sickle cell trait or disease)
- a 50% chance of inheriting one hemoglobin A gene and one hemoglobin S gene (the child has sickle cell trait)
- 25% percent chance of inheriting two hemoglobin S genes (the child has sickle cell disease)
Figure 6. Sickle cell anemia genetics
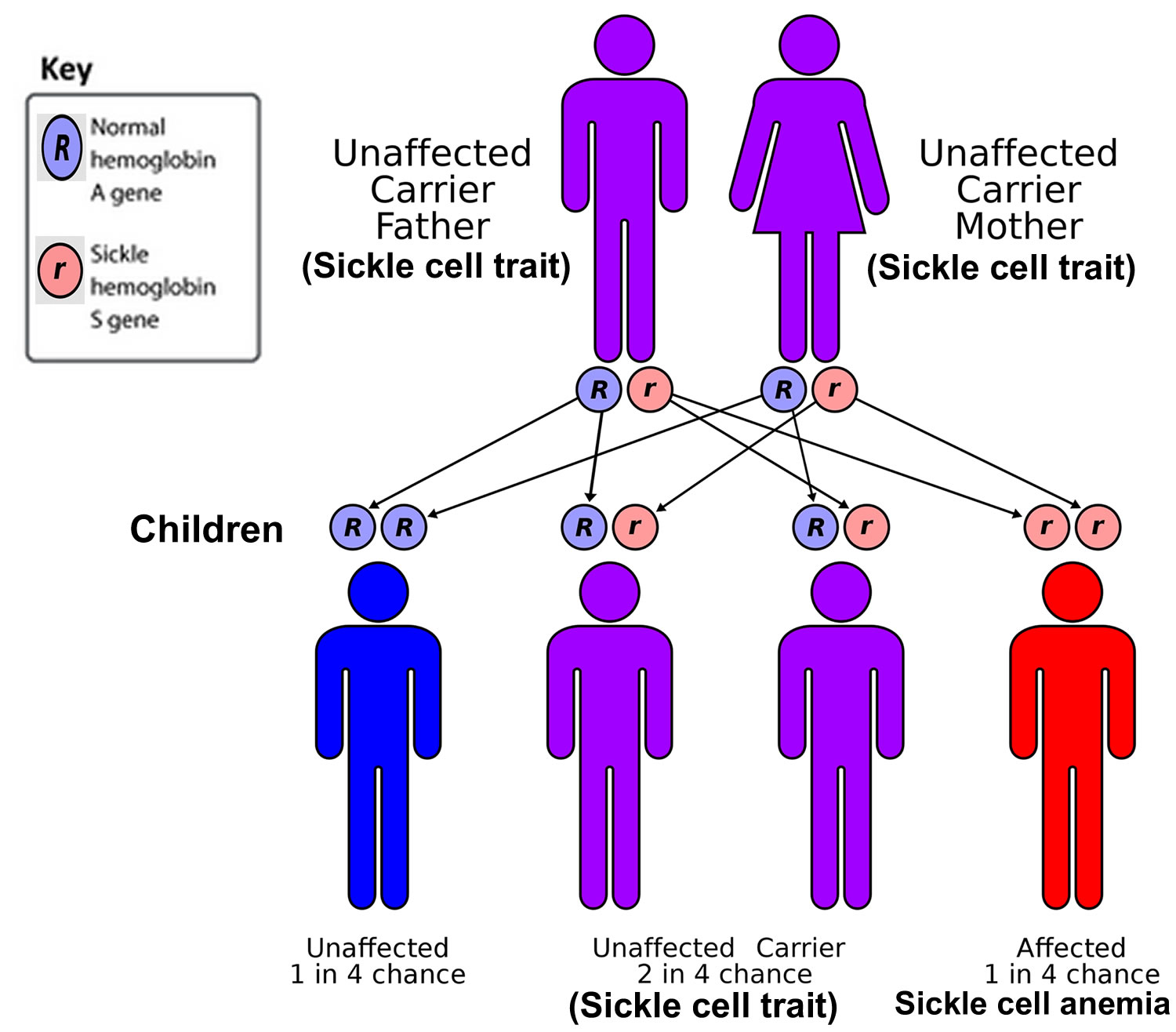
People who have sickle cell disease inherit two abnormal hemoglobin beta (HBB) genes, one from each parent. Sickle cell disease is caused by a point mutation in the hemoglobin-Beta gene (HBB) found on chromosome 11. The hemoglobin beta (HBB) gene provides instructions for making a protein called beta-globin. Beta-globin is a component (subunit) of a larger protein called hemoglobin, which is located inside red blood cells (see Figure 4 above). This point mutation replaces A with T at codon 6 of hemoglobin-Beta chain. This causes the switch from glutamic acid to valine amino acid at position 6 in beta-globin, written as Glu6Val or E6V. Replacing glutamic acid with valine causes the abnormal hemoglobin S (Hb S) subunits to stick together and form long, rigid molecules that bend red blood cells into a sickle (crescent) shape when exposed to a low oxygen level.
Mutations in the hemoglobin-Beta gene (HBB) gene can also cause other abnormalities in beta-globin (β globin), leading to other types of sickle cell disease. These abnormal forms of beta-globin are often designated by letters of the alphabet or sometimes by a name. In these other types of sickle cell disease, just one beta-globin subunit is replaced with hemoglobin S (Hb S). The other beta-globin subunit is replaced with a different abnormal variant, such as hemoglobin C (Hb C) or hemoglobin E (Hb E).
In hemoglobin SC (HbSC) disease, the beta-globin subunits are replaced by hemoglobin S and hemoglobin C (Hb C). Hemoglobin C (Hb C) results when the amino acid lysine replaces the amino acid glutamic acid at position 6 in beta-globin (written Glu6Lys or E6K) 46. The severity of hemoglobin SC (HbSC) disease is variable, but it can be as severe as sickle cell anemia (HbSS). Hemoglobin E (HbE) is caused when the amino acid glutamic acid is replaced with the amino acid lysine at position 26 in beta-globin (written Glu26Lys or E26K). In some cases, the hemoglobin E mutation is present with hemoglobin S. In these cases, a person may have more severe signs and symptoms associated with sickle cell anemia, such as episodes of pain, anemia, and abnormal spleen function.
Other conditions, known as hemoglobin sickle-beta thalassemias (HbSBetaThal), are caused when mutations that produce hemoglobin S (Hb S) and beta thalassemia occur together. Mutations that combine sickle cell disease with beta-zero (β0) thalassemia lead to severe disease, while sickle cell disease combined with beta-plus (β+) thalassemia is generally milder.
About 100,000 Americans have sickle cell disease. In the United States, most people who have sickle cell disease are of African ancestry or identify themselves as Black.
- About 1 in 13 Black or African American babies is born with sickle cell trait.
- About 1 in every 365 Black or African American babies is born with sickle cell disease.
There are also many people who have sickle cell disease who come from Hispanic, southern European, Middle Eastern, or Asian Indian backgrounds.
People who do not know whether they carry an abnormal hemoglobin S (Hb S) gene can ask their doctor to have their blood tested.
Couples who are planning to have children and know that they are at risk of having a child with sickle cell disease may want to meet with a genetic counselor. A genetic counselor can answer questions about the risk and explain the choices that are available.
Sickle cell disease genetics
Sickle cell anemia is caused by a mutation in a gene called hemoglobin beta (HBB), located on chromosome 11. The hemoglobin beta (HBB) gene provides instructions for making a protein called beta-globin. Beta-globin (β globin) is a component (subunit) of a larger protein called hemoglobin, which is located inside red blood cells (see Figure 4 above). Normal adult hemoglobin is called hemoglobin A (α2β2) and consists of four protein subunits: two subunits of alpha (α) globin chains, which is produced by hemoglobin alpha (HBA1 or HBA2) genes and two subunits of beta (β) globin chains, which is produced by HBB gene (hemoglobin beta gene). Each of these protein subunits is attached (bound) to an iron-containing molecule called heme; each heme contains an iron molecule in its center that can bind to one oxygen molecule. Hemoglobin within red blood cells binds to oxygen molecules in the lungs. These cells then travel through the bloodstream and deliver oxygen to tissues throughout the body (see Figure 4). Adult red blood cells normally contain the following hemoglobin chain combinations: hemoglobin A (α2β2) >95%; hemoglobin A2 (α2δ2) 2% to 3.4%; fetal hemoglobin F (α2γ2) <1%.
Genes come in pairs. You inherit 1 set from your mother and 1 set from your father. Sickle cell anemia (HbSS) is a recessive genetic disease, which means that both copies of the mutated gene (HbSS) must present for a person to have sickle cell anemia. If an individual has just one copy of the mutated gene (HbS) they are said to be a sickle cell carrier (HbAS) also known as having the sickle cell trait. If you’re a carrier of sickle cell or have sickle cell trait, it means you carry one of the hemoglobin S (Hb S) gene that causes sickle cell disease, but you do not have the condition yourself.
If both parents are sickle cell carriers (sickle cell trait), there’s a:
- 1 in 4 chance each child they have will not inherit any sickle cell genes and will not have sickle cell disease or be able to pass it on
- 1 in 2 chance each child they have will just inherit a copy of the sickle cell gene from 1 parent and be a carrier
- 1 in 4 chance each child they have will inherit copies of the sickle cell gene from both parents and will be born with sickle cell disease
If you carry the sickle cell trait (you’re a sickle cell carrier), you’re at risk of having children with sickle cell disease, although this can only happen if your partner is also a carrier or has sickle cell disease themselves. If you’re planning to have a child and you know you’re a you’re carrier, it’s a good idea for your partner to be tested.
If you and your partner both carry sickle cell, there’s a:
- 1 in 4 chance each child you have will not have sickle cell disease or be a carrier
- 1 in 2 chance each child you have will be a carrier, but will not have sickle cell disease
- 1 in 4 chance each child you have will be born with sickle cell disease
If both of you are you’re carriers and you’re planning to have a baby, talk to your doctor about getting a referral to a genetic counsellor, who can explain the risks to your children and what your options are.
These include:
- having tests during pregnancy to see if your baby will have sickle cell disease
- adopting a child
- trying in-vitro fertilization (IVF) with a donor egg or sperm
- trying pre-implantation genetic diagnosis (PGD)
Pre-implantation genetic diagnosis (PGD) is similar to IVF, but the resulting embryos are tested to check that they do not have sickle cell disease before they’re implanted in the womb.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Sickle cell disease prevention
Sickle cell disease can’t be prevented since it’s genetic. Early diagnosis and treatment can help prevent and manage symptoms of sickle cell disease.
Anyone can ask to have a free blood test to find out if they’re a sickle cell carrier at any point.
This can be useful if:
- you want to find out if you’re at risk of having a child with sickle cell disease
- you have a family history of sickle cell disease or carrying the sickle cell trait
- your partner carries the sickle cell trait
You can request the test from your doctor or nearest genetic counselor, who’ll discuss the result and implications with you if you’re found to carry sickle cell.
If you are a sickle cell carrier, other members of your biological family could be carriers too. Experts recommend you talk to your parents, brothers, sisters, uncles, aunts and cousins, and encourage them to get a test before they start a family or have any more children.
Talk to your doctor if you or your partner has sickle cell disease or if it runs in your family. If you’re trying to get pregnant, your doctor can tell you options for genetic counseling. If you have sickle cell disease and are pregnant, you should receive extra care to monitor your sickle cell disease and treat problems early. Your baby can be tested for sickle cell disease during pregnancy.
Stroke prevention
Transcranial doppler ultrasound (TCD) uses sound waves to create images of the blood flow inside the brain. Transcranial doppler ultrasound is a diagnostic test. It measures blood flow to and within the brain. Robust data exist for primary stroke prevention using transcranial doppler ultrasound (TCD) as a high-quality screening tool 47 and preventative chronic transfusions for persons identified as high risk 48. Analysis of data from The Stroke Prevention Trial in Sickle Cell Anemia also showed that participants with normal internal carotid artery or middle cerebral artery velocity had a higher risk of stroke (10 times greater) if they had an elevated anterior cerebral artery velocity compared with those with normal anterior cerebral artery velocity 49. Discontinuing chronic transfusion led to a resurgence of stroke risk and subsequent strokes within 1 year 50, so there remains a strong evidence-based recommendation of continuing transfusions to prevent stroke recurrence in children with sickle cell disease 51.
Lifestyle modifications
While intense, episodic exercise may pose risks to patients with sickle cell disease 52, research has demonstrated that regular, moderate exercise training can be beneficial and may contribute to overall wellness and improved quality of life. Data indicate that regular training reduces oxidative stress and thereby decreases the risks of developing chronic and acute complications 53. More and larger studies are needed to determine the best exercise training routines for providing functional benefits.
Another healthy lifestyle recommendation is to optimize water intake to maintain adequate hydration 54 because people living with sickle cell disease are more prone to dehydration. Individuals with sickle cell should drink enough fluids to avoid dehydration and decrease sickling of the red blood cells. The increased fluids will help the blood carry the red blood cells through blood vessels. The amount of fluid should be increased during hot weather, exercise or illness. Westcott et al. 55 found that only 31.8 percent of young adults with sickle cell disease were meeting fluid intake guidelines.
Optimizing nutritional intake is also paramount, although studies about the effects of specific micronutrients and macronutrients and dietary regimens in sickle cell disease are limited.
Cognitive interventions
Screening for specific cognitive deficits in individuals with sickle cell disease may help predict later academic outcomes and stroke risk 56 and may make it possible to deploy targeted cognitive interventions. Memory training programs are a non-pharmacological approach to improving academic outcomes. One study demonstrated that individuals with sickle cell disease who completed a working memory training program exhibited improved visual and working memory compared with non-completers 57. Additionally, a small cohort of children with sickle cell disease with cerebral infarcts who completed weekly combined tutoring and memory/learning strategies had improved memory and academic achievement compared with controls at 2 years of age 58.
Attention is another cognitive domain of focus in sickle cell disease. Although the current literature is limited, children with sickle cell disease in the United States have rates of attention deficit hyperactivity disorder (ADHD) prevalence that are between 19 and 40 percent 59, which are much higher than the general pediatric population (approximately 10 percent) 60. Thus, specific treatments to improve attention may also be beneficial in sickle cell disease.
Sickle cell disease symptoms
If a person has sickle cell disease, it is present at birth. The severity of the symptoms of sickle cell disease can vary greatly depending on the specific genetic type and even within those of the same type. Each child may experience symptoms differently, and symptoms can be very difficult to predict. Sickle cell disease is usually diagnosed at birth with standard newborn screening. Newborns have high levels of protective fetal hemoglobin (HbF), so babies that have sickle cell disease usually do not have any symptoms they are about 4 to 6 months of age.
Sickle cell disease can cause a wide range of symptoms and can change over time. These can start from a few months of age, although many children have few or no symptoms if treatment is started early on. Over time, you may experience symptoms depending on how sickle cell disease affects your health.
Sickle cell disease main symptoms are:
- painful episodes (sickle cell pain crises or vaso-occlusive crisis)
- getting infections often
- anemia
Sickle cell disease symptoms range from mild to severe, and environment and conditions influence your symptoms. Sickle cell disease can worsen in extreme conditions, such as:
- High altitude
- Dehydration
- Illness
- Stress
- Sleep apnea
- Menstruation
- Exposure to cold temperatures
- Intense exercise
Anemia
Nearly all people with sickle cell disease have anemia, where the hemoglobin in the blood is low. Hemoglobin is the substance found in red blood cells that’s used to transport oxygen around the body. Sickled red blood cells do not live as long as healthy red blood cells, and people with sickle cell disease have lower red blood cell counts than those without sickle cell disease, so there are not enough healthy cells to take oxygen to the tissues. Anemia does not usually cause many symptoms or it may delay normal growth and development and decrease energy and endurance. Sometimes anemia can get worse if you become infected with the virus that causes slapped cheek syndrome (parvovirus). This can lead to a sudden drop in the number of red blood cells and symptoms of severe anemia include extreme tiredness (fatigue), shortness of breath, dizziness and fainting, headaches, rapid heartbeat or irregular heartbeat.
Splenic sequestration crisis or an aplastic crisis can also cause severe anemia symptoms. These conditions can be life-threatening.
Anemia is usually treated with a blood transfusion.
Vaso-occlusive crisis or painful episodes
Episodes of pain known as sickle cell crises or vaso-occlusive crisis, are one of the most common and distressing symptoms of sickle cell disease. They happen when blood vessels to part of the body become blocked. The pain can be severe and lasts for up to 7 days on average.
A sickle cell crisis often affects a particular part of the body, such as the:
- hands or feet (particularly in young children)
- ribs and breastbone
- spine
- pelvis
- tummy
- legs and arms
Pain can occur anywhere but most often occurs in the bones of the arms, legs, chest, and spine.
Painful swelling of the small bones of the hands and feet, also known as dactylitis, occurs mostly in infants and toddlers with sickle cell disease. This condition occurs when blood flow is blocked in the small bones of the hands and feet.
Priapism (a persistent and painful erection of the penis) results from sickling that occurs in the penis. This results in a painful and unwanted erection. Priapism (a persistent and painful erection of the penis) if not promptly treated, it can result in impotence. If you experience an erection that lasts for 4 hours or more, go to the hospital to see a hematologist and urologist.
Any interruption in blood flow to the body can result in pain, swelling, dysfunction, and possible death of the surrounding tissue not receiving adequate blood and oxygen.
How often someone with sickle cell disease gets episodes of pain varies a lot. Some people may have one every few weeks, while others may have less than 1 a year. The average is 1 bad episode a year.
It’s not always clear what triggers bad pain, but sometimes painful episodes can be caused by the weather (such as wind, rain or cold), dehydration, stress or strenuous exercise.
Sickle cell crisis
Sickle cell crisis is a term used to describe several acute conditions such as the vaso-occlusive crisis (acute painful crisis), aplastic crisis, splenic sequestration crisis, hyperhemolytic crisis, hepatic crisis, dactylitis, and acute chest syndrome 61. However, the usage of the term ‘sickle cell crisis’ is more commonly associated with sudden pain affecting different parts of the body caused by sickled red blood cells forming clumps in the bloodstream (vaso-occlusive crisis). Other cells also may play a role in this clumping process. These clumps of cells block blood flow through the small blood vessels to your bones and organs. This can cause pain and organ damage. You might have pain in your back, knees, legs, arms, chest or stomach. The pain can be throbbing, sharp, dull or stabbing. How often and how bad the pain gets varies a lot from person to person and from crisis to crisis. Other acute complications include pneumonia, meningitis, sepsis and osteomyelitis, stroke, avascular necrosis, priapism, and venous thromboembolism 62.
The pain from sickle cell crisis can be acute (sudden) or chronic (long-lasting), but acute pain is more common. Acute pain comes suddenly and can range from mild to very severe. The pain usually lasts from hours to a few days. Chronic pain often lasts for weeks to months. Chronic pain can be hard to bear and mentally draining. This pain may severely limit daily activities, work and education.
Almost all people who have sickle cell disease have painful crises at some point in their lives. Some have these crises less than once a year. Others may have 15 or more pain crises in a year. Epidemiologic data indicate that 5.2 percent of patients with sickle cell disease have three to 10 episodes of severe pain every year 63. In most patients, a pain crisis resolves within five to seven days. A severe crisis may cause pain that persists for weeks to months 64.
The frequency, severity, location and duration of pain crises can vary considerably, even within a specific disease subtype 65. Patients with homozygous sickle cell and sickle cell–β°-thalassemia have a higher frequency of vaso-occlusive pain crises than patients with hemoglobin SC and sickle cell–β+-thalassemia genotype 66. Disease severity is thought to depend on a complex interaction of genetic, rheologic and hematologic factors, as well as microvascular and endothelial factors 67.
Severe blockages cause episodes of acute pain or ‘sickle cell crisis’, which may be triggered by a range of physical and psychological stresses, including but not limited to infection, pregnancy, surgery, anxiety, or depression. If not treated promptly, sickle cell crisis can result in internal organ and tissue damage, particularly to the lungs, kidneys, liver and bones. The frequent recurrence of sickle cell crisis can lead to chronic complications such as leg ulcers, blindness, and stroke 68. Acute chest syndrome or chest crisis, is a common and particularly dangerous complication that is currently the leading cause of death among sickle cell disease patients 69. Ambulatory care strategies such as nutritional counseling, folic acid supplementation, pain medication protocols, vaccinations and antibiotics for the prevention and treatment of infection, are essential to sickle cell disease management 70.
A sickle cell crisis pain can begin suddenly and last several hours to several days. You might be able to treat your pain crisis at home with medicines that you take by mouth. If these medicines don’t control your pain, you can’t keep fluids down or you know that you’re having severe pain, you might need to be treated in the emergency department. If your pain still isn’t controlled or you have other problems, you might need to be treated in the hospital.
Types of sickle cell crisis
Vaso-occlusive crisis
The vaso-occlusive crisis or sickle cell crisis, is the most common presentation of sickle cell disease. The vaso-occlusive crisis is initiated and sustained by interactions among sickle cells, endothelial cells and plasma constituents 71. Microvascular occlusion (the cardinal pathophysiologic cause of acute pain) is responsible for a wide variety of clinical complications of sickle cell disease, including pain syndromes, stroke, leg ulcers, spontaneous abortion and renal insufficiency. Reperfusion intensifies the inflammation and resultant pain. Patients complain of severe debilitating pain, which has variable intensity and frequency, in any part of the body but typically in the long bones, back, pelvis, chest and the abdomen. Symptoms may start as early as six months of age with pain and swelling in both hands and feet (dactylitis). In most instances, there are no reliable signs or tests to indicate the presence or absence of pain associated with vaso-occlusive crisis.
Most patients with sickle cell disease experience pain by the age of 6 years. Pain can begin from any part of the body but frequently affects the extremities and back and chest areas. Fever can accompany vaso-occlusive crisis in some patients. Although pain in patients with sickle cell disease is likely to be due to vaso-occlusive crisis, it is prudent to perform a thorough evaluation for other life-threatening causes that can be misattributed to sickle cell pain 72. There is no objective measure or lab test to determine the quality and severity of pain in sickle cell disease, and therefore, patient report is the only available guide.
Acute pain in patients with sickle cell disease is caused by ischemic tissue injury resulting from the occlusion of microvascular beds by sickled erythrocytes during an acute crisis. Chronic pain occurs because of the destruction of bones, joints and visceral organs as a result of recurrent crises. The effect of unpredictable recurrences of acute crises on chronic pain creates a unique pain syndrome 73.
Acute bone pain from microvascular occlusion is a common reason for emergency department visits and hospitalizations in patients with sickle cell disease 65. Obstruction of blood flow results in regional hypoxemia and acidosis, creating a recurrent pattern of further sickling, tissue injury and pain. The severe pain is believed to be caused by increased intra-medullary pressure, especially within the juxta-articular areas of long bones, secondary to an acute inflammatory response to vascular necrosis of the bone marrow by sickled eythrocytes 74. The pain may also occur because of involvement of the periosteum or periarticular soft tissue of the joints.
When a vaso-occlusive crisis lasts longer than seven days, it is important to search for other causes of bone pain, such as osteomyelitis, avascular necrosis and compression deformities 75. When a recurrent bone crisis lasts for weeks, an exchange transfusion may be required to abort the cycle 75.
The approach to pain control must include measures to treat acute pain crises, prevent future vaso-occlusive crises and manage the long-term sequelae of chronic pain that can result from multiple recurrent bony infarctions.
Splenic sequestration crisis
Patients with sickle cell disease have spleen infarction before the end of childhood. The spleen is affected due to its narrow vessels and its role as a key player in the lymphoreticular system. Splenic sequestration crisis causes acute, painful enlargement of the spleen due to intrasplenic trapping of red cells. Patients with splenic sequestration crisis may have a sudden drop in hemoglobin levels, and one should be vigilant about hypovolemic shock. If not treated promptly, this can be a life-threatening situation 72.
Aplastic crisis
Aplastic crisis presents with sudden pallor and weakness confirmed by rapidly dropping hemoglobin levels that are accompanied by reticulocytopenia. The usual trigger for aplastic crisis is parvovirus B19 that directly suppresses the bone marrow affecting red blood cell production, but it can also be caused by other viral infections. The shortened lifespan of red blood cell in sickle cell disease results in worsening of the patient’s baseline anemia, which can dip to dangerously low levels. The infection is self-limited, typically lasting 7 to 10 days 72.
Acute chest syndrome
Acute chest syndrome is defined as the appearance of a new pulmonary infiltrate on chest radiography accompanied by a fever and respiratory symptoms, including a cough, tachypnea, and chest pain 76. Acute chest syndrome is complication of sickle cell disease accounting for 25% of deaths and can follow vaso-occlusive crises. It is hypothesized that acute chest syndrome is the result of hypoxia and an inflammatory mediator-induced increase in adhesion of the pulmonary microvasculature to sickled red blood cells. This process is coupled with a reduction in nitric oxide (NO), which would normally counteract it. The most common symptoms in patients with acute chest syndrome are fever, cough, chest pain, dyspnea, and lung exam may show reduced air entry, rales and sometimes wheeze. acute chest syndrome can progress rapidly to hypoxemia and respiratory failure if not treated promptly. When possible to identify infectious organisms, chlamydia, Streptococcus pneumonia, and Mycoplasma predominate.
Acute chest syndrome could also occur as a result of fat embolism originating from the distal bone in vaso-occlusive crisis. The hypoxia leads to adhesion of sickled erythrocytes to pulmonary microvasculature, setting up local hypoxia in the lungs and causing sickling of more red blood cells; this sets up a vicious cycle. Any pulmonary infiltrate on chest radiography accompanied by abnormal lung findings should raise the suspicion of acute chest syndrome. Affected patients can rapidly progress to worsening respiratory failure and death if not aggressively treated and monitored 77.
Hemolytic crisis
An acute drop in hemoglobin level marks this crisis. It is common in patients with coexistent G6PD deficiency 72.
Others
Femoral/humeral head osteonecrosis due to vaso-occlusion along with increased pressure from increased erythrocyte marrow, priapism, proliferative retinopathy, and renal complications are often due to vaso-occlusion.
Acute chest syndrome
Acute chest syndrome is a life-threatening complication in people living with sickle cell disease that can result in lung injury, breathing difficulty, and low oxygen to the rest of the body. Acute chest syndrome occurs when sickle-shaped cells stick together and block the flow of oxygen in the vessels in the lungs. Acute chest syndrome can be life-threatening and is the leading cause of death in children and adults with sickle cell disease. Acute chest syndrome can be triggered by asthma crisis, infection (viral or bacterial), or pain (particularly in the chest) and can progress rapidly to respiratory failure. In children, acute chest syndrome is usually caused by an infection. Acute chest syndrome resembles pneumonia and includes fever and breathing symptoms such as cough or difficulty catching breath. Acute chest syndrome often occurs suddenly, when the body is under stress from infection, fever, or dehydration, and multiple episodes can cause permanent lung damage.
Acute chest syndrome is a medical emergency and should be treated in the hospital right away. Signs and symptoms are similar to pneumonia and can include:
- Chest pain
- Coughing
- Difficulty breathing
- Fever
Chest pain when breathing is the most common presenting complaint in adults. Fever, cough, tachypnea (abnormally rapid breathing), hypoxemia (an unusually low concentration of oxygen in the blood), or abdominal pain are common presentations for infants and children.
It is always best to rule out infection in these cases and obtain appropriate blood cultures and serologic studies. There may or may not be radiographic evidence (X-ray) of pulmonary infiltrates at the initial time of symptoms. Rib infarction, stomach ulcer, or gallbladder problems can also result in chest pain and should be checked as well.
Infections
People with sickle cell disease are more vulnerable to infections and sepsis, particularly when they’re young. Infections can range from mild, such as colds, to much more serious and potentially life threatening, such as meningitis.
In most children with sickle cell disease, by toddlerhood, the spleen becomes scarred and permanently damaged and no longer has full function. The spleen is important in the body’s defense against serious bacterial infections; therefore, children with sickle cell disease are at risk for life-threatening bacterial infections. Fever (more than 38.5⁰C or 101.5⁰F) is a symptom that must be evaluated immediately by a doctor to rule out a life-threatening bacterial infection and treated with antibiotics right away if required. Some people will need to be hospitalized.
Vaccinations and daily doses of antibiotics can help reduce the risk of many infections. Immunizations with conjugate vaccines against Streptococcus pneumoniae (pneumococcus) and Haemophilus influenzae type b (Hib) have also been critically important at significantly reducing the presence of viable bacteria in the circulating blood (bacteremia) in sickle cell disease 78; the introduction of pneumococcal vaccines led to a drop of the incidence of invasive pneumococcal disease by 90.8 percent in children less than 2 years old and 93.4 percent in children older than 5 years 79.
Splenic sequestration
Sickle cells can block the exit of blood from the spleen resulting in pooling of sickle-shaped cells in the spleen, causing a sudden worsening of the anemia. The spleen becomes enlarged and painful from the increase in trapped blood volume. It can be life threatening if not treated promptly. A severe episode of splenic sequestration requires surgical removal of the spleen.
Stroke
Stroke is a serious life-threatening medical emergency that happens when the blood supply to part of the brain is cut off. Stroke is a sudden and severe complication that can occur in children with sickle cell disease. Sickle-shaped cells can block the major blood vessels that supply the brain with oxygen. Interruption in the flow of blood and oxygen to the brain can result in devastating damage to the brain. Symptoms of a stroke can include weakness, particularly on one side of the body; slurred speech; seizure; confusion; dizziness or loss of coordination; or a severe headache.
The sooner a person receives treatment for a stroke, the less damage is likely to happen. If you suspect that you or someone else is having a stroke, phone your local emergency services number immediately and ask for an ambulance. Immediate treatment may save someone’s life and increase the chances for successful rehabilitation and recovery.
Having had one stroke, a child is much more likely to have more strokes and requires preventative therapy with chronic repeated transfusion for life.
Jaundice
Jaundice is when your skin or the whites of your eyes turn yellow. Jaundice is a common sign and symptom of sickle cell disease. Sickle red blood cells are destroyed prematurely in the spleen. The recycling of sickle hemoglobin from these cells produces increased levels of the yellow bilirubin protein that discolors your skin and eyes. Chronic high bilirubin levels can lead to gallstone formation.
Other problems
Sickle cell disease can also sometimes cause a wide range of other problems which may include:
- delayed growth during childhood and delayed puberty
- gallstones, which can cause tummy (abdominal) pain and yellow skin and eyes (jaundice)
- painful open sores on the lower legs (leg ulcers)
- strokes or transient ischemic attacks, where the flow of blood to the brain is blocked or interrupted
- a serious lung condition called acute chest syndrome, which can cause a fever, cough, chest pain and breathing difficulties. You will need to be admitted to the hospital, where you may receive antibiotics, oxygen therapy, or a blood transfusion.
- swelling of the spleen, which can cause shortness of breath, a rapid heartbeat, tummy pain, a swollen tummy and anemia
- eyesight problems, such as floaters, blurred or patchy vision, reduced night vision and occasionally sudden vision loss
- high blood pressure in the blood vessels that carry blood from the heart to the lungs (pulmonary hypertension)
- kidney or urinary problems, including blood in the urine and bedwetting
Sickle cell disease complications
Sickle cell disease affects multiple organs over the life span. Some complications may be systemwide (affecting many parts of your body at the same time). Other complications affect specific parts of the body. Sickle cell disease can lead to pain throughout the body and serious damage to organs such as the heart and kidneys. The earliest complications develop at the age of 6 months and coincide with the almost complete replacement of fetal hemoglobin with adult hemoglobin in red blood cells 80. Some of the most common complications in children are splenomegaly (an enlarged spleen), dactylitis (painful inflammation of fingers and toes), and jaundice. Some complications may appear in childhood and persist through adulthood, while others, especially those pertaining to organ failure, may manifest later in adulthood. Sickle cell disease complications are best understood when grouped according to whether they are acute or chronic and based on the systems they affect. General medical comorbidities can have a negative effect on sickle cell disease outcomes, with the reverse being also true. Very little data have been published on the impact of general medical comorbidities in sickle cell disease, particularly among adults.
Table 1 summarizes the complications of sickle cell disease by the affected organ system, describing the signs and symptoms experienced acutely and chronically. The table also highlights the comorbidities that often occur as a cause or consequence of these complications and must be considered in the overall management of sickle cell disease. The table is followed by a brief description of available evidence on some of the most common complications. It is important to be aware of the possible complications of sickle cell disease and know when to seek emergency care.
Table 1. Summary of sickle cell disease complications by affected organ or system (in alphabetical order)
| Organ System | Manifestations (Signs/Symptom Burden) | Comorbid Conditions | |
|---|---|---|---|
| Acute | Chronic | ||
| Cardiovascular | Sudden death Fatigue Dyspnea Syncope Relative systolic hypertension Myocardial infarction | Sickle cardiomyopathy Left ventricular hypertrophy Diastolic dysfunction Heart failure with preserved ejection fraction Iron-induced cardiomyopathy and dysrhythmias Endothelial dysfunction/autonomic dysfunction Prolonged QT interval Pulmonary hypertension | Cardiac iron toxicity Methadone related prolonged QT interval a Hyperlipidemia Obesity-related cardiovascular complications Venous thromboembolism |
| Central Nervous System | Headache Infarctive stroke Hemorrhagic stroke Ruptured aneurysms Moyamoya syndrome Silent cerebral infarcts Sino-venous thrombosis | Chronic headaches Neurocognitive disorders due to silent cerebral infarcts/overt cerebrovascular accidents or strokes and chronic anemia Poor executive functioning Memory deficits Increased cerebral blood flow Cerebral vasculopathy and Moyamoya syndrome Cerebral aneurysms | Posterior reversible encephalopathy syndrome Pre-/post-eclampsia Arnold Chiari malformation Cerebral aneurysms |
| Dental | Dental abscess Dental crown fracture Dental pulp fracture | Dental caries Gingivitis Cracked teeth Early dental loss Misaligned dentition | Cardiovascular risk Dental cavities, gingival disease, malocclusion |
| Endocrine | Pain around menses, pregnancy, and menopause | Growth hormone deficiency Hypogonadism Disturbances in cortisol levels Delayed puberty Premature menopause | Diabetes and thyroid disease from iron overload Early menopause and bone health Hypo/hyperthyroidism |
| Gallbladder/Pancreas | Cholelithiasis Cholecystitis Common bile duct obstruction Acute pancreatitis | Chronic gallbladder sludge Dyspepsia Chronic cholecystitis Chronic pancreatitis | Pancreatitis with comorbid alcohol misuse |
| General Gastrointestinal | Mesenteric infarcts | Chronic abdominal pain Constipation Irritable bowel syndrome gastroesophageal reflux disease Increased abdominal girth due to shortened trunk and barrel chest (sickle–habitus) | Constipation (opioid induced and vaso-occlusive episode related) Ileus Cyclic vomiting syndrome Drug-induced nausea and vomiting |
| Genitourinary | Priapism Enuresis Hematuria Menses-induced vaso-occlusive episode | Erectile (sexual) dysfunction Postcoital pain Enuresis/nocturia Hematuria | Menorrhagia leading to worsening anemia Dysmenorrhea with increased acute care use |
| Hematopoietic System (excluding spleen) | Acute anemia Aplastic crisis Sequestration crises Functional asplenia Indirect hyperbilirubinemia Scleral icterus Hemostatic activation b | Chronic hemolysis c Chronic anemia Extramedullary hematopoiesis Leukocytosis Thrombocytosis Splenomegaly Hypersplenism Conjunctival pallor Scleral icterus Hemostatic activation b Thrombophilia | Delayed hemolytic transfusion reactions Parvovirus B19 infection chronic kidney disease, suppressing erythropoiesis Hypoplastic anemia from chronic kidney disease |
| Hepatic | Hyperbilirubinemia Hepatic sequestration Hepatitis Acute intrahepatic cholelithiasis/cholestasis Transaminitis | Hepatomegaly Hepatic congestion/chronic congestive hepatopathy Portal hypertension | Hepatic hemosiderosis and fibrosis Infectious hepatitis Hepatorenal syndrome Autoimmune/chemical (drug-induced) hepatitis Gilberts Syndrome |
| Immune System | Bacteremia/sepsis Meningitis Hepatitis Osteomyelitis Pyelonephritis Influenza | Osteomyelitis Hepatitis Dental abscesses Gingivitis Leg ulcer super infection | Transfusion-associated infection—babesiosis, parvovirus, hepatitis, HIV Salmonella osteomyelitis Sexually transmitted infections |
| Musculoskeletal | Bony infarction Dactylitis Acute vaso-occlusive episode | Avascular necrosis Vertebral body endplate changes, including vertebral compression fractures Maxillary hyperplasia and bony changes associated with extramedullary hematopoiesis Gout Osteopenia/osteoporosis from increased bone turnover Vitamin D deficiency/rickets | Hypovitaminosis D Orbital bone infarction, mental nerve impingement (numb chin syndrome) Osteonecrosis or avascular necrosis of the jaw, particularly when exposed to bisphosphonates Increased risk of pathological fractures |
| Ophthalmic | Retinal detachment Retinal artery occlusion Vitreous hemorrhage Macular infarction | Sickle retinopathy (proliferative and nonproliferative) Maculopathy | Early cataracts Early glaucoma Increased intraocular pressure with posttraumatic hyphema |
| Pulmonary | Acute chest syndrome Pneumonia Pulmonary fat embolism syndrome Airway hyperreactivity Atelectasis from hypoventilation Pulmonary embolism | Chronic lung disease Chronic hypoxemia/hypoxia Nocturnal hypoxemia Chronic pulmonary embolism | In situ pulmonary thrombosis Asthma Adenotonsillar enlargement Obstructive sleep apnea Right middle lobe syndrome |
| Renal | Acute kidney injury (recurrent) Hematuria Papillary necrosis | Hypertension Glomerular hyperfiltration Proteinuria/microalbuminuria Hyposthenuria chronic kidney disease End-stage renal disease Renal tubular acidosis Renal osteodystrophy | Acute increase in blood pressure with acute pain Susceptibility to dehydration NSAID (non-steroidal anti-inflammatory drug) and contrast nephropathy |
| Reproductive | Spontaneous abortion/miscarriages Intrauterine growth retardation Early fetal demise Pre- and post-eclampsia Severe dilutional anemia Other maternal–fetal complications | Low sperm counts/poor sperm function Post-pregnancy chronic pain | Hypospermia from hydroxyurea use Infertility from conditioning regimens from sickle cell trait |
| Skin | Leg ulcers | Leg ulcers Varicosity | Melanonychia and hyperpigmentation from hydroxyurea use |
| Spleen | Acute splenic sequestration Acute splenic infarction Splenic abscesses Traumatic spleen rupture | Splenomegaly Functional asplenia or hyposplenia due to auto-infarction of spleen leading to increased risk for infection with encapsulated organisms Splenic infarction Hypersplenism | Risk of splenic rupture with contact sports in patients with splenomegaly Impact of early and/or prolonged hydroxyurea therapy b |
Footnotes:
a A prolonged QT interval is an electrical impulse that is measured by an electrocardiogram. The QT are the waves displayed on the paper results from the electrical impulses through the heart.
b Hemostatic activation refers to the hypercoagulable state that occurs downstream from the vaso-occlusive process in sickle cell disease (De Franceschi et al., 2011).
c Hydroxyurea, chronic transfusion therapy, and other disease-modifying therapies, when initiated early in life, may alter the natural history of sickle cell disease phenotype.
Complications affecting your whole body
- Acute pain crisis: Also known as sickle cell or vaso-occlusive crisis, this can happen without warning when sickle cells block blood flow. People describe this pain as sharp, intense, stabbing, or throbbing. Pain can strike almost anywhere in the body and in more than one spot at a time. Common areas affected by pain include the abdomen, chest, lower back, or arms and legs. A crisis can be brought on by high altitudes, dehydration, illness, stress, or temperature changes. Often a person does not know what triggers the crisis.,
- Chronic (long-term) pain: Chronic pain is common, but it can be hard to describe. It is usually different from crisis pain or the pain that results from organ damage.
- Delayed growth and puberty: Because of anemia, children who have sickle cell disease may grow and develop more slowly than their peers. They will reach full sexual maturity, but this may be delayed.
- Infections: The spleen is important for protection against certain kinds of infections. If you have sickle cell disease, a damaged spleen raises the risk for certain infections, including chlamydia, Haemophilus influenzae type B, salmonella, and staphylococcus.
- Joint problems: Sickling in the hip bones and, less commonly, the shoulder joints, knees, and ankles can lower oxygen flow and result in a condition called avascular or aseptic necrosis, which severely damages the joints. Symptoms include pain and problems with walking and joint movement. Over time, you may need pain medicines, surgery, or joint replacement.
- Pregnancy problems: Pregnancy can raise the risk of high blood pressure and blood clots in people who have sickle cell disease. The condition also increases the risk of miscarriage, premature birth, and low birth weight babies.
- Sudden death: Vaso-occlusive episode is the most common presentation associated with death in those with sickle cell disease 82. Death can be sudden and unexpected, often occurring at home following a recent discharge from the hospital (approximately 40 percent) or within 24 hours of presentation to the hospital (28 percent) 83. Infection is a leading cause of death (33–48 percent) 84. Other causes of death include overt organ failure, acute chest syndrome, and stroke 85. Evidence of bone marrow fat emboli is common in many autopsy cases of sudden death. In a large autopsy study, there was significantly more organ injury than recognized before death, so the clinical presentation often does not reflect the severity of hidden chronic end-organ damage 84.
Complications affecting specific parts of your body
- Acute chest syndrome: Sickling in blood vessels of the lungs can deprive lungs of oxygen. This can damage lung tissue and cause chest pain, fever, and difficulty breathing. Acute chest syndrome is a medical emergency.
- Aplastic crisis: Aplastic crisis occurs when the bone marrow stops making new red blood cells. An aplastic crisis is usually caused by a parvovirus B19 infection, also called fifth disease or slapped cheek syndrome. Parvovirus B19 is a very common infection, but in sickle cell disease, it can cause the bone marrow to stop producing new red cells for a while, leading to severe anemia. Severe anemia can be life-threatening.
- Enlarged spleen: The spleen is an organ that helps your body fight infection and remove unwanted material. It filters your blood and destroys old blood cells. In people who have sickle cell disease, red blood cells may get trapped in the spleen. This makes the spleen quickly grow larger than normal. With red blood cells trapped in the spleen, fewer are available to circulate in the blood, and this can lead to severe anemia. A large spleen may also cause pain in the left side of the belly. A parent can usually feel a spleen that is larger than normal in a child’s belly.
- Splenic sequestration crisis: Your is an organ that helps your body fight infection and remove unwanted material. It filters your blood and destroys old blood cells. In people who have sickle cell disease, red blood cells may get trapped in the spleen, making it quickly grow larger than normal. With red blood cells trapped in the spleen, fewer are available to circulate in the blood, and this can lead to severe anemia. A large spleen may also cause pain in the left side of the belly. A parent can usually feel a spleen that is larger than normal in a child’s belly.
- Eye problems: Sickle cell disease can injure blood vessels in the eye, most often in the retina. Blood vessels in the retina can overgrow, get blocked, or bleed. This can cause the retina to detach, which means it is lifted or pulled from its normal position. These problems can lead to vision loss.
- Gallstones: When red blood cells break down, in a process called hemolysis, they release hemoglobin. Hemoglobin then gets broken down into a substance called bilirubin. Bilirubin can form stones called gallstones that get stuck in the gallbladder. The gallbladder is a small sac-shaped organ beneath the liver that helps with digestion.
- Heart problems: These can include coronary heart disease and pulmonary hypertension. Frequent blood transfusions can lead to heart damage from iron overload.
- Kidney problems: Sickle cell disease may cause the kidneys to have trouble making the urine as concentrated as it should be. This may lead to a need to urinate often and to bedwetting or uncontrolled urination during the night. These problems often start in childhood.
- Leg ulcers: Sickle cell ulcers are sores that usually start small and then get larger and larger. Some ulcers will heal quickly, but others may not heal and may last for long periods of time. Some ulcers return even after healing. People who have sickle cell disease usually do not get ulcers until after age 10.
- Liver problems: Sickle cell intrahepatic cholestasis is an uncommon but severe type of liver damage that happens when sickled red cells block blood vessels in the liver. This blockage prevents adequate levels of oxygen from reaching liver tissue. These episodes are usually sudden and may happen more than once. Children often recover, but some adults may have chronic problems that lead to liver failure. Frequent blood transfusions can lead to liver damage from iron overload.
- Priapism: Priapism is an unwanted and sometimes prolonged painful erection. This happens when blood flow out of the erect penis is blocked by sickled cells. Over time, priapism can cause permanent damage to the penis and lead to impotence. Priapism that lasts for more than 4 hours is a medical emergency.
- Stroke or silent brain injury: Silent brain injury, also called silent stroke, is damage to the brain without outward signs of stroke. This injury is common and can be detected on magnetic resonance imaging (MRI) scans. Silent brain injury can lead to difficulty in learning, making decisions, or holding down a job.
Aplastic crisis and splenic sequestration crisis most commonly occur in newborns and children who have sickle cell disease. Adults who have sickle cell disease may also experience episodes of severe anemia, but these usually have other causes. Babies and newborns who have severe anemia may not want to eat and may seem very sluggish.
Sickle cell disease diagnosis
All babies born in the United States, regardless of ethnicity, are tested for sickle cell disease as a part of regular newborn screenings. Sickle cell disease is diagnosed with a simple blood test that checks for hemoglobin S (HbS) – the defective form of hemoglobin. To confirm the diagnosis, a sample of blood is examined under a microscope to check for large numbers of sickled red blood cells – the hallmark trait of the disease. Hemoglobin electrophoresis is a blood test that can determine if a person is a carrier of sickle cell, or has any of the diseases associated with the sickle cell gene.
In more than 40 states, testing for the defective sickle cell gene is routinely performed on newborns. It often is included in routine newborn screening tests at birth in the hospital. It’s important to test babies for sickle cell disease. Early diagnosis and treatment can help prevent and manage symptoms of sickle cell disease.
Talk to your doctor if you or your partner has sickle cell disease or if it runs in your family. If you’re trying to get pregnant, your doctor can tell you options for genetic counseling. If you have sickle cell disease and are pregnant, you should receive extra care to monitor your sickle cell disease and treat problems early. Your baby can be tested for sickle cell disease during pregnancy.
Newborn screening
When a child has sickle cell disease, early diagnosis is important to better prevent complications. Every state in the United States, the District of Columbia, and the U.S. territories require that every baby be tested for sickle cell disease as part of a newborn screening program. In newborn screening programs, blood from a heel prick is collected in “spots” on a special paper. The hemoglobin from this blood is then analyzed in special labs. Newborn screening results are sent to the doctor who ordered the test and to the child’s primary doctor.
If a baby is found to have sickle cell disease, health providers from a special follow-up newborn screening group contact the family directly to make sure that the parents know the results. The child is always retested to be sure that the diagnosis is correct.
Newborn screening programs also find out whether the baby has an abnormal hemoglobin trait. If so, the parents are informed, and counseling is offered. Remember that when a child has sickle cell trait or sickle cell disease, a future sibling or the child’s own future child may be at risk.
Prenatal screening
Doctors can also diagnose sickle cell disease before a baby is born. This is done using a sample of amniotic fluid, the liquid in the sac surrounding a growing embryo, or of tissue taken from the placenta, the organ that attaches the umbilical cord to the mother’s womb.
Testing before birth can be done as early as 8 to 10 weeks into the pregnancy. This testing looks for the sickle hemoglobin gene rather than the abnormal hemoglobin.
Sickle cell carriers testing
Screening for sickle cell disease is offered to all pregnant women, although most women will be at low risk and will not need to have a blood test to check if they’re a carrier.
A new technique used in conjunction with in-vitro fertilization (IVF), called pre-implantation genetic diagnosis (PGD), enables parents who carry the sickle cell trait to test embryos for the defective gene before implantation, and to choose to implant only those embryos free of the sickle cell gene.
Sickle cell disease treatment
Sickle cell disease usually requires lifelong treatment. Children and adults with sickle cell disease are supported by a team of different healthcare professionals working together at a specialist sickle cell center. Babies who have sickle cell disease may see a hematologist, a doctor who specializes in blood diseases such as sickle cell disease. For newborns, the first sickle cell disease visit should take place before 8 weeks of age.
A blood and bone marrow transplant is currently the only cure for some patients who have sickle cell disease 86. After early diagnosis, your doctor may recommend medicines or blood transfusions to manage complications, including chronic pain.
Medicine to prevent the sickling of red blood cells
The U.S. Food and Drug Administration (FDA) approved Voxelotor (Oxbryta) in 2019 to treat sickle cell disease in adults and children 12 years and older 87. The oral medicine prevents red blood cells from forming the sickle shape and binding together. This may decrease the destruction of some red blood cells, which in turn lowers the risk for anemia and improves blood flow to your organs.
By increasing the oxygen affinity of hemoglobin, voxelotor maintains hemoglobin in the oxygenated state and reduces polymerization and sickling 87. Clinical trials in adults and children have shown reduced hemolysis and improved hemoglobin, although these changes were not accompanied by a statistically significant reduction in vaso occlusive crisis 87. While voxelotor does not appear to adversely affect oxygen delivery, according to measurements of erythropoietin levels in study patients, more research may be needed in children at a high risk of stroke, in whom cerebral oxygenation partly relies on increased oxygen extraction. There are also lingering concerns that increasing the total hemoglobin without reducing the intracellular concentration of hemoglobin S (HbS) may lead to hyperviscosity and its attendant complications 88.
Possible side effects include headache, diarrhea, abdominal pain, nausea, fatigue, and fever. Rarely, allergic reactions may occur, causing rashes, hives, or mild shortness of breath. Talk to your doctor about other medicines you take.
Medicine to reduce vaso-occlusive and pain crises
In 2019, the FDA approved crizanlizumab-tmca (ADAKVEO®) to reduce the number of pain crises experienced by adults and children 16 years and older who have sickle cell disease 89. Crizanlizumab-tmca (ADAKVEO®) is a monoclonal antibody that targets P-selectins, has been tested in humans and found to prevent vaso occlusive crises, as predicted by the evidence in mouse models. High-dose crizanlizumab reduced the rate of vaso-occlusive episodes to a median of 1.63/year compared with 2.98/year with placebo (a 45.3 percent lower rate) 90. In addition, “the median time to the first crisis was significantly longer with high-dose crizanlizumab than with placebo (4.07 versus 1.38 months), as was the median time to the second crisis (10.32 versus 5.09 months)” 91. The medicine, which is given through an IV in the vein, helps prevent blood cells from sticking to blood vessel walls and causing blood flow blockage, inflammation, and pain crises.
Possible side effects include nausea, joint pain, back pain, and fever.
Hydroxyurea
Hydroxyurea (Hydrea) is an oral medicine that has been shown to reduce or prevent several sickle cell disease complications 92, 93, 94.
- Use in adults: Many studies of adults with hemoglobin SS (also known as sickle cell anemia, which is the most common and most severe type of sickle cell disease) or hemoglobin Sβ thalassemia (Sickle Beta Thalassemia) showed that hydroxyurea reduced the number of episodes of pain crises and acute chest syndrome. It also improved anemia and decreased the need for transfusions and hospital admissions.
- Use in children: Studies in children with severe hemoglobin SS (sickle cell anemia) or Sβ thalassemia (Sickle Beta Thalassemia) showed that hydroxyurea reduced the number of vaso-occlusive crises and hospitalizations. A study of children between the ages of 9 and 18 months with hemoglobin SS or Sβ thalassemia also showed that hydroxyurea reduced the number of pain episodes and dactylitis. There is no information about how safe or effective hydroxyurea is in children under 9 months of age
- Pregnancy: Pregnant people should not use hydroxyurea.
Since hydroxyurea can decrease several complications of sickle cell disease, most experts recommend that children and adults with hemoglobin SS or Sβ0 thalassemia (Sickle Beta Zero Thalassemia) who have frequent painful episodes, recurrent chest crises, or severe anemia take hydroxyurea daily.
Hydroxyurea is initiated in a dosage of 500 mg per day. The dosage is increased to 1,000 mg per day after six to eight weeks, with the patient monitored for a decline in granulocyte or platelet counts. The maintenance dosage is between 1,000 and 2,000 mg per day, depending on the balance between hematologic toxicity and increases in hemoglobin F values 71. Blood counts should be followed every four to six weeks to detect longer term hematologic toxicities.
Possible side effects include decreased white cell count or platelet count. Rarely, it can worsen anemia. These side effects usually go away quickly if a patient stops taking the medicine. When the patient restarts it, the doctor usually prescribes a lower dose.
It is still unclear whether hydroxyurea can cause problems later in life in people who have sickle cell disease and take the medicine for many years. Studies so far suggest that it does not put people at a higher risk of cancer and does not affect growth in children, but further studies are needed.
Medicine to treat pain
The FDA approved L-glutamine (Endari) for people age 5 and older to lower the number of pain crises. L-glutamine is a natural amino acid that your body makes. L-glutamine is also found in meat or plants such as red cabbage, beans, and raw spinach. Research showed that patients taking L-glutamine (Endari) had fewer hospital admissions than patients taking a placebo 95. L-glutamine medicine is prescribed as a powder that is mixed into liquids or foods like applesauce or cereal 96. L-glutamine can be used in addition to hydroxyurea.
The way L-glutamine works is not yet fully understood. L-glutamine helps protect red blood cells by supporting the NAD (nicotinamide adenine dinucleotide) system. The NAD (nicotinamide adenine dinucleotide) system can protect your red blood cells against this destruction. L-glutamine may also make RBCs less sticky, which could reduce the number of painful crises.
Possible side effects of L-glutamine (Endari) medicine include nausea, fatigue, chest pain, and musculoskeletal pain 95. L-glutamine has not yet been tested in older adults. L-glutamine should not be used in people who have problems with their liver or kidney function.
Over-the-counter pain medicine, such as acetaminophen or ibuprofen, can be used to treat mild to moderate pain. Stronger or more serious pain may need to be treated in a clinic or hospital. Your provider may prescribe stronger medicines called opioids for severe pain.
How do you take L-glutamine?
L-glutamine is a powder. It must be mixed well in 8 ounces of a cold beverage, yogurt, or applesauce. It is taken twice a day and the number of packets depends on your weight:
- A person less than 66 pounds needs to take 1 packet twice a day.
- A person 66 to 123 pounds needs to take 2 packets twice a day.
- A person over 143 pounds needs to take 3 packets twice a day.
Medicine to reduce risk of infections
Individuals with sickle cell disease are at higher risk of infection. The spleen is a vital organ which helps fight infection. Due to repeated vaso-occlusion in the tissue of the spleen, this organ either does not work correctly or stops working (functional asplenia). In children who have sickle cell disease, taking penicillin two times a day has been shown to reduce the chance of having a severe infection in the bloodstream 97. Newborns need to take liquid penicillin. Older children can take tablets.
The usual dose of oral penicillin is 125 mg twice daily until age three years, then 250 mg twice daily. For penicillin-allergic patients, use erythromycin 250 mg twice daily. Amoxicillin is not a good choice as it has not been studied to see effectiveness at preventing infection in sickle cell disease; it causes more side effects and puts patients at increased risk for development of resistant organisms.
Many doctors will stop prescribing penicillin after a child has reached the age of 5. A 1995 study of 400 patients with sickle cell disease evaluated discontinuing penicillin therapy in children over 5 years old who had taken penicillin for at least 2 years and also received pneumococcal 23-valent vaccination 98. The equivalency of infection rates on and off penicillin between the study groups led to the recommendations that some people with sickle cell disease may be able to discontinue penicillin therapy safely after age 5, while continuing to be monitored for infection 98.
However, some doctors prefer to continue penicillin antibiotic throughout life, particularly if a person has sickle cell anemia (hemoglobin SS) or hemoglobin Sβ0 thalassemia (Sickle Beta Zero Thalassemia), since people who have sickle cell disease are still at risk. All people who have had surgical removal of the spleen, called a splenectomy, or a past infection with pneumococcus should keep taking penicillin throughout life.
Immunizations
Immunizations are an essential way to prevent infection. Individuals with sickle cell disease are at increased risk for infectious organisms, such as pneumococcus, meningococcus, and Haemophilus influenza. The Advisory Committee on Immunization Practices (ACIP) recommends all infants with sickle cell disease should receive the complete series of the 13-valent conjugate pneumococcal vaccine series beginning shortly after birth (children aged 6 to 18 years with functional or anatomic asplenia should receive one dose of Pneumococcal (PCV13) vaccine) and the 23-valent pneumococcal polysaccharide vaccine (PPSV23) at age 2 years, with a second dose at age 5 years 99. Your healthcare provider will discuss the vaccines needed at every life stage.
Blood transfusions
Your doctor may recommend blood transfusion to treat and prevent certain sickle cell disease complications.
These transfusions may include:
- Acute transfusions treat complications that cause severe anemia. Doctors may also use transfusions when a patient has an acute stroke, in many cases of acute chest crises, and in multi-organ failure. A patient who has sickle cell disease usually receives blood transfusions before surgery, to prevent complications.
- Red blood cell transfusions increase the number of red blood cells and provide normal red blood cells that are more flexible than red blood cells with sickle hemoglobin.
- Regular or ongoing blood transfusions may help lower the chances of another stroke in people who have had an acute stroke.
Doctors also recommend blood transfusions for children who have abnormal transcranial Doppler ultrasound results, because transfusions can reduce the chance of having a first stroke. Some doctors use this approach to treat complications that do not improve with hydroxyurea. Doctors may also use transfusions in people who have too many side effects from hydroxyurea. Possible complications of blood transfusions include alloimmunization, which can make it hard to find a matching unit of blood for a future transfusion; infection; and iron overload.
Blood and bone marrow transplant
Red blood cells are made in the bone marrow. A blood and bone marrow transplant is currently the only cure for sickle cell disease, but it is not for everyone. Most patients who have sickle cell disease either are too old for a transplant or do not have a relative who is a good enough genetic match to be a donor. A well-matched donor is needed for a patient to have the best chance for a successful transplant.
Most sickle cell disease transplants are currently performed in children who have had complications such as strokes, acute chest crises, and recurring pain crises. These transplants usually use a matched donor. Blood and bone marrow transplants are riskier in adults.
Several medical centers are looking into new ways to help more people who have sickle cell disease get a transplant. These include blood and bone marrow transplant techniques in children and adults who do not have a matched donor in the family or who are older than most recipients.
Blood and bone marrow transplants are successful in about 85% of children when the donor is related and HLA (human leukocyte antigen)-matched. Even with this high success rate, transplants still have risks. Complications can include severe infections, seizures, and other clinical problems. About 5% of people who have received such transplants have died. Sometimes transplanted cells attack the recipient’s organs. This is called graft-versus-host disease. You will get medicine to prevent many of the complications, but they still can happen.
Medicine to increase fetal hemoglobin (HbF) production
High intracellular fetal hemoglobin (HbF) levels prevent or reduce hemoglobin S (HbS) polymerization, as evidenced by individuals with congenital hereditary persistence of HbF co-inherited with sickle cell disease, although the protection from sickling is usually not complete.
Drugs, such as decitabine 100, histone deacetylase inhibitors 101, sodium 2,2-dimethylbutyrate 102 and metformin 103, have been found to boost fetal hemoglobin (HbF) levels and are under investigation for sickle cell disease.
Sickle cell disease gene therapy
Researchers are also exploring genetic therapies. Genetic therapies aim to repair a faulty gene or add a missing or new gene. These may help lead to new treatments or help cure sickle cell disease.
Researchers are experimenting with attempts to cure sickle cell disease by correcting the defective gene and inserting it into the bone marrow of those with sickle cell to stimulate production of normal hemoglobin.
Researchers used bioengineering to create mice with a human gene that produces the defective hemoglobin causing sickle cell disease. Bone marrow containing the defective hemoglobin gene is then removed from the mice and genetically “corrected” by the addition of the anti-sickling human beta-hemoglobin gene. The corrected bone marrow is then transplanted into other mice with sickle cell disease. The genetically corrected mice began producing high levels of normal red blood cells and showed a dramatic reduction in sickle cells. Scientists are hopeful that these techniques can be applied to human gene transplantation using autologous transplantation, in which some of the patient’s own bone marrow cells would be removed and genetically corrected.
Sickle cell crisis treatment
Acute sickle cell crises are managed primarily with drug therapy. Psychologic supportive care is also important. The standard treatment approach for sickle cell crisis includes opioid analgesics, adequate hydration, rest, and cognitive and behavioral therapies 65. The management of acute pain in sickle cell crises is summarized in Table 2. In addition, the American Pain Society recently released a comprehensive guideline on pain management in sickle cell disease 104.
Optimal management requires a multidisciplinary team that includes a family physician, a hematologist, nurses, a psychiatrist, a physical therapist, a pain specialist and social workers. These team members work together to provide empathetic, consistent, longitudinal care in a trustworthy environment 65. Discussions of coping mechanisms, reassurance about pain management and the presence of a cohesive family unit are all important in preventing psychologic instability and the development of a chronic pain syndrome.
Therapeutic red cell exchange transfusion is advised as an adjuvant, for the management of sickle cell crisis, and it is mainly practiced in the pediatric population. Red blood cell transfusion is recommended as the first line of management, for sickle cell disease to keep the HbS levels below 30% 105.
Table 2. Treatment Principles for Acute Pain Management in Patients with Sickle Cell Crises
| General principles |
| If possible, identify and treat underlying precipitating factors. |
| If needed, administer fluids orally, or intravenously as 5 percent dextrose in water or in a 25 percent normal saline solution. |
| Use oxygen therapy only if hypoxemia is present. |
| Acute pain management |
| Avoid delays in administering analgesia. |
| Administer an opioid analgesic parenterally (preferably intravenously) on a regular basis in a full therapeutic dosage or by patient-controlled analgesia. Avoid “as-needed” dosing. |
| Reassess the patient every 30 minutes for pain severity, sedation, vital signs and respiratory rate. |
| Use pain measurement scales as an objective guide to titrate the maintenance dosage of an analgesic and to determine treatment effects. |
| For breakthrough pain, administer one fourth to one half of the maintenance dosage, depending on the degree of sedation. |
| If three or more rescue doses are needed within a 24-hour period, increase the maintenance dosage by 25 to 50 percent, and repeat the same steps until analgesia is achieved. |
| Pain management after an acute crisis |
| Begin tapering the parenterally administered analgesic when the pain severity score is less than 5 on the visual analog scale or verbal pain scale and the patient’s mood improves. Reduce the maintenance dosage by 25 percent every 24 hours, and replace the parenterally administered drug with an equianalgesic oral agent given in divided doses. |
| Consider hospital discharge when the patient’s pain is controlled with an orally administered analgesic or no analgesia is needed. |
| If the patient still has pain at the time of hospital discharge, provide a prescription for a sufficient quantify of analgesic drug to treat resolving or relapsing pain until the patient’s next office appointment. |
Pain management
Pain from a vaso-occlusive crisis is often undertreated because of concerns about narcotic addiction and tolerance, perceived drug-seeking behavior, excessive sedation, respiratory depression and lack of specific findings on the physical examination 65.
Physicians often fail to prescribe narcotics appropriately and tend to overestimate opioid dependence in patients with pain crises. Yet the incidence of opioid analgesic addiction in patients with sickle cell disease has been reported to be no higher than 3 percent 106.
Many drug regimens have been effective in the treatment of acute pain in sickle cell disease. Pain management should follow the three-step “analgesic ladder” recommended by the World Health Organization for the treatment of cancer-related pain 107. The choice of analgesic and the dosage used should be based on the severity of pain in the individual patient.
Patients with mild pain can often be treated at home with oral fluids and nonnarcotic analgesics (Table 3). Patients can also be started on acetaminophen with or without codeine or oxycodone (Roxicodone), depending on pain severity. Nonsteroidal anti-inflammatory drugs can be used unless they are specifically contraindicated because of peptic ulcer disease, renal disease or hepatic dysfunction.
Table 3. Nonnarcotic Analgesics for Mild Pain in Sickle Cell Disease
| Drug | Usual starting dosage in adults |
|---|---|
| Acetaminophen (Tylenol) | 500 to 1,000 mg every 4 to 6 hours (maximum < 4,000 mg per day) |
| Acetylsalicylic acid (aspirin) | 650 to 1,000 mg every 4 to 6 hours (maximum < 4,000 mg per day) |
| Diflunisal (Dolobid) | 1,000 mg initially, then 500 mg every 8 to 12 hours |
| Choline magnesium trisalicylate (Trilisate) | 1,000 to 1,500 mg every 12 hours |
| Ibuprofen (Advil) | 200 to 400 mg every 4 to 6 hours |
| Naproxen (Naprosyn) | 500 mg initially, then 250 mg every 6 to 8 hours |
| Fenoprofen (Nalfon) | 200 mg every 4 to 6 hours |
| Ketoprofen (Orudis) | 25 to 75 mg, then 250 mg every 6 to 8 hours (maximum < 300 mg per day) |
Narcotic analgesics can be used in patients with moderate to severe pain. The dosage of the selected narcotic should be titrated to achieve effective pain control. Because of the dose-limiting side effects of weak opioids (codeine and oxycodone), which include sedation, nausea and vomiting, these drugs are best used to manage moderate pain (Table 4). Pain that is sufficiently severe to require an emergency department visit or hospitalization should be treated with stronger opioids (Table 5).
If a patient has poor venous access and is unable to take enteral narcotics because of vomiting, the subcutaneous route can be employed, using morphine or its equivalent. It is important to remember that subcutaneous administration may result in prolonged absorption if a patient is dehydrated.
Adjuvant nonopioid agents such as antihistamines and antiemetics can be helpful for preventing or relieving opioid-related side effects 108. The use of adjuvant analgesics such as tricyclic antidepressants should be considered in patients with a chronic pain syndrome resulting from recurrent acute pain crises 109.
Nonpharmacologic techniques can also be tried. These measures include physical therapy, rest, heat application, transcutaneous electrical nerve stimulation (TENS), self-hypnosis and diversional techniques 65.
Table 4. Common Opioids Used to Treat Mild to Moderate Pain in Sickle Cell Disease
| Drug | Usual oral starting dosage in adults | Comments and precautions |
|---|---|---|
| Codeine | 30 to 60 mg Every 3 or 4 hours | Available in liquid or tablet form, alone or in combination with acetaminophen |
| Side effects: impaired ventilation (histamine release possibly triggering bronchospasm) and increased intracranial pressure as a result of carbon dioxide retention | ||
| Oxycodone (Roxicodone) | 10 to 30 mg every 4 hours | Often used in combination with acetaminophen, which limits safe dosage to 12 tablets per day (about 4 g of acetaminophen) |
| Side effects: similar to those of codeine |
Table 5. Opioids Used to Treat Severe Pain in Sickle Cell Disease
| Drug | Oral/IM potency | Equianalgesic dosages | Usual starting dosage in adults | ||
|---|---|---|---|---|---|
| IM | Oral | Oral | Parenteral | ||
| Morphine (Duramorph) | 6* | 10 mg | 60 mg | 15 to 30 mg every 4 hours | 0.1 to 0.15 mg per kg every 3 or 4 hours |
| Hydromorphone (Dilaudid-Hp) | 5 | 1.5 mg | 7.5 mg | 2 to 4 mg every 4 to 6 hours | 1 to 2 mg every 4 to 6 hours |
| Meperidine (Demerol) | 4 | 75 mg | 300 mg | 50 to 150 mg every 3 or 4 hours | 75 to 100 mg every 3 or 4 hours |
| Levorphanol (Levo-Dromoran) | 2 | 2 mg | 4 mg | 2 to 4 mg every 6 to 8 hours | Up to 1 mg IV every 3 to 6 hours; 1 to 2 mg IM or SC every 6 to 8 hours |
Footnote: *—Single-dose studies determined that the relative potency is 6:1; with repetitive doses, this ratio changes to 3:1.
Abbreviations: IM = intramuscular; IV = intravenous; SC = subcutaneous.
[Source 75 ]Meperidine vs. morphine
In the past, moderate to severe pain in sickle cell disease was usually treated with meperidine (Demerol) administered parenterally or, more commonly, intramuscularly. Compared with morphine, however, meperidine has a number of properties that make it a poor opioid analgesic for repeated use in most patients with sickle cell disease.
Meperidine is a weak opioid analgesic with a short half-life (two to three hours). Thus, frequent dosing is required to maintain a sustained analgesic effect. In addition, normeperidine, a metabolite of meperidine, has been associated with seizures, particularly in patients with impaired renal function who are being given high doses at frequent intervals 65. Repeated injections of meperidine can lead to fibrosis, infection and sterile abscess formation at the injection site.
For these reasons, parenterally administered morphine should be considered the treatment of choice for moderate to severe pain in vaso-occlusive crises. Morphine’s side effects include pruritus, nausea, vomiting and rash. In addition, dosage adjustments are necessary in patients with liver dysfunction 110.
Some patients prefer meperidine for the treatment of pain crises and may be reluctant to change to morphine. Analgesia should be discussed when patients are not in pain.
Regardless of the type of opioid analgesic used, respiratory rate and oxygen saturation must be closely monitored because of the potential for respiratory depression. If the respiratory rate is less than 10 per minute or excessive sedation occurs, the opiate should be discontinued, the dosage should be reduced or the dosing frequency should be lengthened 111.
Methods of narcotic analgesics delivery
With parenteral administration, narcotic analgesics can be given using a fixed schedule (with rescue doses administered for breakthrough pain), continuous infusion or patient-controlled administration. Dosing on an “as-needed” basis should be avoided because it does not confer an adequate sustained level of analgesia.
Patient-controlled analgesia offers several unique advantages in the treatment of severe pain occurring in a vaso-occlusive crisis 112. One study 113 found that the intermittent fixed schedule and the patient-controlled method were equally efficacious in providing adequate analgesia. Patient-controlled analgesia prevents fluctuation in blood drug levels and may reduce the time between the perception of pain and the administration of the analgesic. This approach reduces overmedication and excessive sedation. It also provides patient autonomy and decreases the nursing time required for analgesic administration.
Oxygen therapy
Oxygen therapy is often used routinely in the management of vaso-occlusive crises, despite lack of evidence supporting the effectiveness of these measures in all patients.25,26 Oxygen therapy has not been shown to affect the duration of a pain crisis or to be useful in patients with acute chest syndrome whose partial pressure of arterial oxygen (PaO2) is in the normal range. Hence, oxygen should be administered only if hypoxemia is present 114. Oxygen may also suppress erythrocyte production, depress reticulocytosis and cause rebound sickle cell crises on discontinuation of therapy when the arterial oxygen tension is raised above the normal range 115.
Pulse oximetry may not be a reliable method of determining the PaO2 in patients with sickle cell disease.28 One reason may be the differences in the oxygen dissociation curve between normal hemoglobin and sickle cell hemoglobin (hemoglobin S) 116. Sickle cell erythrocytes have decreased oxygen affinity and increased unsaturated hemoglobin in the arterial blood. All low pulse-oximetry saturation values should be compared with values obtained at steady state, if available, or should be confirmed by measuring the PaO2 directly with an arterial blood gas determination 117.
Blood transfusion
Most patients with sickle cell anemia have hemoglobin values of 6 to 10 g per dL (60 to 100 g per L). The hemoglobin S molecule has a low affinity for oxygen (which allows for adequate tissue oxygenation). During a vaso-occlusive crisis, a patient’s hemoglobin level often declines by at least 1 g per dL (10 g per L).
A hemoglobin value of 5 g per dL (50 g per L) or less or a decline in the hemoglobin value of greater than 2 g per dL (20 g per L) from the patient’s baseline level has been used as a guide for considering simple transfusion therapy 118. Patients should be transfused to their baseline hemoglobin level. A higher hematocrit may make the blood more viscous and further increase sickling.
Other indications for transfusion include acute chest syndrome with hypoxia and the need for surgery using general anesthesia.
Exchange transfusions should be considered only in patients who have a prolonged refractory vaso-occlusive crisis with a stable baseline hemoglobin concentration. The goal is to reduce sickling by reducing the hemoglobin S level to below 20 percent 118.
Fluid replacement
Increased plasma osmolarity from a reduced plasma volume can worsen a vaso-occlusive crisis by causing intracellular dehydration, hemoglobin polymerization and further sickling. During hyponatremia, the affinity of hemoglobin S for oxygen is increased. Therefore, at any given PaO2, less oxygen is in the deoxygenated state, which is the form most susceptible to polymerization. Patients with sickle cell disease have isosthenuria, which leads to difficulty in excreting a sodium load 116.
Fluids should be administered in a quantity sufficient to correct existing deficits and replace ongoing losses in order to maintain a euvolemic state. Large fluid volumes may decrease plasma oncotic pressure and increase hydrostatic pressure. This can lead to pulmonary edema, especially in patients with impaired renal function, cardiac failure or pulmonary vascular injury.
If tolerated, oral rehydration should be used in patients with milder vaso-occlusive crises. The parenteral route of rehydration is indicated in patients with severe pain, vomiting or volume depletion. After existing volume deficits have been corrected with normal saline, fluid replacement should consist of 5 percent dextrose in water or in a 25 percent normal saline solution 116.
Vaso-occlusive crisis management
Vaso-occlusion is an emergency condition requiring intensive care admission and carries a high mortality 119. Rapid pain assessment and initiation of analgesia should be undertaken promptly. Depending on the degree and severity of pain, an analgesic administration can be given intravenously (IV) or intranasally. For patients who are not in severe pain and can tolerate oral medications, oral analgesics can be used. Generally, the type, route, and dose of the analgesic should be individualized to the patient. Most guidelines recommend early initiation of parenteral opioid analgesics, usually with morphine at 0.1 mg/kg IV or subcutaneously (SC) every 20 minutes and maintaining this analgesia with morphine at doses of 0.05 to 0.1 mg/ kg every 2 to 4 hrs (SC/IV or PO) 61. Those with persistent pain benefit from a patient-controlled analgesia pump. Close monitoring of vital signs including oxygen saturation should be maintained with frequent reassessments of pain severity or resolution 120. If the pain is controlled, the patient may be ready for discharge with a home care plan and oral analgesia 121. If the pain is uncontrolled despite the above treatment plan, consider hospitalization and the use of stronger forms of analgesia or higher doses titrated to the patient’s needs. Simple or exchange transfusion may be warranted 122. Low molecular weight heparin tinzaparin has been found to shorten the course of pain. A randomized, controlled, double-blind study has suggested that the clinical effects of tinzaparin are due to its effect on cellular factors. No special monitoring is needed for once-daily dosing 122. Adjuvant therapy includes hydroxyurea, antihistamines, anxiolytics, and antiemetics 122. It is prudent to maintain adequate hydration and be vigilant in identifying other causes of pain that may need additional treatment.
For other complications like acute chest syndrome, splenic sequestration-supportive care with oxygen, judicious fluid administration, and transfusion therapy is needed. Close monitoring of oxygen saturation and respiratory status, with particular attention to excessive sedation, is also necessary 123. For acute chest syndrome, empiric antibiotics, adequate analgesics, simple or exchange transfusion, may be considered. Incentive spirometry, oral hydration, and comfort measures are recommended. Patients with splenic sequestration crisis resulting in hypovolemic shock, if not treated aggressively, have higher mortality. Management requires aggressive supportive care and blood transfusion 124. Aplastic crisis is treated with supportive care and simple transfusions as needed.
Management after hospital discharge
Most patients have residual pain at the time they are discharged from the hospital. Therefore, they should be given an oral narcotic analgesic in a dosage equivalent to the dosage that was necessary to control their pain while they were hospitalized. They should be given enough of the narcotic analgesic to last until the next scheduled outpatient follow-up visit 108.
Patients should be instructed to use the visual analog scale or the verbal categorical scale as a guide for self-tapering of the analgesic dosage based on their level of pain.
Living with sickle cell disease
Follow these steps to help relieve symptoms and help you manage your condition at home:
- See your healthcare provider regularly. Most people who have sickle cell disease should see their provider every 3 to 12 months, depending on their age.
- Get regular vaccines, including an influenza or flu shot every year. Your doctor may also recommend a second pneumococcus (PPSV23) vaccination, in addition to the pneumococcus (PCV13) vaccination that all children get as part of their regular immunizations. This second vaccine is given after 24 months of age and again 5 years later. Adults who have sickle cell disease and have not received any pneumococcal vaccine should get a dose of the PCV13 vaccine. They should later receive the PPSV23 if they have not already received it or if it has been more than 5 years since they did. Follow these guidelines even if you or your child is still taking penicillin.
- Avoid situations that may set off a vaso occlusive crisis. Extreme heat or cold, as well as sudden changes in temperature, are often triggers. When going swimming, ease into the water rather than jumping right in. Do not travel in an aircraft cabin that is unpressurized. If you experience priapism (a prolonged, painful erection), you may be able to relieve your symptoms by doing light exercise, emptying your bladder by urinating, drinking more fluids, and taking medicine recommended by your healthcare provider.
- When an acute crisis is just starting, drink lots of fluids and take a nonsteroidal anti-inflammatory (NSAID) pain medicine, such as ibuprofen. If you have kidney problems, acetaminophen is often preferred.
- If you cannot manage the pain at home, go to a sickle cell disease day hospital or outpatient unit or an emergency room to receive additional, stronger medicines and IV fluids. You may need to be admitted to the hospital to fully control an acute pain crisis. You may be able to return home once your pain is under better control.
- Adopt a healthy lifestyle
- Get regular physical activity can help manage risk factors for heart disease such as high blood cholesterol, high blood pressure, and overweight and obesity. Before starting any exercise program, ask about what level of physical activity is right for you.
- Choose heart-healthy foods. A heart-healthy eating plan includes fruits, vegetables, and whole grains and limits saturated fats, sodium (salt), added sugars, and alcohol.
- If you smoke, quit smoking.
- Learn how to palpate (feel) your child’s spleen. Because of the risk of splenic sequestration crisis, caretakers should learn how to palpate a child’s spleen. They should try to feel for the spleen daily and more often when the child is ill. If the spleen feels larger than usual, they should call the care provider.
- Take care of your mental health. Talk to your family and friends about how you are feeling. Talk to your provider if you have feelings of depression or anxiety. Supportive counseling and, sometimes, antidepressant medicines may help.
Sickle cell disease life expectancy
Until recently, people with sickle cell disease were not expected to survive childhood. But today, due to preventive drug treatment, improved medical care and aggressive research, life expectancy in sickle cell disease is 20 to 30 years and half of sickle cell patients live beyond 50 years 125, 3. Most children with sickle cell anemia (HbSS) or sickle β0-thalassemia (HbSβ0) (93.9%) and nearly all children with sickle hemoglobin C disease (HbSC) or sickle β+-thalassemia (HbSβ+) (98.4%) now live to become adults 125. Factors that predict poor prognosis include painful swelling of the small bones of the hands and feet (dactylitis) in infancy, hemoglobin level less than 7 g/dL, and higher white cell count in the absence of infection.
References- Sickle Beta Plus Thalassemia. https://dshs.texas.gov/newborn/pdf/FactSBPlusThal.doc
- Sangani V, Pokal M, Balla M, Merugu GP, Khokher W, Gayam V, Konala VM. Fat Embolism Syndrome in Sickle Cell β-Thalassemia Patient With Osteonecrosis: An Uncommon Presentation in a Young Adult. J Investig Med High Impact Case Rep. 2021 Jan-Dec;9:23247096211012266. doi: 10.1177/23247096211012266
- Sedrak A, Kondamudi NP. Sickle Cell Disease. [Updated 2021 Nov 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482384
- Sickle cell disease. https://medlineplus.gov/genetics/condition/sickle-cell-disease/#frequency
- Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484. doi: 10.1371/journal.pmed.1001484
- Ashorobi D, Ramsey A, Yarrarapu SNS, et al. Sickle Cell Trait. [Updated 2022 Apr 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537130
- Sickle Cell Trait. https://www.ncaa.org/sports/2016/7/27/sickle-cell-trait.aspx
- El Ariss AB, Younes M, Matar J, Berjaoui Z. Prevalence of Sickle Cell Trait in the Southern Suburb of Beirut, Lebanon. Mediterr J Hematol Infect Dis. 2016 Feb 20;8(1):e2016015. doi: 10.4084/MJHID.2016.015
- Gibson JS, Rees DC. How benign is sickle cell trait? EBioMedicine. 2016 Sep;11:21-22. doi: 10.1016/j.ebiom.2016.08.023
- Sickle-cell disease: one amino acid makes a difference. https://www.labxchange.org/library/items/lb:LabXchange:b1217566:html:1
- Barfield WD, Barradas DT, Manning SE, Kotelchuck M, Shapiro-Mendoza CK. Sickle cell disease and pregnancy outcomes: women of African descent. Am J Prev Med. 2010 Apr;38(4 Suppl):S542-9. doi: 10.1016/j.amepre.2009.12.020
- Oteng-Ntim E. Pregnancy in women with sickle cell disease is associated with risk of maternal and perinatal mortality and severe morbidity. Evid Based Nurs. 2017 Apr;20(2):43. doi: 10.1136/eb-2016-102450
- Ngô C, Kayem G, Habibi A, Benachi A, Goffinet F, Galactéros F, Haddad B. Pregnancy in sickle cell disease: maternal and fetal outcomes in a population receiving prophylactic partial exchange transfusions. Eur J Obstet Gynecol Reprod Biol. 2010 Oct;152(2):138-42. doi: 10.1016/j.ejogrb.2010.05.022
- Bonnin M, Mercier FJ, Sitbon O, Roger-Christoph S, Jaïs X, Humbert M, Audibert F, Frydman R, Simonneau G, Benhamou D. Severe pulmonary hypertension during pregnancy: mode of delivery and anesthetic management of 15 consecutive cases. Anesthesiology. 2005 Jun;102(6):1133-7; discussion 5A-6A. doi: 10.1097/00000542-200506000-00012
- Kiely DG, Condliffe R, Webster V, Mills GH, Wrench I, Gandhi SV, Selby K, Armstrong IJ, Martin L, Howarth ES, Bu’lock FA, Stewart P, Elliot CA. Improved survival in pregnancy and pulmonary hypertension using a multiprofessional approach. BJOG. 2010 Apr;117(5):565-74. doi: 10.1111/j.1471-0528.2009.02492.x
- Wilson S, Ellsworth P, Key NS. Pregnancy in sickle cell trait: what we do and don’t know. Br J Haematol. 2020 Aug;190(3):328-335. doi: 10.1111/bjh.16518
- Petersen EE, Davis NL, Goodman D, Cox S, Syverson C, Seed K, Shapiro-Mendoza C, Callaghan WM, Barfield W. Racial/Ethnic Disparities in Pregnancy-Related Deaths – United States, 2007-2016. MMWR Morb Mortal Wkly Rep. 2019 Sep 6;68(35):762-765. doi: 10.15585/mmwr.mm6835a3
- Oteng-Ntim E, Meeks D, Seed PT, Webster L, Howard J, Doyle P, Chappell LC. Adverse maternal and perinatal outcomes in pregnant women with sickle cell disease: systematic review and meta-analysis. Blood. 2015 May 21;125(21):3316-25. doi: 10.1182/blood-2014-11-607317
- Baill IC, Witter FR. Sickle trait and its association with birthweight and urinary tract infections in pregnancy. Int J Gynaecol Obstet. 1990 Sep;33(1):19-21. doi: 10.1016/0020-7292(90)90649-6
- Thurman AR, Steed LL, Hulsey T, Soper DE. Bacteriuria in pregnant women with sickle cell trait. Am J Obstet Gynecol. 2006 May;194(5):1366-70. doi: 10.1016/j.ajog.2005.11.022
- Tita AT, Biggio JR, Chapman V, Neely C, Rouse DJ. Perinatal and maternal outcomes in women with sickle or hemoglobin C trait. Obstet Gynecol. 2007 Nov;110(5):1113-9. doi: 10.1097/01.AOG.0000285995.41769.83
- Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications associated with sickle cell trait: a brief narrative review. Am J Med. 2009 Jun;122(6):507-12. doi: 10.1016/j.amjmed.2008.12.020
- ACOG technical bulletin. Hemoglobinopathies in pregnancy. Number 220–February 1996 (replaces no. 185, October 1993). Committee on Technical Bulletins of the American College of Obstetricians and Gynecologists. Int J Gynaecol Obstet. 1996 May;53(2):184-94. https://doi.org/10.1016/S0020-7292(96)90113-7
- ACOG Committee on Obstetrics. ACOG Practice Bulletin No. 78: hemoglobinopathies in pregnancy. Obstet Gynecol. 2007 Jan;109(1):229-37. doi: 10.1097/00006250-200701000-00055
- Larrabee KD, Monga M. Women with sickle cell trait are at increased risk for preeclampsia. Am J Obstet Gynecol. 1997 Aug;177(2):425-8. doi: 10.1016/s0002-9378(97)70209-6
- Taylor MY, Wyatt-Ashmead J, Gray J, Bofill JA, Martin R, Morrison JC. Pregnancy loss after first-trimester viability in women with sickle cell trait: time for a reappraisal? Am J Obstet Gynecol. 2006 Jun;194(6):1604-8. doi: 10.1016/j.ajog.2006.02.027
- O’Hara C, Singer DE, Niebuhr DW. The Risk of Pregnancy Related Hypertension Disorder Associated with Sickle Cell Trait in U.S. Service Women. Mil Med. 2020 Feb 12;185(1-2):e183-e190. doi: 10.1093/milmed/usz143
- Hu J, Nelson DA, Deuster PA, Marks ES, O’Connor FG, Kurina LM. Sickle cell trait and renal disease among African American U.S. Army soldiers. Br J Haematol. 2019 May;185(3):532-540. doi: 10.1111/bjh.15820
- Naik RP, Irvin MR, Judd S, Gutiérrez OM, Zakai NA, Derebail VK, Peralta C, Lewis MR, Zhi D, Arnett D, McClellan W, Wilson JG, Reiner AP, Kopp JB, Winkler CA, Cushman M. Sickle Cell Trait and the Risk of ESRD in Blacks. J Am Soc Nephrol. 2017 Jul;28(7):2180-2187. doi: 10.1681/ASN.2016101086
- WHALLEY PJ, PRITCHARD JA, RICHARDS JR Jr. SICKLE CELL TRAIT AND PREGNANCY. JAMA. 1963 Dec 28;186:1132-5. doi: 10.1001/jama.1963.03710130028008
- Taylor MY, Wyatt-Ashmead J, Gray J, Bofill JA, Martin RW, Morrison JC. Pregnancy loss after first trimester viability in women with sickle cell trait: a preliminary report. South Med J. 2008 Feb;101(2):150-1. doi: 10.1097/SMJ.0b013e31816122ea
- Bryant AS, Cheng YW, Lyell DJ, Laros RK, Caughey AB. Presence of the sickle cell trait and preterm delivery in African-American women. Obstet Gynecol. 2007 Apr;109(4):870-4. doi: 10.1097/01.AOG.0000258275.24990.9c
- Tan TL, Seed P, Oteng-Ntim E. Birthweights in sickle cell trait pregnancies. BJOG. 2008 Aug;115(9):1116-21. doi: 10.1111/j.1471-0528.2008.01776.x
- Naik RP, Haywood C Jr. Sickle cell trait diagnosis: clinical and social implications. Hematology Am Soc Hematol Educ Program. 2015;2015(1):160-7. doi: 10.1182/asheducation-2015.1.160
- Naik RP, Smith-Whitley K, Hassell KL, Umeh NI, de Montalembert M, Sahota P, Haywood C Jr, Jenkins J, Lloyd-Puryear MA, Joiner CH, Bonham VL, Kato GJ. Clinical Outcomes Associated With Sickle Cell Trait: A Systematic Review. Ann Intern Med. 2018 Nov 6;169(9):619-627. doi: 10.7326/M18-1161
- Taylor SM, Parobek CM, Fairhurst RM. Haemoglobinopathies and the clinical epidemiology of malaria: a systematic review and meta-analysis. Lancet Infect Dis. 2012 Jun;12(6):457-68. doi: 10.1016/S1473-3099(12)70055-5
- Ojodu J, Hulihan MM, Pope SN, Grant AM; Centers for Disease Control and Prevention (CDC). Incidence of sickle cell trait–United States, 2010. MMWR Morb Mortal Wkly Rep. 2014 Dec 12;63(49):1155-8.
- Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med. 1987 Sep 24;317(13):781-7. doi: 10.1056/NEJM198709243171301
- Harmon KG, Drezner JA, Klossner D, Asif IM. Sickle cell trait associated with a RR of death of 37 times in National Collegiate Athletic Association football athletes: a database with 2 million athlete-years as the denominator. Br J Sports Med. 2012 Apr;46(5):325-30. doi: 10.1136/bjsports-2011-090896
- Naik RP, Derebail VK, Grams ME, Franceschini N, Auer PL, Peloso GM, Young BA, Lettre G, Peralta CA, Katz R, Hyacinth HI, Quarells RC, Grove ML, Bick AG, Fontanillas P, Rich SS, Smith JD, Boerwinkle E, Rosamond WD, Ito K, Lanzkron S, Coresh J, Correa A, Sarto GE, Key NS, Jacobs DR, Kathiresan S, Bibbins-Domingo K, Kshirsagar AV, Wilson JG, Reiner AP. Association of sickle cell trait with chronic kidney disease and albuminuria in African Americans. JAMA. 2014 Nov 26;312(20):2115-25. doi: 10.1001/jama.2014.15063
- Li EJ, Carroll VG. Sickle cell trait and renal papillary necrosis. Clin Pediatr (Phila). 2014 Sep;53(10):1013-5. doi: 10.1177/0009922814533418
- Yanamandra U, Das R, Malhotra P, Varma S. A Case of Autosplenectomy in Sickle Cell Trait Following an Exposure to High Altitude. Wilderness Environ Med. 2018 Mar;29(1):85-89. doi: 10.1016/j.wem.2017.08.021
- Goenaga-Vázquez Y, Colón G, Barrios N, Correa M. Renal medullary carcinoma: a nearly fatal malignancy specifically affecting patients with a so-called benign condition. CEN Case Rep. 2018 May;7(1):121-126. doi: 10.1007/s13730-018-0308-3
- Tarini BA, Brooks MA, Bundy DG. A policy impact analysis of the mandatory NCAA sickle cell trait screening program. Health Serv Res. 2012 Feb;47(1 Pt 2):446-61. doi: 10.1111/j.1475-6773.2011.01357.x
- Nelson DA, Deuster PA, Carter R 3rd, Hill OT, Wolcott VL, Kurina LM. Sickle Cell Trait, Rhabdomyolysis, and Mortality among U.S. Army Soldiers. N Engl J Med. 2016 Aug 4;375(5):435-42. doi: 10.1056/NEJMoa1516257
- HBB gene. https://medlineplus.gov/genetics/gene/hbb/#conditions
- Krejza J, Chen R, Romanowicz G, Kwiatkowski JL, Ichord R, Arkuszewski M, Zimmerman R, Ohene-Frempong K, Desiderio L, Melhem ER. Sickle cell disease and transcranial Doppler imaging: inter-hemispheric differences in blood flow Doppler parameters. Stroke. 2011 Jan;42(1):81-6. doi: 10.1161/STROKEAHA.110.591818
- Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Chevret S, Hau I, Coïc L, Leveillé E, Lemarchand E, Lesprit E, Abadie I, Medejel N, Madhi F, Lemerle S, Biscardi S, Bardakdjian J, Galactéros F, Torres M, Kuentz M, Ferry C, Socié G, Reinert P, Delacourt C. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood. 2011 Jan 27;117(4):1130-40; quiz 1436. doi: 10.1182/blood-2010-06-293514
- Kwiatkowski JL, Granger S, Brambilla DJ, Brown RC, Miller ST, Adams RJ; STOP Trial Investigators. Elevated blood flow velocity in the anterior cerebral artery and stroke risk in sickle cell disease: extended analysis from the STOP trial. Br J Haematol. 2006 Aug;134(3):333-9. doi: 10.1111/j.1365-2141.2006.06193.x
- Adams RJ, Brambilla D; Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial Investigators. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005 Dec 29;353(26):2769-78. doi: 10.1056/NEJMoa050460
- Clinical Alert: Periodic Transfusions Lower Stroke Risk in Children with Sickle Cell Anemia. National Heart, Lung, and Blood Institute (NHLBI) September 18, 1997 https://wayback.archive-it.org/org-350/20211101182421/https://www.nlm.nih.gov/databases/alerts/sickle97.html
- Chirico EN, Faës C, Connes P, Canet-Soulas E, Martin C, Pialoux V. Role of Exercise-Induced Oxidative Stress in Sickle Cell Trait and Disease. Sports Med. 2016 May;46(5):629-39. doi: 10.1007/s40279-015-0447-z
- Connes P, Machado R, Hue O, Reid H. Exercise limitation, exercise testing and exercise recommendations in sickle cell anemia. Clin Hemorheol Microcirc. 2011;49(1-4):151-63. doi: 10.3233/CH-2011-1465
- Okomo U, Meremikwu MM. Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. Cochrane Database Syst Rev. 2017 Jul 31;7(7):CD005406. doi: 10.1002/14651858.CD005406.pub5
- Westcott, E. (2017). Healthy Behaviors of Adolescents and Young Adults with Sickle Cell Disease [Master’s thesis, University of Cincinnati]. OhioLINK Electronic Theses and Dissertations Center. http://rave.ohiolink.edu/etdc/view?acc_num=ucin1504800267776992
- Schatz J, Schlenz AM, Smith KE, Roberts CW. Predictive validity of developmental screening in young children with sickle cell disease: a longitudinal follow-up study. Dev Med Child Neurol. 2018 May;60(5):520-526. doi: 10.1111/dmcn.13689
- Hardy SJ, Hardy KK, Schatz JC, Thompson AL, Meier ER. Feasibility of Home-Based Computerized Working Memory Training With Children and Adolescents With Sickle Cell Disease. Pediatr Blood Cancer. 2016 Sep;63(9):1578-85. doi: 10.1002/pbc.26019
- King, A., White, D., Armstrong, M. et al. An Educational Remediation Program Benefits Children With Sickle Cell Disease and Cerebral Infarcts.. Pediatr Res 56, 668 (2004). https://doi.org/10.1203/00006450-200410000-00034
- Acquazzino MA, Miller M, Myrvik M, Newby R, Scott JP. Attention Deficit Hyperactivity Disorder in Children With Sickle Cell Disease Referred for an Evaluation. J Pediatr Hematol Oncol. 2017 Jul;39(5):350-354. doi: 10.1097/MPH.0000000000000847
- Xu G, Strathearn L, Liu B, Yang B, Bao W. Twenty-Year Trends in Diagnosed Attention-Deficit/Hyperactivity Disorder Among US Children and Adolescents, 1997-2016. JAMA Netw Open. 2018 Aug 3;1(4):e181471. doi: 10.1001/jamanetworkopen.2018.1471
- Borhade MB, Kondamudi NP. Sickle Cell Crisis. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK526064
- Hiran S. Multiorgan dysfunction syndrome in sickle cell disease. J Assoc Physicians India. 2005 Jan;53:19-22.
- Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse WF, Vichinsky E, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325:11–6.
- Shapiro BS. The management of pain in sickle cell disease. Pediatr Clin North Am. 1989;36:1029–45.
- Ballis SK, Carlos TM, Dampier C, and Guidelines Committee. Guidelines for standard of care of acute painful episodes in patients with sickle cell disease. Harrisburg, Pa.: Commonwealth of Pennsylvania Department of Health, 1996.
- Nagel RL. Sickle cell anemia is a multigene disease: sickle cell painful crises, a case in point. Am J Hematol. 1993;42:96–101.
- Solovey A, Lin Y, Browne P, Choong S, Wayner E, Hebbel RP. Circulating activated endothelial cells in sickle cell anemia. N Engl J Med. 1997;337:1584–90.
- Green SA, Aljuburi G, Majeed A, et al. Characterizing emergency admissions of patients with sickle cell crisis in NHS brent: observational study. JRSM Short Rep. 2012;3(6):37. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3386659/
- Moore F, Mortimer P, Wooller J Updated Sickle Cell Crisis Guidelines. Joint Royal Colleges Ambulance Liaison Committee. 2009
- Sickle-cell disease and other haemoglobin disorders. Fact sheet N°308. WHO, January 2011
- Steinberg MH. Management of sickle cell disease. N Engl J Med. 1999;340:1021–30.
- Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N. Engl. J. Med. 1980 May 01;302(18):992-5. https://www.ncbi.nlm.nih.gov/pubmed/7366623
- Payne R. Pain management in sickle cell disease. Ann N Y Acad Sci. 1989;565:189–206.
- Serjeant GR, Ceulaer CD, Lethbridge R, Morris J, Singhal A, Thomas PW. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol. 1994;87:586–91.
- Approach to the Vaso-occlusive Crisis in Adults with Sickle Cell Disease. Am Fam Physician. 2000 Mar 1;61(5):1349-1356. https://www.aafp.org/afp/2000/0301/p1349.html
- Sedrak A, Kondamudi NP. Sickle Cell Disease. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482384
- Simon E, Long B, Koyfman A. Emergency Medicine Management of Sickle Cell Disease Complications: An Evidence-Based Update. J Emerg Med. 2016 Oct;51(4):370-381. https://www.ncbi.nlm.nih.gov/pubmed/27553919
- Knight-Madden J, Serjeant GR. Invasive pneumococcal disease in homozygous sickle cell disease: Jamaican experience 1973-1997. J Pediatr. 2001 Jan;138(1):65-70. doi: 10.1067/mpd.2001.109709
- Halasa NB, Shankar SM, Talbot TR, Arbogast PG, Mitchel EF, Wang WC, Schaffner W, Craig AS, Griffin MR. Incidence of invasive pneumococcal disease among individuals with sickle cell disease before and after the introduction of the pneumococcal conjugate vaccine. Clin Infect Dis. 2007 Jun 1;44(11):1428-33. doi: 10.1086/516781
- Kanter J, Kruse-Jarres R. Management of sickle cell disease from childhood through adulthood. Blood Rev. 2013 Nov;27(6):279-87. doi: 10.1016/j.blre.2013.09.001
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Population Health and Public Health Practice; Committee on Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action; Martinez RM, Osei-Anto HA, McCormick M, editors. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. Washington (DC): National Academies Press (US); 2020 Sep 10. 4, Complications of Sickle Cell Disease and Current Management Approaches. Available from: https://www.ncbi.nlm.nih.gov/books/NBK566466
- Rizio AA, Bhor M, Lin X, McCausland KL, White MK, Paulose J, Nandal S, Halloway RI, Bronté-Hall L. The relationship between frequency and severity of vaso-occlusive crises and health-related quality of life and work productivity in adults with sickle cell disease. Qual Life Res. 2020 Jun;29(6):1533-1547. doi: 10.1007/s11136-019-02412-5
- Niraimathi M, Kar R, Jacob SE, Basu D. Sudden Death in Sickle Cell Anaemia: Report of Three Cases with Brief Review of Literature. Indian J Hematol Blood Transfus. 2016 Jun;32(Suppl 1):258-61. doi: 10.1007/s12288-015-0571-9
- Manci EA, Culberson DE, Yang YM, Gardner TM, Powell R, Haynes J Jr, Shah AK, Mankad VN; Investigators of the Cooperative Study of Sickle Cell Disease. Causes of death in sickle cell disease: an autopsy study. Br J Haematol. 2003 Oct;123(2):359-65. doi: 10.1046/j.1365-2141.2003.04594.x
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994 Jun 9;330(23):1639-44. doi: 10.1056/NEJM199406093302303
- Sickle Cell Disease. https://www.nhlbi.nih.gov/health/sickle-cell-disease/treatment
- Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, Hassab H, Achebe MM, Alkindi S, Brown RC, Diuguid DL, Telfer P, Tsitsikas DA, Elghandour A, Gordeuk VR, Kanter J, Abboud MR, Lehrer-Graiwer J, Tonda M, Intondi A, Tong B, Howard J; HOPE Trial Investigators. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med. 2019 Aug 8;381(6):509-519. doi: 10.1056/NEJMoa1903212
- Estepp JH. Voxelotor (GBT440), a first-in-class hemoglobin oxygen-affinity modulator, has promising and reassuring preclinical and clinical data. Am J Hematol. 2018 Mar;93(3):326-329. doi: 10.1002/ajh.25042
- FDA approves crizanlizumab-tmca for sickle cell disease. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-crizanlizumab-tmca-sickle-cell-disease
- Kutlar A, Kanter J, Liles DK, Alvarez OA, Cançado RD, Friedrisch JR, Knight-Madden JM, Bruederle A, Shi M, Zhu Z, Ataga KI. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am J Hematol. 2019 Jan;94(1):55-61. doi: 10.1002/ajh.25308
- Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro S, Colella MP, Smith WR, Rollins SA, Stocker JW, Rother RP. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017 Feb 2;376(5):429-439. doi: 10.1056/NEJMoa1611770
- Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, Ataga K, Swerdlow P, Kutlar A, DeCastro L, Waclawiw MA; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia and MSH Patients’ Follow-Up. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010 Jun;85(6):403-8. doi: 10.1002/ajh.21699
- Tshilolo L, Tomlinson G, Williams TN, Santos B, Olupot-Olupot P, Lane A, Aygun B, Stuber SE, Latham TS, McGann PT, Ware RE; REACH Investigators. Hydroxyurea for Children with Sickle Cell Anemia in Sub-Saharan Africa. N Engl J Med. 2019 Jan 10;380(2):121-131. doi: 10.1056/NEJMoa1813598
- Nevitt SJ, Jones AP, Howard J. Hydroxyurea (hydroxycarbamide) for sickle cell disease. Cochrane Database Syst Rev. 2017 Apr 20;4(4):CD002202. doi: 10.1002/14651858.CD002202.pub2
- Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, Gordeuk VR, Viswanathan K, Sarnaik S, Osunkwo I, Guillaume E, Sadanandan S, Sieger L, Lasky JL, Panosyan EH, Blake OA, New TN, Bellevue R, Tran LT, Razon RL, Stark CW, Neumayr LD, Vichinsky EP; Investigators of the Phase 3 Trial of l-Glutamine in Sickle Cell Disease. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med. 2018 Jul 19;379(3):226-235. doi: 10.1056/NEJMoa1715971
- Quinn CT. l-Glutamine for sickle cell anemia: more questions than answers. Blood. 2018 Aug 16;132(7):689-693. doi: 10.1182/blood-2018-03-834440
- Gaston MH, Verter JI, Woods G, Pegelow C, Kelleher J, Presbury G, Zarkowsky H, Vichinsky E, Iyer R, Lobel JS, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med. 1986 Jun 19;314(25):1593-9. doi: 10.1056/NEJM198606193142501
- Falletta JM, Woods GM, Verter JI, Buchanan GR, Pegelow CH, Iyer RV, Miller ST, Holbrook CT, Kinney TR, Vichinsky E, et al. Discontinuing penicillin prophylaxis in children with sickle cell anemia. Prophylactic Penicillin Study II. J Pediatr. 1995 Nov;127(5):685-90. doi: 10.1016/s0022-3476(95)70154-0
- https://www.nhlbi.nih.gov/sites/default/files/media/docs/sickle-cell-disease-report%20020816_0.pdf
- Molokie R, Lavelle D, Gowhari M, Pacini M, Krauz L, Hassan J, Ibanez V, Ruiz MA, Ng KP, Woost P, Radivoyevitch T, Pacelli D, Fada S, Rump M, Hsieh M, Tisdale JF, Jacobberger J, Phelps M, Engel JD, Saraf S, Hsu LL, Gordeuk V, DeSimone J, Saunthararajah Y. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized phase 1 study. PLoS Med. 2017 Sep 7;14(9):e1002382. doi: 10.1371/journal.pmed.1002382
- Shearstone JR, Golonzhka O, Chonkar A, Tamang D, van Duzer JH, Jones SS, Jarpe MB. Chemical Inhibition of Histone Deacetylases 1 and 2 Induces Fetal Hemoglobin through Activation of GATA2. PLoS One. 2016 Apr 13;11(4):e0153767. doi: 10.1371/journal.pone.0153767
- Kutlar A, Reid ME, Inati A, Taher AT, Abboud MR, El-Beshlawy A, Buchanan GR, Smith H, Ataga KI, Perrine SP, Ghalie RG. A dose-escalation phase IIa study of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. Am J Hematol. 2013 Nov;88(11):E255-60. doi: 10.1002/ajh.23533
- Eapen M, Brazauskas R, Walters MC, Bernaudin F, Bo-Subait K, Fitzhugh CD, Hankins JS, Kanter J, Meerpohl JJ, Bolaños-Meade J, Panepinto JA, Rondelli D, Shenoy S, Williamson J, Woolford TL, Gluckman E, Wagner JE, Tisdale JF. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol. 2019 Nov;6(11):e585-e596. doi: 10.1016/S2352-3026(19)30154-1
- Preboth M. Management of pain in sickle cell disease. Am Fam Physician. 2000;61:1541–2.
- Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014 Sep 10;312(10):1033-48. doi: 10.1001/jama.2014.10517. https://jamanetwork.com/journals/jama/article-abstract/1902235
- Pathophysiology and management of sickle cell pain crisis. Lancet. 1995;346:1408–11.
- Cancer pain relief. Geneva: World Health Organization, 1986.
- Ballas SK. Management of sickle pain. Curr Opin Hematol. 1997;4:104–11
- Pollack CV Jr, Sanders DY, Severance HW Jr. Emergency department analgesia without narcotics for adults with acute sickle cell pain crisis: case reports and review of crisis management. J Emerg Med. 1991;9:445–52.
- Ballas SK. Management of sickle pain. Curr Opin Hematol. 1997;4:104–11.
- Ballas SK Complications of sickle cell anemia in adults: guidelines for effective management. Clev Clin J Med. 1999;66:48–58.
- Shapiro BS, Cohen DE, Howe CJ. Patient-controlled analgesia for sickle-cell-related pain. J Pain Symptom Manage. 1993;8:22–8.
- Gonzalez ER, Bahal N, Hansen LA, Ware D, Bull DS, Ornato JP, et al. Intermittent injection vs. patient-controlled analgesia for sickle cell crisis pain. Arch Intern Med. 1991;151:1373–8.
- Zipursky A, Robieux IC, Brown EJ, Shaw D, O’Brodovich H, Kellner JD, et al. Oxygen therapy in sickle cell disease. Am J Pediatr Hematol Oncol. 1992;14:222–8.
- Embury SH, Garcia JF, Mohandas N, Pennathur-Das R, Clark MR. Effects of oxygen inhalation on endogenous erythropoietin kinetics, erythropoiesis, and properties of blood cells in sickle-cell anemia. N Engl J Med. 1984;311:291–5.
- Okpala I. The management of crisis in sickle cell disease. Eur J Haematol. 1998;60:1–6.
- Homi J, Levee L, Higgs D, Thomas P, Serjeant G. Pulse oximetry in a cohort study of sickle cell disease. Clin Lab Haematol. 1997;19:17–22.
- Brozovic M, Davies S. Management of sickle cell disease. Postgrad Med J. 1987;63:605–9.
- Mamdapur AB, Sagar MS, Madhusudan R, Samir M. Therapeutic Red Cell Exchange Transfusion as an Adjuvant Therapy for Management of Sickle Cell Crisis in Adults. Indian J Crit Care Med. 2018;22(6):457-459. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6020634/
- Chou ST, Fasano RM. Management of Patients with Sickle Cell Disease Using Transfusion Therapy: Guidelines and Complications. Hematol. Oncol. Clin. North Am. 2016 Jun;30(3):591-608. https://www.ncbi.nlm.nih.gov/pubmed/27112998
- Ballas SK. Current issues in sickle cell pain and its management. Hematology Am Soc Hematol Educ Program. 2007:97-105.
- Robieux IC, Kellner JD, Coppes MJ, Shaw D, Brown E, Good C, O’Brodovich H, Manson D, Olivieri NF, Zipursky A. Analgesia in children with sickle cell crisis: comparison of intermittent opioids vs. continuous intravenous infusion of morphine and placebo-controlled study of oxygen inhalation. Pediatr Hematol Oncol. 1992 Oct-Dec;9(4):317-26.
- Porter M. Rapid Fire: Sickle Cell Disease. Emerg. Med. Clin. North Am. 2018 Aug;36(3):567-576.
- Chou ST, Fasano RM. Management of Patients with Sickle Cell Disease Using Transfusion Therapy: Guidelines and Complications. Hematol. Oncol. Clin. North Am. 2016 Jun;30(3):591-608.
- Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010 Apr 29;115(17):3447-52. doi: 10.1182/blood-2009-07-233700





{kind=link}