Tumoral calcinosis
Tumoral calcinosis also known as familial tumoral calcinosis, hyperphosphatemic familial tumoral calcinosis or hyperostosis-hyperphosphatemia syndrome, is a rare inherited disease characterized by solitary or multiple painless periarticular calcium salt deposition (calcinosis) without joint involvement 1. These lesions consist of crystals of calcium hydroxy apatite and amorphous calcium phosphate which are commonly located around extensor surface of large joints, hip, elbow, shoulder, small joints of hands and foot. Adolescents and young adults are commonly affected 2. The term tumoral calcinosis should be strictly used to refer to a disease caused by a hereditary metabolic dysfunction of phosphate regulation associated with massive periarticular calcinosis and should not be used to refer to soft-tissue calcification in general 3. Tumoral calcinosis is usually found in African-American females and commonly presenting in childhood or early adolescence. Regions most commonly involved by familial tumoral calcinosis are soft tissues of peri-articular upper limb (shoulder and elbow) and hip regions 4. Still, spinal 5, temporo-mandibular joint 6, metacarpals or metatarsals 7 and popliteal space 8 involvement has also been reported.
Unfortunately, the term tumoral calcinosis has been liberally and imprecisely used to describe any massive collection of periarticular calcification, although the term tumoral calcinosis actually refers to a hereditary condition associated with massive periarticular calcification. The inconsistent use of this term has created confusion throughout the medical literature. Inclan et al 2 described this condition in the American literature in 1943; this publication became the pivotal article in developing a standard by which to diagnose the disorder and in coining the term tumoral calcinosis. Inclan et al 2 differentiated tumoral calcinosis from the dystrophic and metabolic (also known as “metastatic”) calcifications previously described in the literature, specifically calcifications associated with renal osteodystrophy, connective tissue disease, and hormonal imbalance. In addition to outlining the metabolic features of tumoral calcinosis, namely normal serum calcium levels with elevated serum phosphate levels, Inclan et al 2 were the first to publish radiographs of the condition (see Figure 1). Radiography will demonstrate a cloud-like soft-tissue mass.
Ectopic calcifications in the skin and subcutaneous tissue are a classic and potentially morbid feature of hyperphosphatemic familial tumoral calcinosis (Figures 1 to 3). Lesions consist of hydroxyapatite and/or calcium carbonate (Figure 1C) 9 and typically occur in peri-articular locations that are exposed to repeated pressure or trauma 10. The lateral hips are the most frequently affected site, but a wide range of areas may be involved, including the elbows, shoulders, hands, Achilles tendons, and others 11. Calcifications typically onset during the first two decades of life, and have been reported in children as young as 6 weeks 12. Once present, lesions may grow slowly over time. Patients present along a broad spectrum, ranging from no involvement to lesions that are large, painful, and debilitating 11. Calcifications that occur around joint spaces can impair mobility and physical function. In severe cases, calcific tumors perforate the skin and drain liquid hydroxyapatite, which may be confused with purulent drainage. Ulceration may also be accompanied by recurrent secondary infections.
Hyperphosphatemic familial tumoral calcinosis or hyperostosis-hyperphosphatemia syndrome is a disorder of fibroblast growth factor 23 (FGF23) deficiency or resistance 13. Fibroblast growth factor 23 (FGF23) is a phosphate- and 1,25(OH)2 vitamin D (also known as “calcitriol”) regulating hormone produced by osteoblasts and osteocytes 14. FGF23 acts via FGF receptor 1 (FGFR1) coupled with the coreceptor αKlotho to reduce expression of sodium phosphate cotransporters (NaPi-2a and -2c) and renal 25-hydroxyvitamin D 1-α-hydroxylase 15. FGF23 lowers serum phosphorus and 1,25-dihydroxyvitamin D levels by its actions on the kidney to reduce renal tubular reabsorption of phosphate and 1,25-dihydroxyvitamin D (calcitriol) production. Excess FGF23 has been implicated in a number of hypophosphatemic disorders, such as tumor-induced osteomalacia 16, X-linked hypophosphatemia 17 and autosomal dominant hypophosphatemic rickets 18.
Individuals with familial tumoral calcinosis develop ectopic calcifications called tumoral calcinosis and/or diaphyseal hyperostosis, which manifests clinically in the long bones as diaphyseal pain and swelling 19. Characteristic dental findings of familial tumoral calcinosis include shortened roots with dilacerations, thistle-shaped dental pulps, pulp chamber and root canal obliteration, and pulp stones 20. In addition, some patients experience systemic inflammation.
Familial tumoral calcinosis, hyperphosphatemic familial tumoral calcinosis or hyperostosis-hyperphosphatemia syndrome, is an autosomal recessive disease and to date, causal mutations in 3 genes have been identified: FGF23 (12p13.3) 21, polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3) (2q24-q31) 22 and Klotho (KL) (13q12) 23. Mutations in FGF23 and GALNT3 result in premature cleavage of biologically active intact FGF23 (iFGF23) into inactive fragments, while mutations in KL interrupt FGF23 signaling, causing FGF23 resistance. Lack of iFGF23 results in hyperphosphatemia, due to increased tubular reabsorption of phosphate and elevated or inappropriately normal 1,25-dihydroxyvitamin D production, which promotes gastrointestinal absorption of phosphorus and calcium. The net effect is an increase in the calcium and phosphate product, leading to tumoral calcinosis.
Figure 1. Tumoral calcinosis
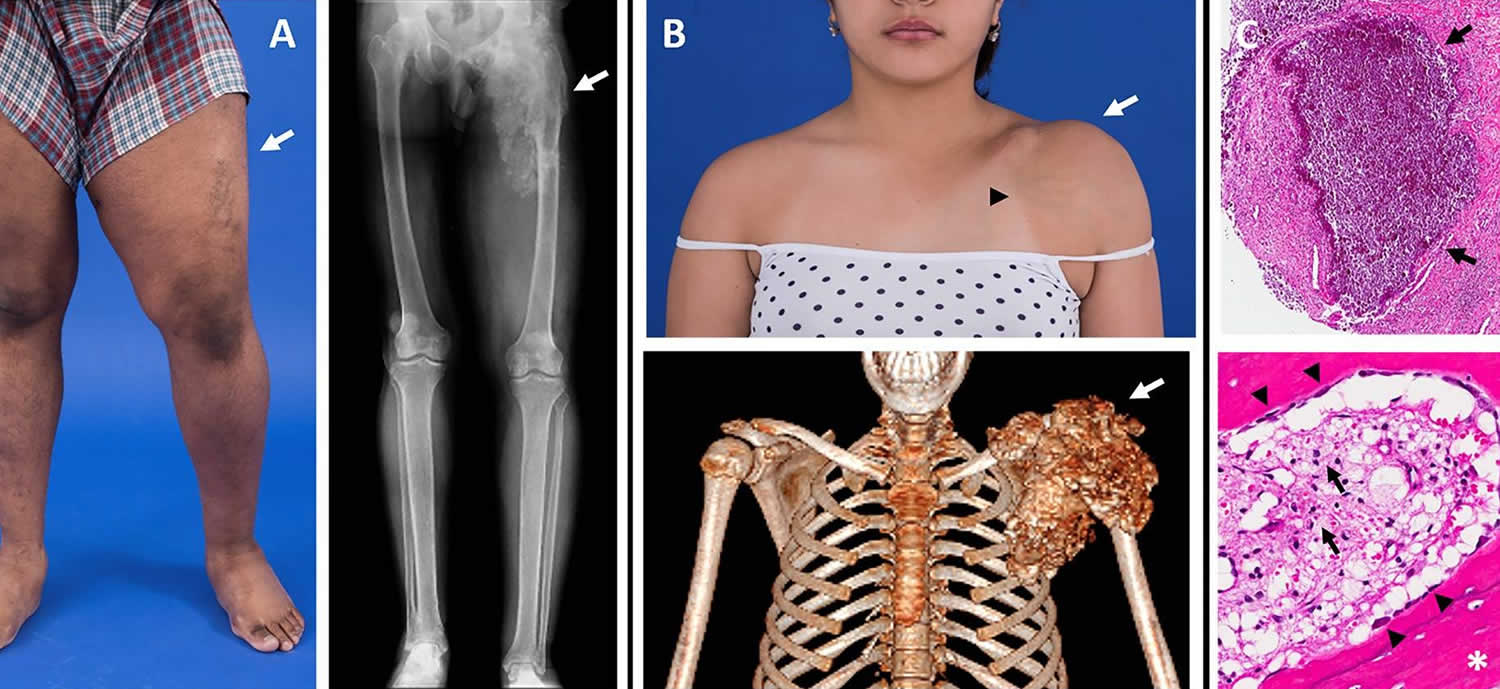
Footnote: Images of tumoral calcinosis lesions. (A) Photograph (left panel) of a patient with swelling and decreased range-of-motion of the left lower extremity (arrow). The corresponding radiograph (right panel) shows a large area of tumoral calcinosis involving the left proximal femur. (B) Photograph (upper panel) of a patient with painful swelling and reduced range-of-motion of the left shoulder (white arrow). Note the overlying skin pigmentation and increased vascularity (black arrowhead). A corresponding three-dimensional computed tomography scan shows a large calcified mass involving the left shoulder (arrow). (C) Hematoxylin and eosin stained sections from resected tumoral calcinosis lesions. The upper panel shows a subcutaneous calcification (arrows). The lower panel shows heterotopic ossification (white asterisk) with active osteoblasts laying down new bone (black arrowheads). Note the presence of chronic inflammation with visible foamy macrophages (black arrows).
[Source 24 ]Figure 2. Familial tumoral calcinosis
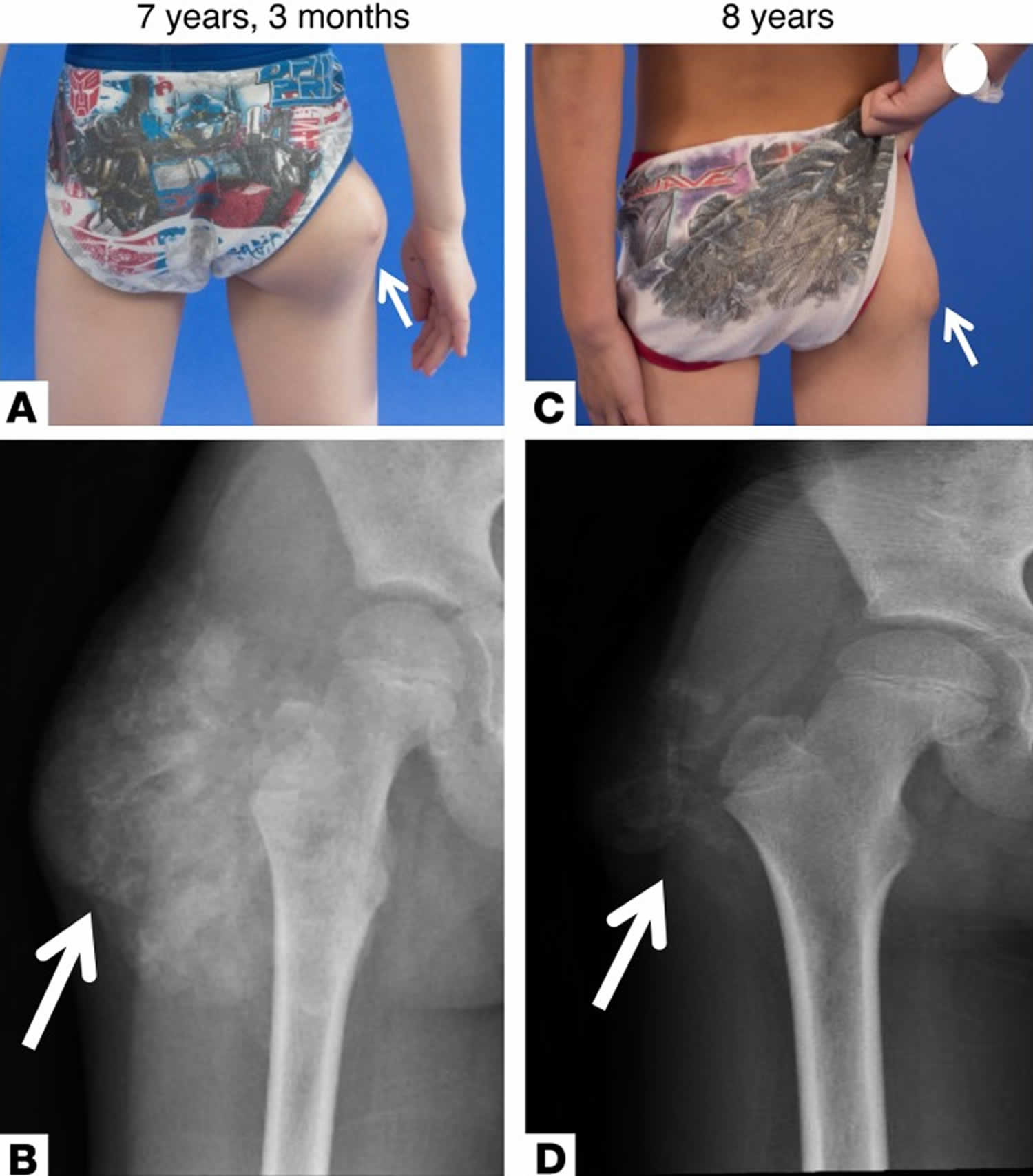
Footnote: Clinical presentation and radiographs of patient with tumoral calcinosis. (A) Tumoral calcinosis (arrows) of the lateral right hip at initial presentation at 7 years, 3 months old. (B) Radiograph of the lesion revealed a soft tissue mass with amorphous calcifications around the right greater trochanter consistent with tumoral calcinosis. Repeat evaluation 9 months after initial presentation while on phosphate-lowering medications (sevelamer and acetazolamide) and a low-phosphate diet showed decrease in the size of the tumoral calcinosis (arrows) on physical exam (C) and repeat radiograph (D).
[Source 13 ]Figure 3. Hyperostosis
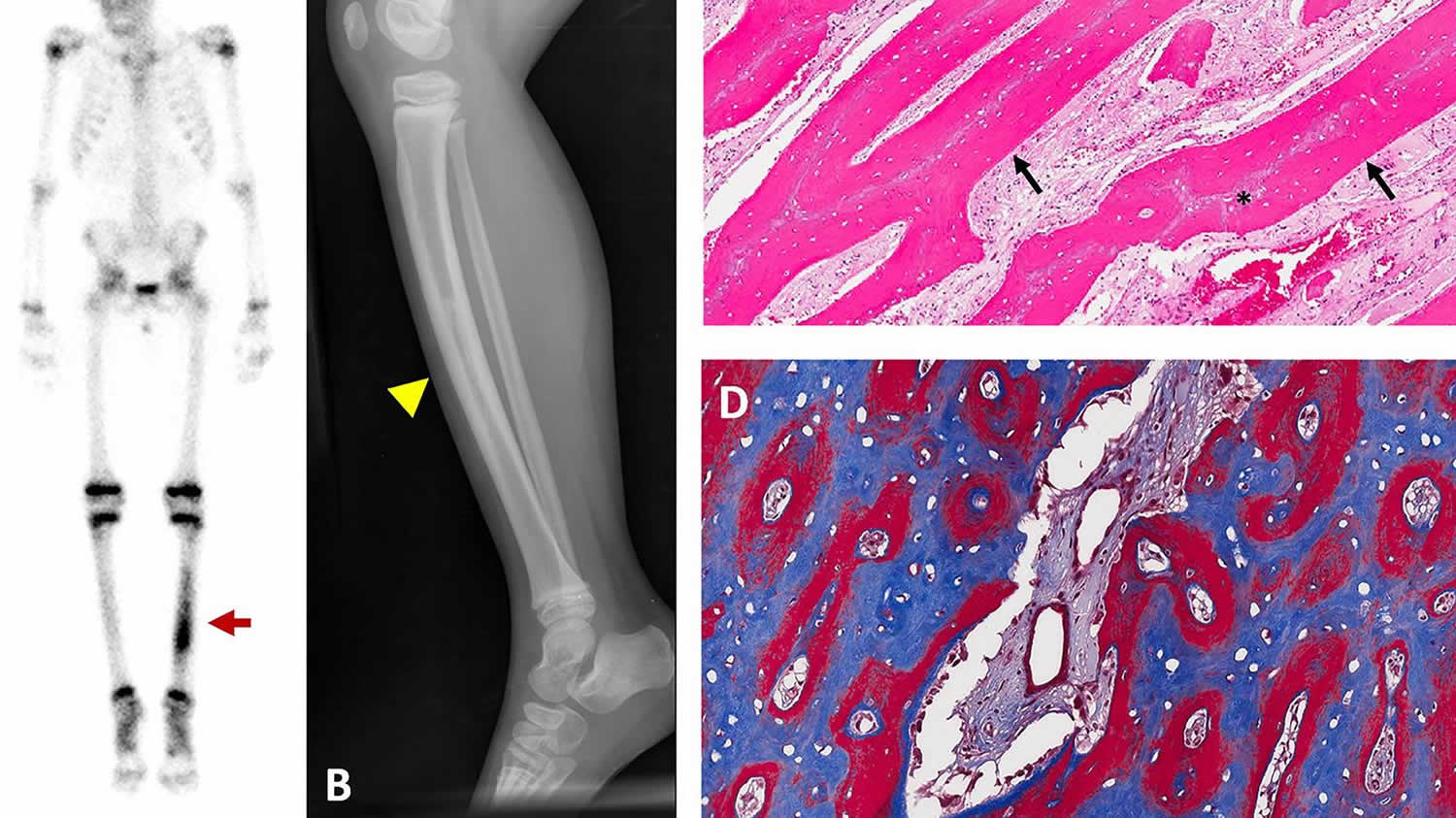
Footnote: Representative images of hyperostosis. Images are from a 4-year-old girl who presented with pain and tenderness over her tibia. (A) Technetium-99 bone scintigraphy shows increased uptake in the bilateral tibias, greater on the left (red arrow). (B) Radiograph of the left tibia shows thickened cortices with patchy radio-opacities in the medullary canal (yellow arrowhead). (C) Hematoxylin and Eosin stained section of periosteal bone from a tibial biopsy shows sheets of mineralized lamellar bone (arrows) replacing central areas of woven bone (asterisks). (D) Goldner trichrome staining in an undecalcified section reveals mature lamellar bone in blue and woven bone in red.
[Source 24 ]Figure 4. Hyperphosphatemic tumoral calcinosis
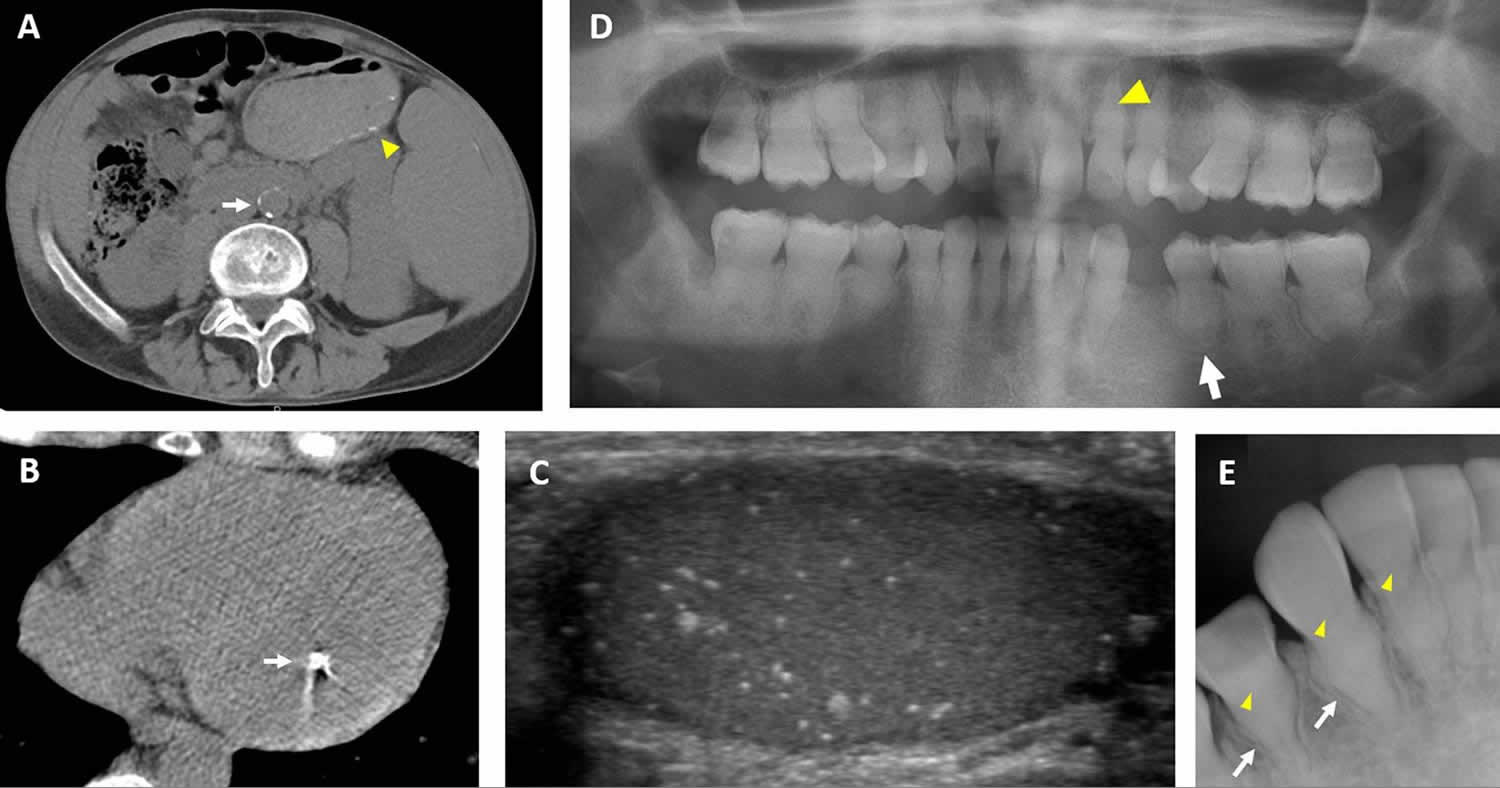
Footnote: Clinical images of extraskeletal features in hyperphosphatemic tumoral calcinosis. (A) Computed tomography scan from a 36-year-old man demonstrates calcification in the abdominal aorta (white arrow). Small submucosal bowel calcifications are also visible (yellow arrowhead). (B) Computed tomography scan of the heart in a 29-year-old man demonstrates a calcified lesion in the papillary muscle (arrow). (C) Testicular ultrasound from a 32-year-old man showing diffuse microlithiasis. (D) Panoramic dental radiograph from an 18-year-old woman shows teeth with short, bulbous roots (white arrow) and obliteration of dental pulp (yellow arrowhead). (E) Periapical dental radiograph from a 12-year-old girl shows thistle-shaped root (white arrows) and obliteration of dental pulp (yellow arrowheads).
[Source 24 ]Tumoral calcinosis causes
Familial tumoral calcinosis is caused by genetic mutations in the FGF23, GALNT3 or KLOTHO gene resulting in inactivation of the phosphaturic protein fibroblast growth factor 23 (FGF23) resulting in deficiency of active, intact FGF23 or a defect in its signaling (Table 1). FGF23 lowers serum phosphorus and 1,25-dihydroxyvitamin D levels by its actions on the kidney to reduce renal tubular reabsorption of phosphate and 1,25-dihydroxyvitamin D (calcitriol) production. FGF23 acts via FGF receptor 1 (FGFR1) coupled with the coreceptor αKlotho to reduce expression of sodium phosphate cotransporters (NaPi-2a and -2c) and renal 25-hydroxyvitamin D 1-α-hydroxylase 15. Familial tumoral calcinosis is typically inherited in an autosomal recessive pattern 10. Biallelic inactivating variants in GALNT3 cause hyperphosphatemic familial tumoral calcinosis by impairing the O-glycosylation of FGF23, leading to its cleavage and inactivation 25. Additionally, pathogenic variants in FGF23 may lead to increased cleavage and decreased circulating intact FGF23 26. Biochemically, patients with GALNT3 and FGF23 variants demonstrate hyperphosphatemia, increased tubular reabsorption of phosphate, inappropriately normal or frankly elevated 1,25-dihydroxyvitamin D (calcitriol), normal or decreased intact FGF23 and markedly elevated C-terminal FGF23 10. Patients may also have high-normal blood calcium and low-normal parathyroid hormone (PTH) levels, secondary to elevated 1,25-dihydroxyvitamin D (calcitriol) with increased intestinal calcium absorption. Recessive variants in the KLOTHO gene, which encodes the co-receptor KLOTHO, have also been shown to cause hyperphosphatemic familial tumoral calcinosis due to FGF23 resistance 27, with elevations in both intact and C-terminal FGF23. Tumoral calcinosis has been reported in one young child with Hartsfield Syndrome due to a heterozygous inactivating variant in FGFR1; however, the degree of hyperphosphatemia and FGF23 resistance in that patient is unclear 28. Interestingly, autoantibodies targeting FGF23 were found to be responsible for the development of hyperphosphatemic tumoral calcinosis in a patient with no identified genetic etiology and subsequent development of type 1 diabetes 13, resulting in an acquired form of FGF23 resistance due to decreased binding of FGF23 to its receptor. Accordingly, in this patient, FGF23 levels were elevated.
The hyperphosphatemia and high-normal calcium seen with intact FGF23 deficiency or resistance leads to an increased calcium and phosphate product, which likely contributes to ectopic calcifications. Calcifications often develop in areas of inflammation, tissue hypoxia, or repetitive trauma, although it is unclear what exactly precipitates their formation.
Table 1. Causes of familial tumoral calcinosis or hyperphosphatemic tumoral calcinosis
| Locus | Gene | OMIM | Inheritance | iFGF23 | cFGF23 | Associated features | |
| Hyperphosphatemic familial tumoral calcinosis type 1 | 2q24.3 | GALNT3 | 211900 | Recessive | Low/normal | High | |
| Hyperphosphatemic familial tumoral calcinosis type 2 | 12p13.32 | FGF23 | 617993 | Recessive | Low/normal | High | |
| Hyperphosphatemic familial tumoral calcinosis type 3 | 13q13.1 | KLOTHO | 617994 | Recessive | High | High | Hyperparathyroidism |
| Autoimmune | NA | NA | NA | Acquired | High | High | Type 1 diabetes mellitus |
Tumoral calcinosis signs and symptoms
Familial tumoral calcinosis mainly manifests in childhood or adolescence as painless, firm, tumor-like masses around the joints that may lead to discomfort, pain and joint movement limitation specially when large in size 29. Growth of such lesions is mostly slow and progressive in nature over several years 30. Sometimes, ulceration of the overlying skin occurs with superadded secondary infection 31. Huge bilateral cases of tumoral calcinosis though rare, have been described in the literature 32.
Tumoral calcinosis symptoms:
- mass or swelling typically around joints
- pain secondary to compression of normal surrounding structures
The most common locations are (in descending order of frequency) 33:
- hip
- elbow
- shoulder
- foot
- wrist.
Inflammatory disease
Patients may exhibit clinical signs of systemic inflammation, including recurrent fevers, fatigue, anemia, and polyarthritis, often accompanied by increased serum levels of C-reactive protein (CRP) and/or erythrocyte sedimentation rate (ESR) 11. The etiology of inflammation in hyperphosphatemic familial tumoral calcinosis is unknown but is speculated to be related to macrophagic engulfment of hydroxyapatite crystals in calcific lesions 11. Preliminary support for this concept is provided by the clinical literature: (1) all reported patients with systemic inflammation also had large calcifications 11, and (2) two patients who had resolution of calcified lesions after treatment also had concomitant improvement in serum inflammatory markers and symptoms of inflammation 34. Further investigation is needed to define the relationship between tumoral calcinosis lesions and inflammation, as well as the clinical sequelae of chronic inflammation in patients with hyperphosphatemic familial tumoral calcinosis.
Hyperostosis
Hyperostosis is a unique and poorly understood characteristic of hyperphosphatemic familial tumoral calcinosis (Figure 3). Patients present with pain and tenderness overlying the diaphyseal regions of long bones, often accompanied by edema, erythema, and warmth 35. The tibias are most commonly affected, but multiple sites may be involved, including the ulnas, radii, and metacarpals 36. Symptoms may onset acutely with a variable duration and may recur episodically, leading to significant pain and functional impairment. A misdiagnosis of osteomyelitis is frequent, particularly if hyperostosis is the initial presenting feature.
Radiographs often show pronounced periosteal reaction with hypermineralized cortical bone and patchy sclerotic areas involving the medullary canal (Figure 3B). Lesions are typically active on nuclear medicine scan (Figure 3A). Biopsies typically reveal areas of reactive bone with fibroblastic stroma, which are infiltrated with inflammatory polymorphonuclear cells and lymphocytes (Figures 3C,D) 11.
Eye involvement
Ocular manifestations are an uncommon but distinct feature of hyperphosphatemic familial tumoral calcinosis. Calcifications may involve the eyelids and/or conjunctiva, which may present with eye itching and irritation 37. Corneal calcifications have led to band keratopathy in several patients 37. Retinal angioid streaks have been reported 38, likely arising from calcification of the elastin-rich membrane between the retina and choriocapillaris 39. One patient with hyperphosphatemic familial tumoral calcinosis developed sudden vision loss as a result of a subretinal hemorrhage from choroidal neovascularization of a retinal angioid streak 40.
Dental involvement
Dental pathology is one of the most penetrant features of hyperphosphatemic familial tumoral calcinosis 10. Prominent dental abnormalities such as shortening of roots and partial obliteration of dental pulp are commonly seen on panoramic and periapical radiographs (Figures 4D,E). Findings are similar to dentin dysplasia, including short bulbous teeth with abnormal calcifications 11. While generalized short roots can be seen in numerous syndromes, teeth of patients with hyperphosphatemic familial tumoral calcinosis reveal a unique thistle-shaped root with gross enlargement in the coronal third of the root and acutely tapering apical third 41. Abnormal curvature of the dental roots, known as dilacerations, is seen in some patients 11. Additionally, varying degrees of obliteration of the pulp chamber and root canal is commonly observed 11. The pulp, normally an unmineralized oral tissue composed of vascular, nervous, and connective tissue, are often obstructed with pulp stones in hyperphosphatemic familial tumoral calcinosis 11. Complete obliteration of the pulp space is also observed in patients 42. The presence of abnormal calcification in the pulp space can hinder root canal therapies 43. Currently, the etiology of dental pulp obliteration and root dysmorphology in hyperphosphatemic familial tumoral calcinosis is unknown. It is important for dentists to be aware of the unique presentation of pulpal obliteration and thistle-shaped roots because the dental radiographic findings can be the first sign of the disease in patients without other systemic manifestations 44.
Clinically, the tooth morphology of patients with hyperphosphatemic familial tumoral calcinosis appear normal in color, size, and shape 45. Most studies have described a healthy oral mucosa in patients with hyperphosphatemic familial tumoral calcinosis 6. Although enamel hypoplasia has been observed in several case reports, enamel is rarely affected in most patients 46. Bilateral maxillary and mandibular tori have been reported in some patients, which can interfere with tongue movement and speech 47. The rare presence of enamel abnormalities can increase caries risk in hyperphosphatemic familial tumoral calcinosis; more commonly found pulp calcifications do not allow endodontic treatment of caries 46. Thus, application of dental sealants for caries prevention is suggested as a better treatment option.
Other calcifications
Calcifications may affect small and large vessels in various locations, including the aorta, iliacs, carotids, cerebral vasculature, and others (Figure 4A) 11. Patients may present with signs of peripheral vascular disease, including pain and diminished peripheral pulses, which in severe cases have necessitated amputations 48. Cardiac calcifications may include the coronary vessels or muscular structures (Figure 4B) 11. Coronary artery calcifications and systemic inflammation are both established risk factors for cardiac disease; prospective studies are needed to evaluate cardiac outcomes in patients with hyperphosphatemic familial tumoral calcinosis and determine therapeutic strategies.
Calcifications may occur in a variety of other extracutaneous tissues, including the dura 35, tongue 49, and submucosal gastrointestinal tract (Figure 4A) 11. Testicular microlithiasis was associated with decreased sperm production in one patient (Figure 4C) 50. Nephrocalcinosis has been reported, which was associated with decreased renal function in one patient 51.
Other clinical manifestations
Phenotyping studies are likely to uncover additional clinical manifestations in patients with hyperphosphatemic familial tumoral calcinosis, particularly features that are less prevalent or more subtle in their presentation. Much interest has been paid to “off-target” effects of FGF23 in other metabolic bone disorders, such as X-linked hypophosphatemia and chronic renal insufficiency, where excess FGF23 has been associated with varying effects on the cardiovascular, gastrointestinal, immune, and central nervous systems 52. Emerging evidence demonstrates that FGF23 processing is affected by inflammation and iron metabolism, which may be relevant for patients with hyperphosphatemic familial tumoral calcinosis who may demonstrate both chronic inflammation and anemia 53. Studies are needed to determine if these and other “off-target” effects affect patients with different forms of hyperphosphatemic familial tumoral calcinosis.
Tumoral calcinosis diagnosis
Diagnosis of tumoral calcinosis is mainly based on imaging modalities. Plain radiographs show the typical appearance of amorphous, multilobulated and cystic calcifications in a peri-articular location most often distributed along the extensor surfaces of large joints 33. Computed tomography (CT) helps in determining the extent and relations of individual lesions, and as a guide for surgical planning. It usually shows cystic loculi with fluid-fluid levels caused by calcium layering giving rise to “the sedimentation sign” 54. In other instances, the lesion may appear homogenous denoting decreased activity in the quiescent stage 55. Erosion or osseous destruction by adjacent soft-tissue masses is consistently absent; another hallmark of tumoral calcinosis 33. Magnetic resonance imaging (MRI) shows inhomogeneous high signal intensity on T2-weighted sequences with two patterns frequently observed; diffuse lower-signal-intensity pattern, or nodular pattern with alternating areas of high signal intensity and signal void. The lesions appear inhomogeneous with low-signal intensity on T1-weighted sequences 55.
Other conditions including calcinosis universalis, calcinosis circumscripta, calcific tendonitis, synovial osteochondromatosis, synovial sarcoma, osteosarcoma, myositis ossificans, tophaceous gout, and calcific myonecrosis can confuse both the radiologists and the clinicians regarding the nature of these lesions. This can be resolved through combining typical radiological features of tumoral calcinosis with the serum biochemical profile (including serum calcium level, serum phosphorus levels, renal function tests , serum parathormone level and 1,25-dihydroxy-vitamin D levels) 56. Detailed family, drug and past history should also be obtained.
It should be emphasized that connective tissue diseases should be excluded before settling the diagnosis as primary tumoral calcinosis specially in the setting of normal calcium and phosphorus levels. This can be achieved with a negative antinuclear, anti-Smith, anti-centromere and anti-scleroderma antibodies profile 33.
Although biopsy is better avoided for fear of infection 32. It may still be done in difficult cases to settle the diagnosis 57. Histopathological examination of tumoral calcinosis lesions after biopsy or surgical excision shows certain characteristic morphologic features differentiating it from other calcifying processes. This includes formation of the characteristic compartments, which contain liquid chalky content together with calcifications. Such compartmentalized configuration frequently remains even in the quiescent stage 58.
Tumoral calcinosis treatment
Treatment of tumoral calcinosis should be tailored according to the type of the lesion, stage of the pathology together with the site, size and relations of the lesion, as well as symptoms of the patient. Current interventions focus on managing blood phosphate, reducing pain and inflammation, and addressing calcifications and their complications.
From a pathogenetic point of view, medical treatment during the active stage maybe superior to surgery which is usually doomed with recurrence in this stage. On the other hand, surgical treatment may be more effective in the relatively quiescent stage where encapsulation occurs and hinders the ion exchange process leading to failure of phosphate depletion treatment 59. Some authors advocate a combination therapy that includes surgical excision and medical treatment as a necessity in some resistant cases 60. Alternative treatment modalities including the administration of steroids, diphosphonates, or calcitonin and radiation therapy have not proven to be effective 61.
Phosphate-lowering therapies
A low phosphate diet is recommended for patients with hyperphosphatemic familial tumoral calcinosis, although there is limited evidence that this alone is sufficient 62. As phosphate is abundant in many foods, particularly those high in protein such as dairy, nuts, and meat, consultation with a dietician may be necessary to assist with meal planning. The United States Recommended Daily Allowance for phosphate ranges from 500 to 1,250 mg/day in children and adolescents and 700 mg/day in adults 63. However, because of the high protein diet in the United States, most individuals consume at least twice the Recommended Daily Allowance 64. Similar to patients with hyperphosphatemia secondary to renal insufficiency, patients with hyperphosphatemic familial tumoral calcinosis are typically recommended to restrict phosphate intake to 600–800 mg/day (less in young children), which is difficult for many to achieve 65.
Medications that inhibit intestinal absorption of dietary phosphate have been tried in patients with hyperphosphatemic familial tumoral calcinosis with varying success, including sevelamer, lanthanum, and aluminum hydroxide 11. To be effective, phosphate binders should be given with all meals and snacks. Common side effects include constipation, nausea, and abdominal pain; in rare cases, intestinal obstruction or perforation can occur. Aluminum toxicity is unlikely as renal function is usually normal in patients with hyperphosphatemic familial tumoral calcinosis, however, this potential complication should always be considered when choosing this therapy. Lanthanum is a soft metal which is radiopaque and, while benign, can be mistaken for intestinal foreign bodies on abdominal radiographs 66. Calcium salts, which are often used to lower blood phosphate in other disorders, should be avoided in patients with hyperphosphatemic familial tumoral calcinosis, as these could potentially increase the calcium-phosphate product and worsen calcifications. As hyperphosphatemic familial tumoral calcinosis is associated with high 1,25 vitamin D, vitamin D supplements should never be administered to these patients, even in the face of a low 25-hydroxyvitamin D or 25-OH-vitamin D (calcidiol) level.
Acetazolamide, a carbonic anhydrase inhibitor which induces a proximal tubular acidosis, is commonly used in hyperphosphatemic familial tumoral calcinosis. Efficacy is variable, with some reporting a decrease in blood phosphate, tubular reabsorption of phosphate, and calcific tumors with others reporting no obvious benefit 67. It has been suggested that its mode of action on calcifications is not through promoting renal phosphate excretion, but rather by increasing calcium-phosphate solubility through lowered serum pH 67. Serum bicarbonate should be monitored periodically; while the lower acceptable limit of bicarbonate is not known, a level of 18–20 mmol/L is likely to be tolerated without significant complications.
Probenecid is a uricosuric agent that also increases renal phosphate excretion and has been tried in tumoral calcinosis 11. It should be used with caution when co-administered with other medications, as probenecid can increase the half-life of many drugs (e.g., certain antibiotics), leading to potential toxicity. Nicotinamide, a drug which downregulates sodium-phosphate co-transporters in the kidney and intestine, was shown to decrease progression of calcifications in Galnt3 knock-out mice without an effect on serum phosphate levels 68. Brief treatments with niacinamide and nicotinamide have been tried in a small number of patients with hyperphosphatemic familial tumoral calcinosis 69, but there are no available long-term data.
Anti-inflammatory therapies
In patients with significant inflammatory disease, often evidenced by erythema, lesional warmth, intermittent fevers, elevated erythrocyte sedimentation rate and/or c-reactive protein, anti-inflammatory medications may be useful. Non-steroidal anti-inflammatory drugs (NSAIDS) and glucocorticoids have been reported to improve symptomatic hyperostosis 70. Blockade of interleukin-1 (IL-1) action with anakinra, an IL-1 receptor antagonist, and canakinumab, a monoclonal antibody against IL-1β, have been shown to reduce inflammation and pain and improve quality of life in a small number of patients 71; inflammatory calcifications resolved in one 11.
Anti-mineralization therapies
Sodium thiosulfate, approved for use as an antidote to cyanide poisoning, also appears to have anti-mineralization properties due to several proposed, yet unconfirmed, mechanisms such as increasing the solubility and excretion of calcium through chelation, and acting as an antioxidant to reduce inflammation 72. As a result, intravenous, oral, topical, and intralesional sodium thiosulfate has been studied in a variety of calcific disorders including nephrolithiasis 73, autoimmune calcinosis cutis 74, dermatomyositis-related tumoral calcinosis 75 and calcific uremic arteriopathy 76. In hyperphosphatemic familial tumoral calcinosis, topical application of sodium thiosulfate decreased calcifications in three patients after many months of therapy 36; intravenous and intralesional applications have not been reported in hyperphosphatemic familial tumoral calcinosis.
Surgical resection
Surgical outcomes are highly variable, with some patients experiencing recurrence of lesions or poor wound healing 11. The high rate of recurrence warrants repeated excisions 77. Given the risks, surgery is often reserved for those with severe lesions affecting activities of daily living or chronic drainage and infections.
During surgery, tumoral calcinosis lesions show a cystic nature with white and yellow chalky material formed by calcium hydroxyapatite crystals, calcium carbonate and calcium phosphate 78. The presence of a hyper-vascular region beyond the periphery of the calcified mass as proven by angiography raises the possibility that a wider surgical resection margin may lead to fewer recurrences. Confirmation of this theory is however, still needed 79. Slavin et al 59, reported that immobilization after resection may also have a role in decreasing new lesion formation in the adjacent tissues.
Huge lesions may require extensive surgical excision and reconstruction 80. Relations to important neurovascular structures may be challenging, resulting mostly in partial excision and rapid recurrence. However, partial excision of large symptomatic lesions can be helpful providing significant pain relief 81. Indications for surgical excision also include recurrent infection, ulceration, and functional impairment 82.
According to a literature review done by King et al 82, surgical complications of tumoral calcinosis excision include postoperative prolonged drainage which can lead to delayed wound healing and sinus tract formation, secondary infections caused by chronic wound problems specially with extensive disease or incomplete resection, and recurrence, which is frequent after incomplete excision and usually has a faster rate of growth.
Physical and occupational therapy
As joints are most commonly affected by tumoral calcinosis, patients often have reduced range of motion and/or pain that can severely impair activities of daily living, including walking, eating, and routine hygiene. Consultation with physiatrists and therapists are an important and often underutilized resource to ensure that patients are taught management strategies and provided with adaptive devices.
Other therapies
Given the inconsistent efficacy of the treatments listed above, there have been attempts to manage hyperphosphatemic familial tumoral calcinosis with calcium-channel blockers, bisphosphonates, ketoconozale, methotrexate, parathyroid hormone, and TNF-blockade, most without demonstrable improvement. Calcitonin appeared to lower phosphate and stabilize calcifications in one case 83. In a patient with ocular compromise due to angioid streaks, intravitreal injections of ranibizumab (an antibody to vascular endothelial growth factor) improved visual acuity 40. Immunomodulatory therapy has not been tried in the one known patient with autoimmune hyperphosphatemic tumoral calcinosis, because he has responded generally well to conventional therapy. Patients with vascular calcifications require evaluation and management by a vascular specialist and/or cardiologist. Consultation with a pain specialist may be needed in severe cases of hyperphosphatemic familial tumoral calcinosis. As with other chronic pain conditions, involvement of a mental health specialist may also be beneficial.
Future treatment options
Until gene therapy become routine practice, hormone replacement therapy with recombinant or synthetic FGF23 would be the optimal treatment for patients with hyperphosphatemic familial tumoral calcinosis due to GALNT3 or FGF23 variants. Other approaches, particularly for autoimmune and FGF23-resistant forms of tumoral calcinosis, include targeted therapies that inhibit renal phosphate reabsorption. For example, a recent study using an oral inhibitor of a renal sodium-phosphate co-transporter increased phosphate excretion and reduced serum phosphate in a murine model of chronic kidney disease 84. Whether this class of drugs will be effective in hyperphosphatemic familial tumoral calcinosis remains to be seen.
There are multiple challenges in designing interventional trials in hyperphosphatemic familial tumoral calcinosis. The development of calcified lesions is by nature an intermittent process, therefore prospective observational studies are critical to define the natural history of the disease, and to understand the phenotypic variability between individuals. Clinically relevant surrogate endpoints are another key area of need for future research. Phosphate levels and inflammatory markers are intuitive potential biomarkers, however more robust studies correlating these levels with clinical outcomes are needed. Investigation is also needed to define optimal imaging strategies to detect, monitor, and predict lesion development.
References- McClatchie S, Bremner AD. Tumoral calcinosis–an unrecognized disease. Br Med J. 1969 Jan 18;1(5637):153-5. doi: 10.1136/bmj.1.5637.142-a
- Inclan A, Leon PP, Camejo M. Tumoral calcinosis. J Am Med Ass. 1943;121:490–495.
- Tumoral calcinosis. https://radiopaedia.org/articles/tumoral-calcinosis
- Fathi I, Sakr M. Review of tumoral calcinosis: A rare clinico-pathological entity. World J Clin Cases. 2014;2(9):409-414. doi:10.12998/wjcc.v2.i9.409 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4163761
- Durant DM, Riley LH 3rd, Burger PC, McCarthy EF. Tumoral calcinosis of the spine: a study of 21 cases. Spine (Phila Pa 1976). 2001 Aug 1;26(15):1673-9. doi: 10.1097/00007632-200108010-00009
- Gal G, Metzker A, Garlick J, Gold Y, Calderon S. Head and neck manifestations of tumoral calcinosis. Oral Surg Oral Med Oral Pathol. 1994 Feb;77(2):158-66. doi: 10.1016/0030-4220(94)90279-8
- Mozaffarian G, Lafferty FW, Pearson OH. Treatment of tumoral calcinosis with phosphorus deprivation. Ann Intern Med. 1972 Nov;77(5):741-5. doi: 10.7326/0003-4819-77-5-741
- Giardina F, Sudanese A, Bertoni F, Guerra E, Paderni S. Tumoral calcinosis of the popliteal space. Orthopedics. 2004 Oct;27(10):1104-7.
- Boskey AL, Vigorita VJ, Sencer O, Stuchin SA, Lane JM. Chemical, microscopic, and ultrastructural characterization of the mineral deposits in tumoral calcinosis. Clin Orthop Relat Res. 1983 Sep;(178):258-69. DOI:10.1097/00003086-198309000-00036
- Ramnitz MS, Gafni RI, Collins MT. Hyperphosphatemic familial tumoral calcinosis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews(R). Seattle, WA: University of Washington; (1993).
- Ramnitz MS, Gourh P, Goldbach-Mansky R, Wodajo F, Ichikawa S, Econs MJ, White KE, Molinolo A, Chen MY, Heller T, Del Rivero J, Seo-Mayer P, Arabshahi B, Jackson MB, Hatab S, McCarthy E, Guthrie LC, Brillante BA, Gafni RI, Collins MT. Phenotypic and Genotypic Characterization and Treatment of a Cohort With Familial Tumoral Calcinosis/Hyperostosis-Hyperphosphatemia Syndrome. J Bone Miner Res. 2016 Oct;31(10):1845-1854. doi: 10.1002/jbmr.2870
- Polykandriotis EP, Beutel FK, Horch RE, Grünert J. A case of familial tumoral calcinosis in a neonate and review of the literature. Arch Orthop Trauma Surg. 2004 Oct;124(8):563-7. doi: 10.1007/s00402-004-0715-0
- Roberts MS, Burbelo PD, Egli-Spichtig D, et al. Autoimmune hyperphosphatemic tumoral calcinosis in a patient with FGF23 autoantibodies. J Clin Invest. 2018;128(12):5368-5373. doi:10.1172/JCI122004 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6264742
- Riminucci M, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112(5):683–692. doi: 10.1172/JCI18399
- Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004 Mar;19(3):429-35. doi: 10.1359/JBMR.0301264
- Chong WH, Molinolo AA, Chen CC, Collins MT. Tumor-induced osteomalacia. Endocr Relat Cancer. 2011;18(3):R53–R77. doi: 10.1530/ERC-11-0006
- Jonsson KB, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348(17):1656–1663. doi: 10.1056/NEJMoa020881
- White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60(6):2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x
- Narchi H. Hyperostosis with hyperphosphatemia: evidence of familial occurrence and association with tumoral calcinosis. Pediatrics. 1997;99(5):745–748. doi: 10.1542/peds.99.5.745
- Dumitrescu CE, et al. A case of familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome due to a compound heterozygous mutation in GALNT3 demonstrating new phenotypic features. Osteoporos Int. 2009;20(7):1273–1278. doi: 10.1007/s00198-008-0775-z
- Benet-Pagès A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14(3):385–390. doi: 10.1093/hmg/ddi034
- Topaz O, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36(6):579–581. doi: 10.1038/ng1358
- Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Musculoskelet Neuronal Interact. 2007 Oct-Dec;7(4):318-9.
- Boyce AM, Lee AE, Roszko KL, Gafni RI. Hyperphosphatemic Tumoral Calcinosis: Pathogenesis, Clinical Presentation, and Challenges in Management. Front Endocrinol (Lausanne). 2020;11:293. Published 2020 May 8. doi:10.3389/fendo.2020.00293 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7225339
- Ichikawa S, Lyles KW, Econs MJ. A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab. 2005 Apr;90(4):2420-3. doi: 10.1210/jc.2004-2302
- Benet-Pagès A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005 Feb 1;14(3):385-90. doi: 10.1093/hmg/ddi034
- Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007 Sep;117(9):2684-91. doi: 10.1172/JCI31330
- Prasad R, Brewer C, Burren CP. Hartsfield syndrome associated with a novel heterozygous missense mutation in FGFR1 and incorporating tumoral calcinosis. Am J Med Genet A. 2016 Aug;170(8):2222-5. doi: 10.1002/ajmg.a.37731
- Pakasa NM, Kalengayi RM. Tumoral calcinosis: a clinicopathological study of 111 cases with emphasis on the earliest changes. Histopathology. 1997 Jul;31(1):18-24. doi: 10.1046/j.1365-2559.1997.6050831.x
- Meltzer CC, Fishman EK, Scott WW Jr. Tumoral calcinosis causing bone erosion in a renal dialysis patient. Clin Imaging. 1992 Jan-Mar;16(1):49-51. doi: 10.1016/0899-7071(92)90091-m
- Geirnaerdt MJ, Kroon HM, van der Heul RO, Herfkens HF. Tumoral calcinosis. Skeletal Radiol. 1995 Feb;24(2):148-51. doi: 10.1007/BF00198081
- Farzan M, Farhoud AR. Tumoral calcinosis: what is the treatment? Report of two cases of different types and review of the literature. Am J Orthop (Belle Mead NJ). 2011 Sep;40(9):E170-6.
- Olsen KM, Chew FS. Tumoral calcinosis: pearls, polemics, and alternative possibilities. Radiographics. 2006 May-Jun;26(3):871-85. doi: 10.1148/rg.263055099
- Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, Cullen MJ, Draman MS, Conlon N, Jain A, Fedarko NS, Dasgupta B, White KE. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab. 2006 Oct;91(10):4037-42. doi: 10.1210/jc.2006-0305
- Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, Austin AM, Sorenson AH, Pejin Z, Topouchian V, Quartier P, Cormier-Daire V, Dechaux M, Malandrinou FCh, Singhellakis PN, Le Merrer M, Econs MJ. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. 2010 Apr;152A(4):896-903. doi: 10.1002/ajmg.a.33337
- Jost J, Bahans C, Courbebaisse M, Tran TA, Linglart A, Benistan K, Lienhardt A, Mutar H, Pfender E, Ratsimbazafy V, Guigonis V. Topical Sodium Thiosulfate: A Treatment for Calcifications in Hyperphosphatemic Familial Tumoral Calcinosis? J Clin Endocrinol Metab. 2016 Jul;101(7):2810-5. doi: 10.1210/jc.2016-1087
- Rafaelsen S, Johansson S, Ræder H, Bjerknes R. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet. 2014 Sep 24;15:98. doi: 10.1186/s12863-014-0098-3
- Yancovitch A, Hershkovitz D, Indelman M, Galloway P, Whiteford M, Sprecher E, Kılıç E. Novel mutations in GALNT3 causing hyperphosphatemic familial tumoral calcinosis. J Bone Miner Metab. 2011 Sep;29(5):621-5. doi: 10.1007/s00774-011-0260-1
- Chatziralli I, Saitakis G, Dimitriou E, Chatzirallis A, Stoungioti S, Theodossiadis G, Theodossiadis P. ANGIOID STREAKS: A Comprehensive Review From Pathophysiology to Treatment. Retina. 2019 Jan;39(1):1-11. doi: 10.1097/IAE.0000000000002327
- McGrath E, Harney F, Kinsella F. An ocular presentation of familial tumoral calcinosis. BMJ Case Rep. 2010 Sep 20;2010:bcr0520103044. doi: 10.1136/bcr.05.2010.3044
- Dumitrescu CE, Kelly MH, Khosravi A, Hart TC, Brahim J, White KE, Farrow EG, Nathan MH, Murphey MD, Collins MT. A case of familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome due to a compound heterozygous mutation in GALNT3 demonstrating new phenotypic features. Osteoporos Int. 2009 Jul;20(7):1273-8. doi: 10.1007/s00198-008-0775-z
- Naikmasur V, Guttal K, Bhargava P, Burde K, Sattur A, Nandimath K. Tumoral calcinosis with dental manifestations–a case report. Dent Update. 2008 Mar;35(2):134-6, 138. doi: 10.12968/denu.2008.35.2.134
- Stewart GG. Gaining access to calcified canals. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995 Jun;79(6):764-8. doi: 10.1016/s1079-2104(05)80314-2
- Yamamoto H, Ramos-Molina B, Lick AN, Prideaux M, Albornoz V, Bonewald L, Lindberg I. Posttranslational processing of FGF23 in osteocytes during the osteoblast to osteocyte transition. Bone. 2016 Mar;84:120-130. doi: 10.1016/j.bone.2015.12.055
- Vieira AR, Lee M, Vairo F, Loguercio Leite JC, Munerato MC, Visioli F, D’Ávila SR, Wang SK, Choi M, Simmer JP, Hu JC. Root anomalies and dentin dysplasia in autosomal recessive hyperphosphatemic familial tumoral calcinosis (HFTC). Oral Surg Oral Med Oral Pathol Oral Radiol. 2015 Dec;120(6):e235-9. doi: 10.1016/j.oooo.2015.05.006
- Favia G, Lacaita MG, Limongelli L, Tempesta A, Laforgia N, Cazzolla AP, Maiorano E. Hyperphosphatemic familial tumoral calcinosis: odontostomatologic management and pathological features. Am J Case Rep. 2014 Dec 24;15:569-75. doi: 10.12659/AJCR.892113
- Yilanci Hoa N, Ozbek M, Celik HH. Dental findings of hyperphosphatemic familial tumoral calcinosis. Oral Radiol. (2017) 33:65–70. 10.1007/s11282-016-0237-9
- Shah A, Miller CJ, Nast CC, Adams MD, Truitt B, Tayek JA, Tong L, Mehtani P, Monteon F, Sedor JR, Clinkenbeard EL, White K, Mehrotra R, LaPage J, Dickson P, Adler SG, Iyengar SK. Severe vascular calcification and tumoral calcinosis in a family with hyperphosphatemia: a fibroblast growth factor 23 mutation identified by exome sequencing. Nephrol Dial Transplant. 2014 Dec;29(12):2235-43. doi: 10.1093/ndt/gfu324
- MCPHAUL JJ Jr, ENGEL FL. Heterotopic calcification, hyperphosphatemia and angioid streaks of the retina. Am J Med. 1961 Sep;31:488-92. doi: 10.1016/0002-9343(61)90131-0
- Campagnoli MF, Pucci A, Garelli E, Carando A, Defilippi C, Lala R, Ingrosso G, Dianzani I, Forni M, Ramenghi U. Familial tumoral calcinosis and testicular microlithiasis associated with a new mutation of GALNT3 in a white family. J Clin Pathol. 2006 Apr;59(4):440-2. doi: 10.1136/jcp.2005.026369
- Chefetz I, Heller R, Galli-Tsinopoulou A, Richard G, Wollnik B, Indelman M, Koerber F, Topaz O, Bergman R, Sprecher E, Schoenau E. A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum Genet. 2005 Nov;118(2):261-6. doi: 10.1007/s00439-005-0026-8
- Bacchetta J, Bardet C, Prié D. Physiology of FGF23 and overview of genetic diseases associated with renal phosphate wasting. Metabolism. 2020 Feb;103S:153865. doi: 10.1016/j.metabol.2019.01.006
- Richter B, Faul C. FGF23 Actions on Target Tissues-With and Without Klotho. Front Endocrinol (Lausanne). 2018 May 2;9:189. doi: 10.3389/fendo.2018.00189
- Hug I, Gunçaga J. Tumoral calcinosis with sedimentation sign. Br J Radiol. 1974 Oct;47(562):734-6. doi: 10.1259/0007-1285-47-562-734.
- Martinez S, Vogler JB 3rd, Harrelson JM, Lyles KW. Imaging of tumoral calcinosis: new observations. Radiology. 1990 Jan;174(1):215-22. doi: 10.1148/radiology.174.1.2294551
- Huang YT, Chen CY, Yang CM, Yao MS, Chan WP. Tumoral calcinosis-like metastatic calcification in a patient on renal dialysis. Clin Imaging. 2006 Jan-Feb;30(1):66-8. doi: 10.1016/j.clinimag.2005.06.024
- Petscavage JM, Richardson ML. Tumoral calcinosis mimicking recurrent osteosarcoma. Radiol Case Rep. 2015 Nov 6;4(4):336. doi: 10.2484/rcr.v4i4.336
- Slavin RE, Wen J, Barmada A. Tumoral calcinosis–a pathogenetic overview: a histological and ultrastructural study with a report of two new cases, one in infancy. Int J Surg Pathol. 2012 Oct;20(5):462-73. doi: 10.1177/1066896912444925
- Slavin RE, Wen J, Kumar D, Evans EB. Familial tumoral calcinosis. A clinical, histopathologic, and ultrastructural study with an analysis of its calcifying process and pathogenesis. Am J Surg Pathol. 1993 Aug;17(8):788-802.
- Hamada J, Tamai K, Ono W, Saotome K. Uremic tumoral calcinosis in hemodialysis patients: clinicopathological findings and identification of calcific deposits. J Rheumatol. 2006 Jan;33(1):119-26.
- Gregosiewicz A, Warda E. Tumoral calcinosis: successful medical treatment. A case report. J Bone Joint Surg Am. 1989 Sep;71(8):1244-9.
- Carmichael KD, Bynum JA, Evans EB. Familial tumoral calcinosis: a forty-year follow-up on one family. J Bone Joint Surg Am. (2009) 91:664–71. 10.2106/JBJS.G.01512
- Institute of Medicine Standing Committee on the Scientific Evaluation of Dietary Reference I The National Academies Collection: Reports Funded by National Institutes of Health. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, DC: National Academies Press (US); National Academy of Sciences; (1997).
- Calvo MS, Lamberg-Allardt CJ. Phosphorus. Adv Nutr. (2015) 6:860–2. 10.3945/an.115.008516
- Ketteler M, Block GA, Evenepoel P, Fukagawa M, Herzog CA, McCann L, et al. . Diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder: synopsis of the kidney disease: improving global outcomes 2017. Clinical practice guideline update. Ann Intern Med. (2018) 168:422–30. 10.7326/M17-2640
- Galo J, Madrid B, Kupin W. Lanthanum-induced radiopaque intestinal precipitates: a potential cause of intestinal foreign bodies. Case Rep Nephrol. (2019) 2019:1298674. 10.1155/2019/1298674
- Finer G, Price HE, Shore RM, White KE, Langman CB. Hyperphosphatemic familial tumoral calcinosis: response to acetazolamide and postulated mechanisms. Am J Med Genet A. (2014) 164a:1545–9. 10.1002/ajmg.a.36476
- Reilly AM, Gray AK, Moe SM, Ichikawa S. Nicotinamide treatment in a murine model of familial tumoral calcinosis reduces serum Fgf23 and raises heart calcium. Bone. (2014) 67:139–44. 10.1016/j.bone.2014.06.036
- Claramunt-Taberner D, Bertholet-Thomas A, Carlier MC, Dijoud F, Chotel F, Silve C, et al. . Hyperphosphatemic tumoral calcinosis caused by FGF23 compound heterozygous mutations: what are the therapeutic options for a better control of phosphatemia? Pediatr Nephrol. (2018) 33:1263–7. 10.1007/s00467-018-3945-z
- Masi L, Beltrami G, Ottanelli S, Franceschelli F, Gozzini A, Zonefrati R, et al. . Human preosteoblastic cell culture from a patient with severe tumoral calcinosis-hyperphosphatemia due to a new GALNT3 gene mutation: study of in vitro mineralization. Calcif Tissue Int. (2015) 96:438–52. 10.1007/s00223-015-9974-8
- Dauchez A, Souffir C, Quartier P, Baujat G, Briot K, Roux C. Hyperphosphatemic familial tumoral calcinosis with Galnt3 mutation: transient response to anti-interleukin-1 treatments. JBMR Plus. (2019) 3:e10185. 10.1002/jbm4.10185
- O’Neill WC. Sodium thiosulfate: mythical treatment for a mysterious disease? Clin J Am Soc Nephrol. (2013) 8:1068–9. 10.2215/CJN.04990513
- Yatzidis H. Successful sodium thiosulphate treatment for recurrent calcium urolithiasis. Clin Nephrol. (1985) 23:63–7.
- Ma JE, Ernste FC, Davis MDP, Wetter DA. Topical sodium thiosulfate for calcinosis cutis associated with autoimmune connective tissue diseases: the Mayo Clinic experience, 2012-2017. Clin Exp Dermatol. (2019) 44:e189–e92. 10.1111/ced.13782
- Goossens J, Courbebaisse M, Caudron E, Bahans C, Vacquerie V, Melchior J, et al. . Efficacy of intralesional sodium thiosulfate injections for disabling tumoral calcinosis: two cases. Semin Arthritis Rheum. (2017) 47:451–5. 10.1016/j.semarthrit.2017.05.013
- Nigwekar SU, Brunelli SM, Meade D, Wang W, Hymes J, Lacson E, Jr. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. (2013) 8:1162–70. 10.2215/CJN.09880912
- Kirk TS, Simon MA. Tumoral calcinosis. Report of a case with successful medical management. J Bone Joint Surg Am. 1981 Sep;63(7):1167-9.
- Smack D, Norton SA, Fitzpatrick JE. Proposal for a pathogenesis-based classification of tumoral calcinosis. Int J Dermatol. 1996 Apr;35(4):265-71. doi: 10.1111/j.1365-4362.1996.tb02999.x
- Neeman Z, Wood BJ. Angiographic findings in tumoral calcinosis. Clin Imaging. 2003 May-Jun;27(3):184-6. doi: 10.1016/s0899-7071(02)00523-5
- Lykoudis EG, Seretis K, Ristanis S. Huge recurrent tumoral calcinosis needing extensive excision and reconstruction: report of a rare case and brief literature review. Aesthetic Plast Surg. 2012 Oct;36(5):1194-7. doi: 10.1007/s00266-012-9923-0
- Tezelman S, Siperstein AE, Duh QY, Clark OH. Tumoral calcinosis. Controversies in the etiology and alternatives in the treatment. Arch Surg. 1993 Jul;128(7):737-44; discussion 744-5. doi: 10.1001/archsurg.1993.01420190027004
- King JJ, Brennan KB, Crawford EA, Fox EJ, Ogilvie CM. Surgical complications associated with extensive tumoral calcinosis. Am J Orthop (Belle Mead NJ). 2011 May;40(5):247-52.
- Candrina R, Cerudelli B, Braga V, Salvi A. Effects of the acute subcutaneous administration of synthetic salmon calcitonin in tumoral calcinosis. J Endocrinol Invest. (1989) 12:55–7. 10.1007/BF03349921
- Thomas L, Xue J, Murali SK, Fenton RA, Dominguez Rieg JA, Rieg T. Pharmacological Npt2a inhibition causes phosphaturia and reduces plasma phosphate in mice with normal and reduced kidney function. J Am Soc Nephrol. (2019) 30:2128–39. 10.1681/ASN.2018121250





{kind=link}