

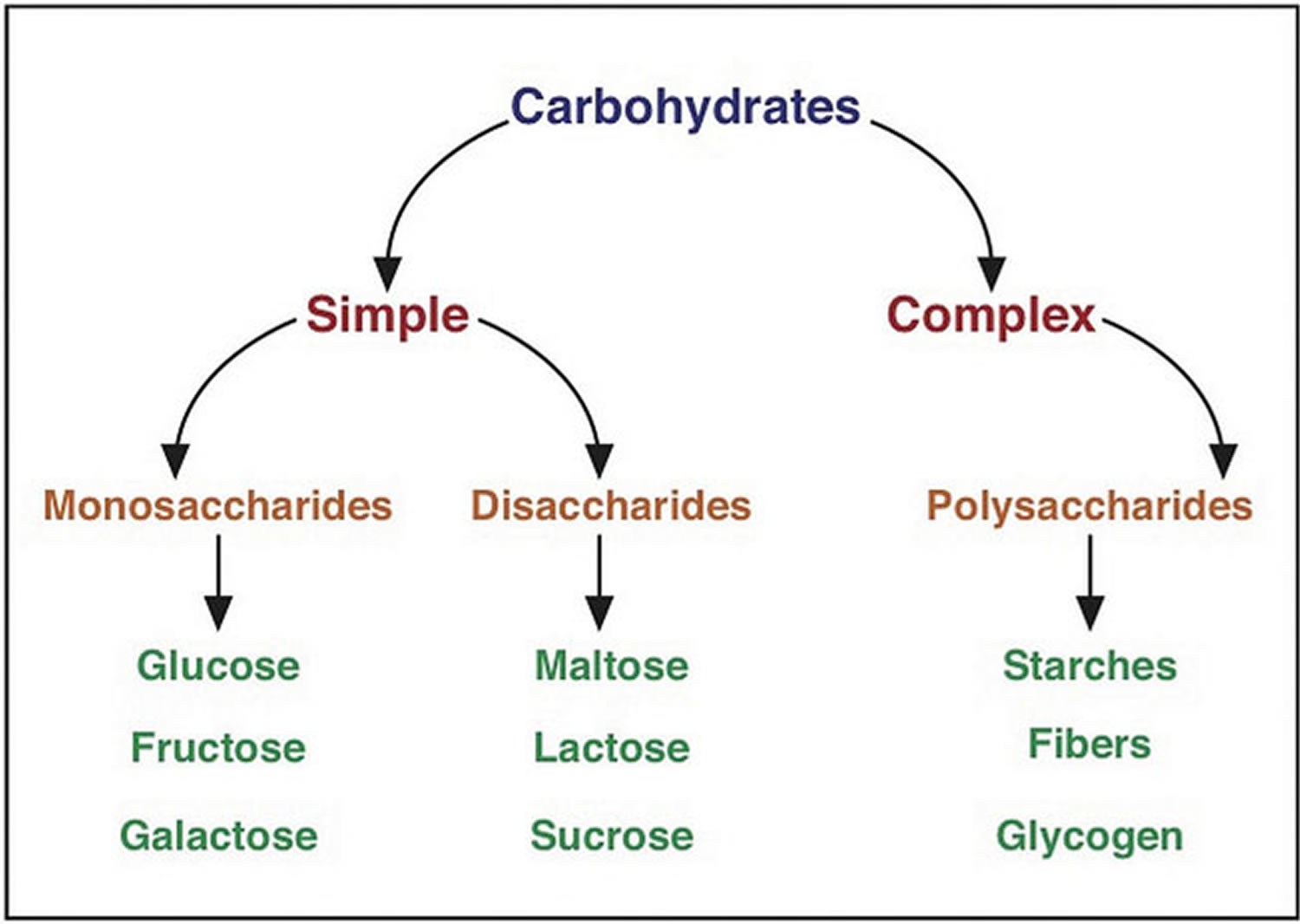
What are Carbohydrates
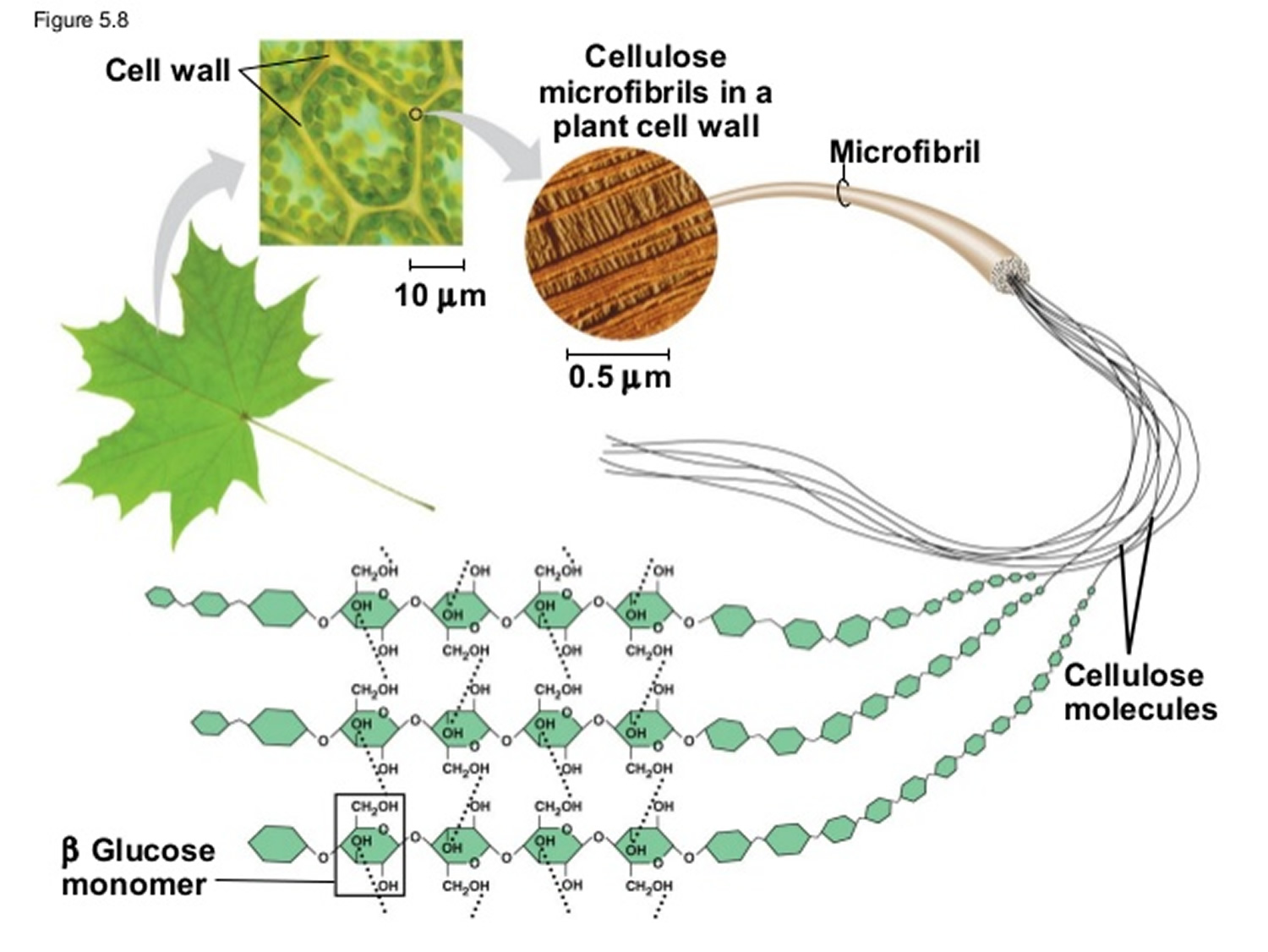
Carbohydrates are by far the most abundant organic molecules found in nature and nearly all organisms synthesize and metabolize carbohydrates 1. The term carbohydrate arose from the fact that most simple sugars have the empirical formula CnH2nOn, where n is ≥ 3, suggesting that carbon atoms are in some way combined with water. Chemists referred to these compounds as “hydrates of carbon” or “carbohydrates” 1. Glucose, for example, is a common monosaccharide that is oxidized to form carbon dioxide and water, providing energy for cellular processes such as protein synthesis, movement and transport. Plants and animals link numerous glucose molecules together to form large energy storing molecules such as starch and glycogen. However, glucose molecules may be linked to form a variety of other macromolecules. Cellulose is a component of the cell wall in plants and it is composed of glucose molecules linked together through β-1,4 glycosidic bonds. The glucose monomers in starch, on the other hand, are linked through α-1,4 glycosidic bonds, and the glycosidic bonds of glycogen are α-1,4 and α-1,6 1.
Figure 1. Glucose
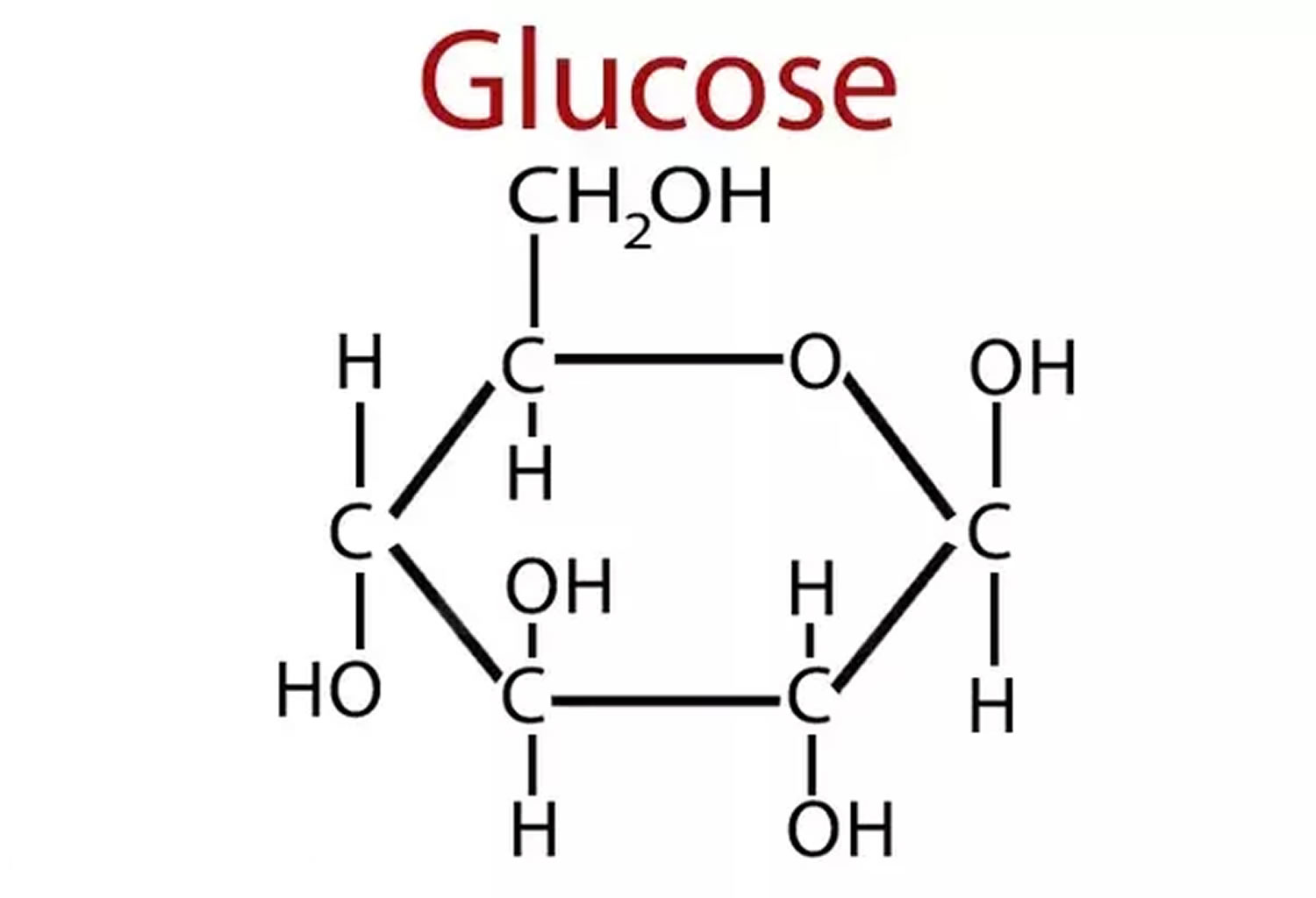
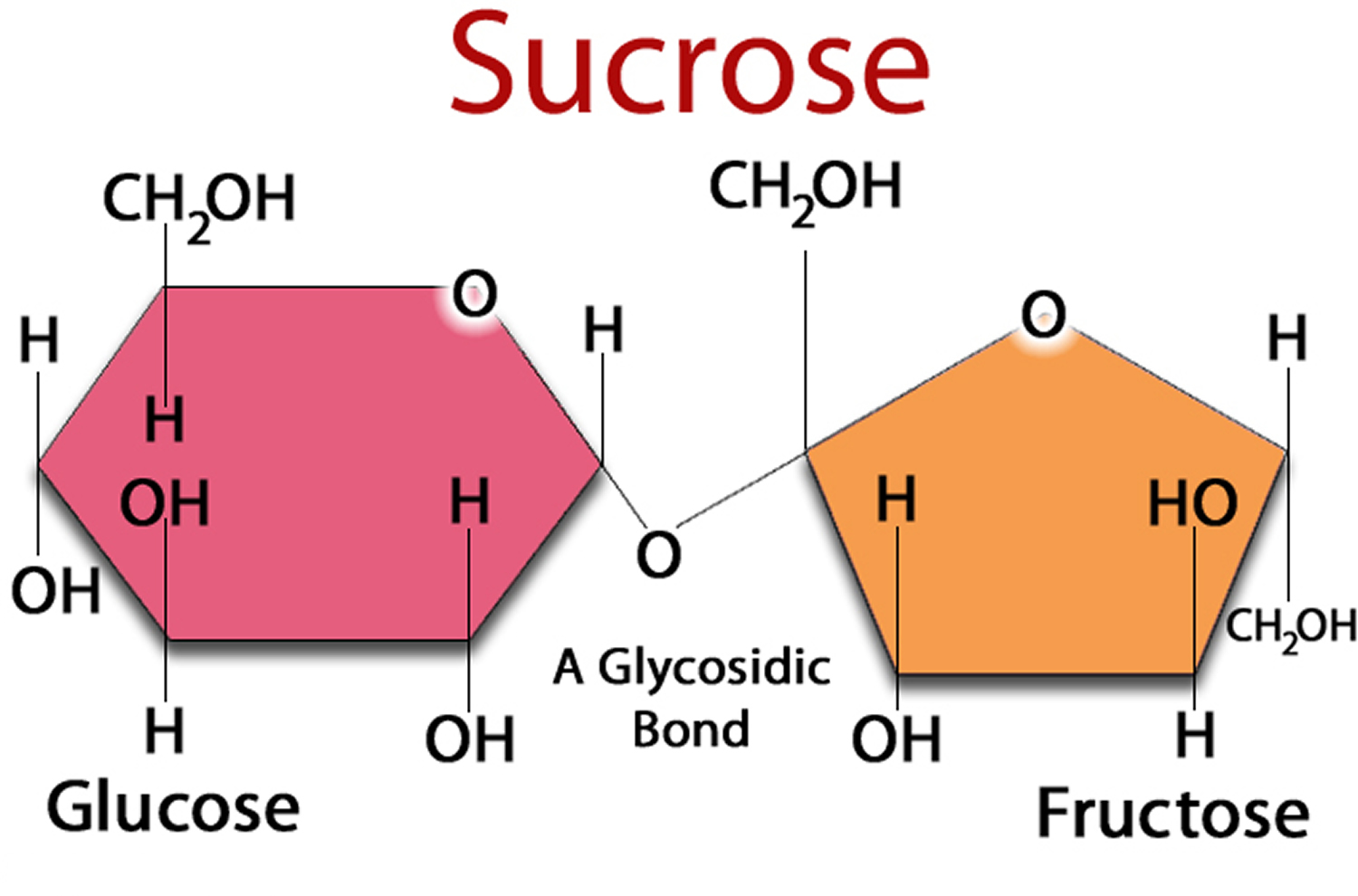
Figure 2. Sucrose (Sugar) made up of one Glucose and one Fructose molecule
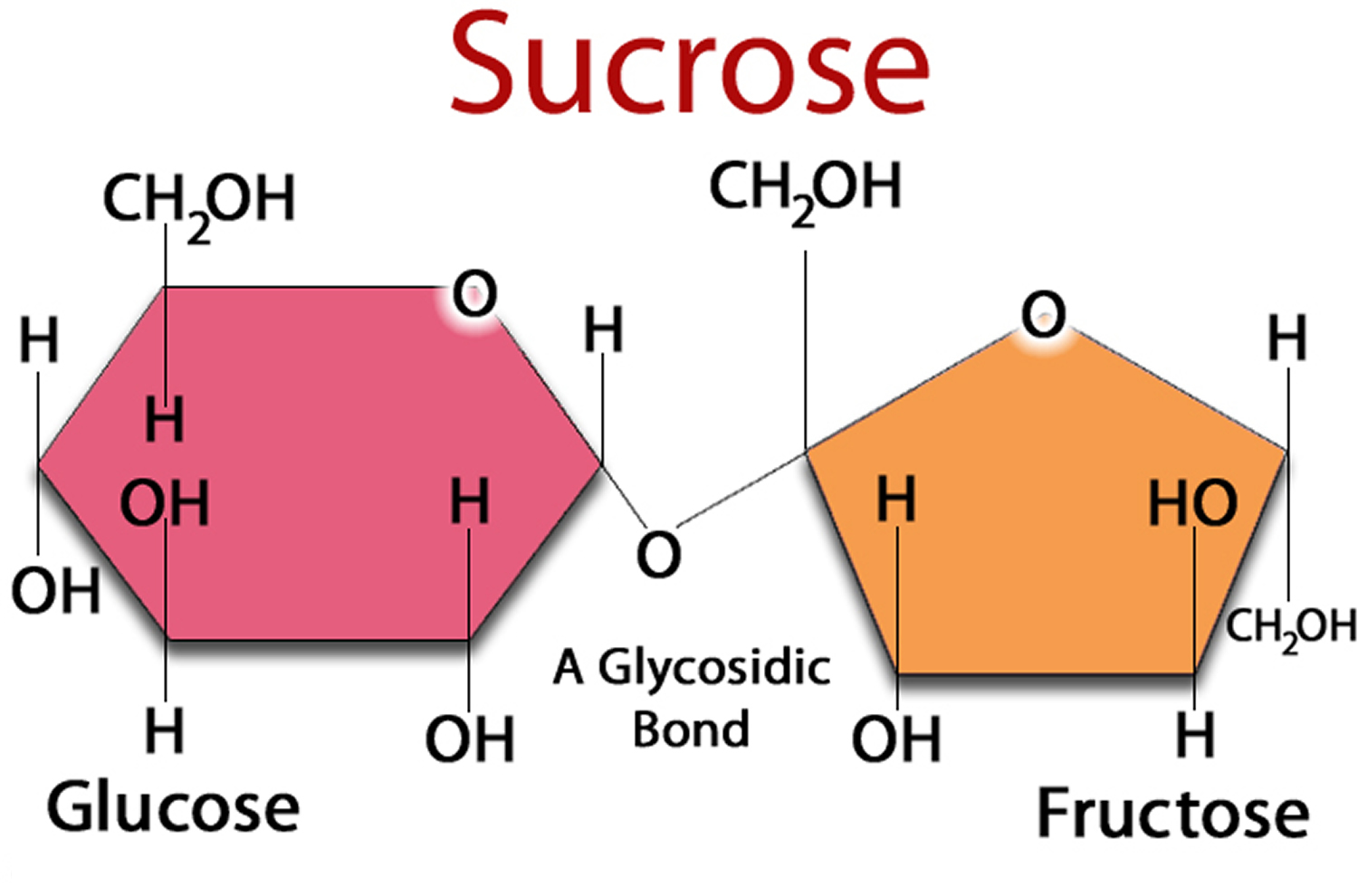
Figure 3. Glycogen is a polymer of glucose (up to 120,000 glucose molecules)
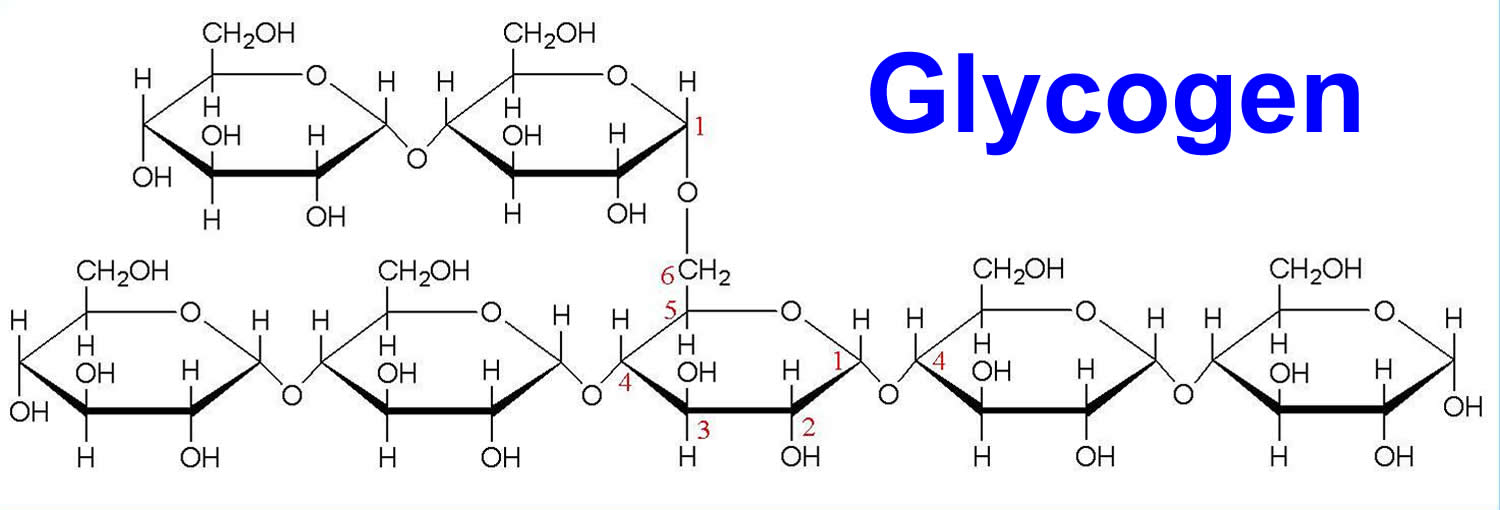
Figure 4. Cellulose is composed of a long chain of at least 500 glucose molecules
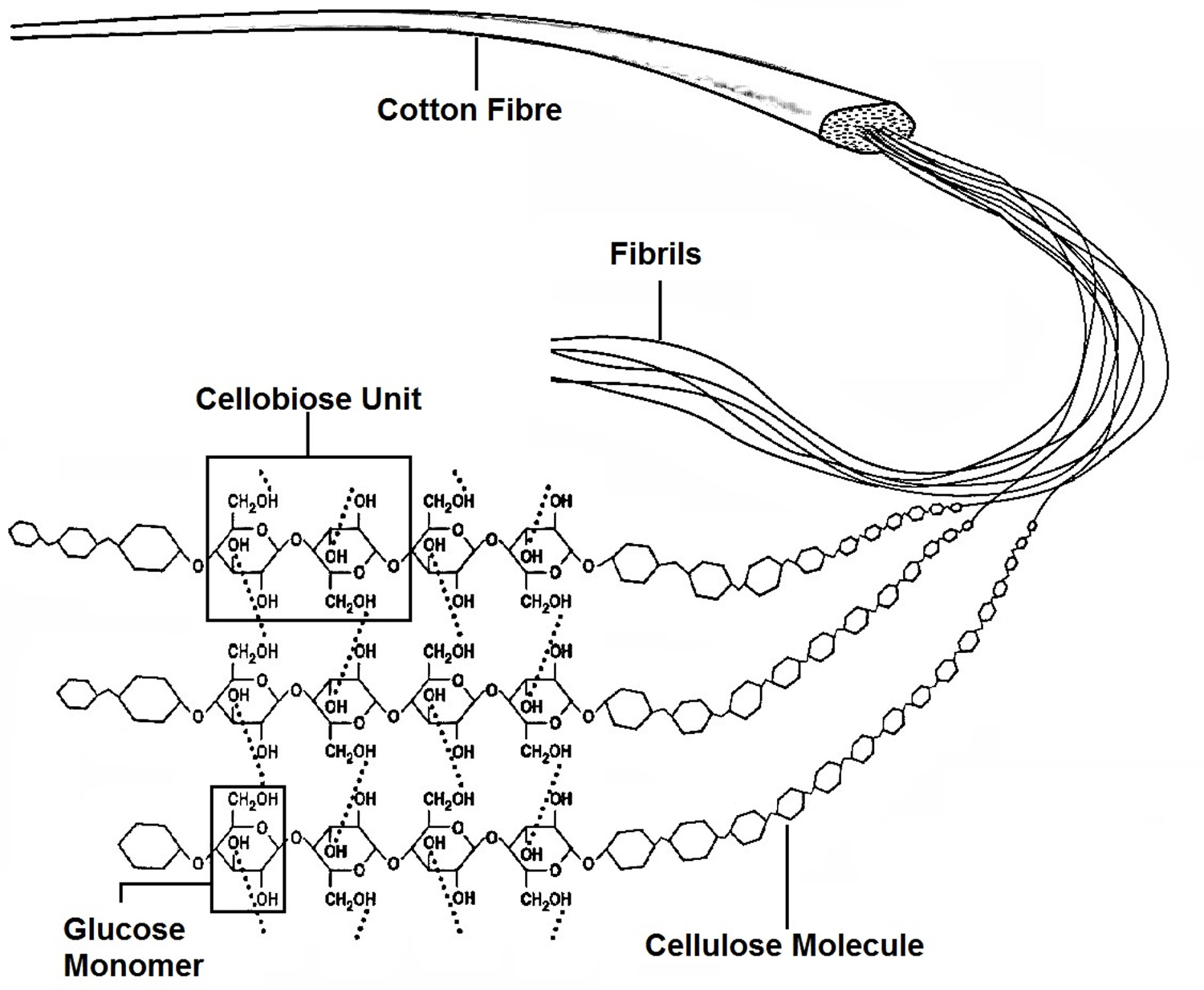
Figure 1 to 4 are just some examples of the complex heterogeneity of carbohydrates in living systems and is a direct result of several carbohydrate characteristics: the ability of different types and numbers of sugar molecules to form glycosidic bonds with one another, the structural characteristics of these molecules, the type of anomeric linkage, the position and the absence or presence of branching 2, 3.
On a larger scale, four different glucose monosaccharides may potentially produce 35,560 unique tetrasaccharides 4, 5. What is intriguing about the ability of glycans (compounds consisting of a large number of monosaccharides linked glycosidically) to encode an immense repertoire of biological information is that they are not encoded by the genome 6. The genome codes for enzymes that act on glycans such as glycosyltransferases and glycosidases. The combined activity levels of these enzymes in the endoplasmic reticulum (ER) and the Golgi apparatus, and perhaps on the cell surface, determine the glycosylation patterns (carbohydrate domains) of glycolipids and glycoproteins 7, 8. For a more complete and in-depth discussion of carbohydrate structure, chemistry, synthesis, and function we highly recommend reviewing Chapter 23 of the Wade Organic Chemistry 1 textbook and Introduction to Glycobiology 9.
Three main types of carbohydrate in food.
- Starches (also known as complex carbohydrates)
- Sugars
- Fiber
You’ll also hear terms like naturally occurring sugar, added sugar, low-calorie sweeteners, sugar alcohols, reduced-calorie sweeteners, processed grains, enriched grains, complex carbohydrate, sweets, refined grains and whole grains.
No wonder knowing what kind and how much carbohydrate to eat can be confusing !
On the nutrition label, the term “total carbohydrate” includes all three types of carbohydrates. This is the number you should pay attention to if you are carbohydrate counting.
What happens when you eat foods containing carbohydrates ?
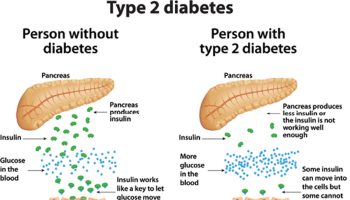
When you eat foods containing carbohydrates, your digestive system breaks down the sugars and starches into glucose. Glucose is one of the simplest forms of sugar. Glucose then enters your bloodstream from your digestive tract and raises your blood glucose levels. The hormone insulin, which comes from the pancreas or from insulin shots, helps cells throughout your body absorb glucose and use it for energy. Once glucose moves out of the blood into cells, your blood glucose levels go back down.
The Glycemic Index – a guide to Carbs
The Glycemic Index (GI) is a relative ranking of carbohydrate in foods according to how they affect blood glucose levels 10. The GI index runs from 0–100 and usually uses glucose, which has a GI of 100, as the reference. Slowly absorbed carbohydrates have a low GI rating (55 or below), and include most fruits and vegetables, milk, some wholegrain cereals and bread, pulses and basmati rice. GI numbers are to be used as a guide only as individual foods do not have the same response in all people.
- Low GI foods are foods with a GI less than 55.
- Intermediate GI foods are foods with a GI between 55 and 70.
- High GI foods are foods with a GI greater than 70.
Meats and fats don’t have a GI because they do not contain carbohydrate.
Research shows that both the amount and the type of carbohydrate in food affect blood glucose levels. Studies also show that the total amount of carbohydrate in food, in general, is a stronger predictor of blood glucose response than the GI.
The GI value relates to the food eaten on its own and in practice we usually eat foods in combination as meals. Bread, for example is usually eaten with butter or margarine, and potatoes could be eaten with meat and vegetables. Therefore relying solely on the glycemic index of foods could result in eating unbalanced and un-healthy diets high in fat, salt and saturated fats.
An additional problem is that GI compares the glycaemic effect of an amount of food containing 50g of carbohydrate but in real life we eat different amounts of food containing different amounts of carbohydrate.
The recommendation is to eat more low and intermediate GI foods, not to exclude high GI foods. By choosing the low glycaemic index foods and thus the minimally processed foods, people can lose more weight, feel fuller longer, and remain healthier.
Some nutrition experts believe that people with diabetes should pay attention to both the glycemic index and glycemic load to avoid sudden spikes in blood sugar. The American Diabetes Association, on the other hand, says that the total amount of carbohydrate in a food, rather than its glycemic index or load, is a stronger predictor of what will happen to blood sugar. And some dietitians also feel that focusing on the glycemic index and load adds an unneeded layer of complexity to choosing what to eat.
How much carbohydrate do you need each day ?
How much carbohydrate you eat is very individual. Finding the right amount of carbohydrate depends on many things including how active you are (sedentary lifestyle or elite athlete), your body weight (healthy weight or overweight or obese) and what, if any, medicines you take. Some people are active and can eat more carbohydrate. You may need to have less carbohydrate to keep your blood glucose in control 11.
The daily amount of carbohydrate ~ 130 grams of carbohydrate / day, (protein, and fat) for Americans can be found in the U.S. Department of Health and Human Services and U.S. Department of Agriculture. 2015–2020 Dietary Guidelines for Americans. 8th Edition. December 2015 12. Experts suggest that carbohydrate intake for most people should be between 45 and 65 percent of total calories. People on low-calorie diets and people who are physically inactive may want to aim for the lower end of that range 12.
A research team led by Dr. Kevin Hall of National Institute of Health’s National Institute of Diabetes and Digestive and Kidney Diseases conducted a study under carefully controlled conditions to determine how the body responds to dietary fat vs. dietary carbohydrate restriction 13. The conclusion from that study, that selective reduction of dietary carbohydrate resulted in decreased insulin secretion and increased fat burning, resulting in significant body fat loss. In contrast, selective reduction of dietary fat led to no significant changes in insulin secretion or fat burning compared to the eucaloric baseline diet, but interestingly, the fat restricted participants lost even more body fat during the fat-restricted diet, as it resulted in a greater imbalance between the fat eaten and fat burned 13.
You and your health care team can figure out the right amount for you. Once you know how much carb to eat at a meal, choose your food and the portion size to match.
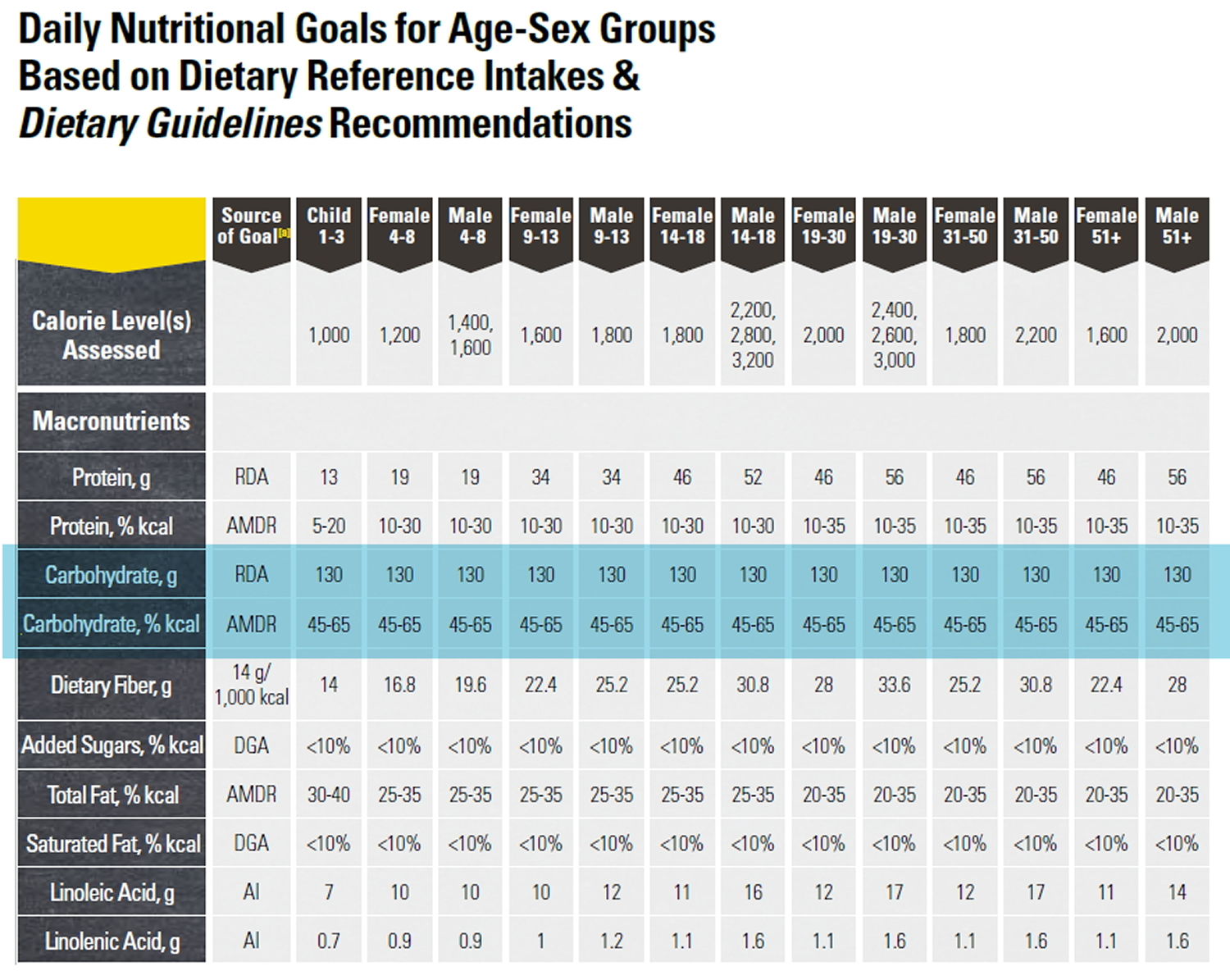
Table of Daily Dietary Reference Intake for Carbohydrate, Protein and Fat (source 12).
However, the daily amount of carbohydrate, protein, and fat for people with diabetes has not been defined—what is best for one person may not be best for another. Everyone needs to get enough carbohydrate to meet the body’s needs for energy, vitamins and minerals, and fiber. Finding the balance for yourself is important so you can feel your best, do the things you enjoy, and lower your risk of diabetes complications. A place to start is at about 45-60 grams of carbohydrate at a meal. You may need more or less carbohydrate at meals depending on how you manage your diabetes 11.
One gram of carbohydrate provides about 4 calories, so you’ll have to divide the number of calories you want to get from carbohydrates by 4 to get the number of grams. For example, if you want to eat 1,800 total calories per day and get 45 percent of your calories from carbohydrates, you would aim for about 200 grams of carbohydrate daily.
You would calculate that amount as follows:
.45 x 1,800 calories = 810 calories
810 ÷ 4 = 202.5 grams of carbohydrate
You’ll need to spread out your carbohydrate intake throughout the day. A dietitian or diabetes educator can help you learn what foods to eat, how much to eat, and when to eat based on your weight, activity level, medicines, and blood glucose targets.
- Nutrition Labels
You can find out how many grams of carbohydrate are in the foods you eat by checking the nutrition labels on food packages.
Nutrition labels tell you
- the food’s serving size––such as one slice or 1/2 cup
- the total grams of carbohydrate per serving
- other nutrition information, including calories and the amount of protein and fat per serving
If you have two servings instead of one, such as one cup of pinto beans instead of 1/2 cup, you multiply the number of grams of carbohydrate in one serving—for example, 15—by two to get the total number of grams of carbohydrate—30.
15 x 2 = 30
Following is an example of a nutrition label:
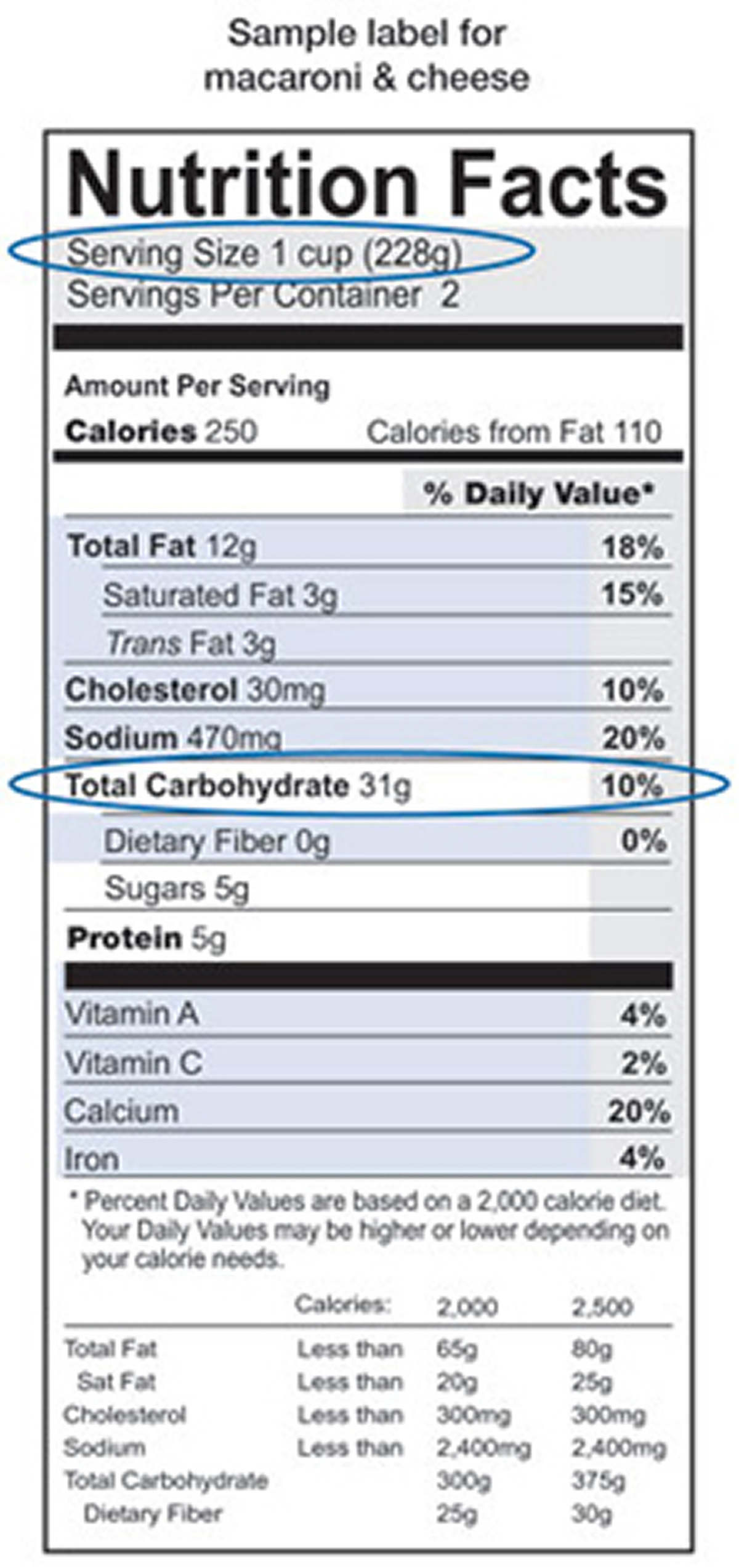 How can you find out how much carbohydrate is in the foods you eat ?
How can you find out how much carbohydrate is in the foods you eat ?
To find out the amount of carbohydrate in homemade foods, you’ll need to estimate and add up the grams of carbohydrate from the ingredients. You can use books or websites that list the typical carbohydrate content of homemade items to estimate the amount of carbohydrate in a serving.
You can also weigh foods with a scale or measure amounts with measuring cups or spoons to estimate the amount of carbohydrate. For example, if a nutrition label shows that 1 1/2 cups of cereal contain 45 grams of carbohydrate, then 1/2 cup will have 15 grams of carbohydrate and 1 cup will have 30 grams of carbohydrate.
You will need to learn to estimate the amount of carbohydrate in foods you typically eat. For example, the following amounts of carbohydrate-rich foods each contain about 15 grams of carbohydrate:
- one slice of bread
- one 6-inch tortilla
- 1/3 cup of pasta
- 1/3 cup of rice
- 1/2 cup of canned or fresh fruit or fruit juice or one small piece of fresh fruit, such as a small apple or orange
- 1/2 cup of pinto beans
- 1/2 cup of starchy vegetables such as mashed potatoes, cooked corn, peas, or lima beans
- 3/4 cup of dry cereal or 1/2 cup cooked cereal
- 1 tablespoon of jelly
- 1 small piece of fresh fruit (4 oz)
- 1/2 cup of canned or frozen fruit
- 1 slice of bread (1 oz) or 1 (6 inch) tortilla
- 1/2 cup of oatmeal
- 1/3 cup of pasta or rice
- 4-6 crackers
- 1/2 English muffin or hamburger bun
- 1/2 cup of black beans or starchy vegetable
- 1/4 of a large baked potato (3 oz)
- 2/3 cup of plain fat-free yogurt or sweetened with sugar substitutes
- 2 small cookies
- 2 inch square brownie or cake without frosting
- 1/2 cup ice cream or sherbet
- 1 Tbsp syrup, jam, jelly, sugar or honey
- 2 Tbsp light syrup
- 6 chicken nuggets
- 1/2 cup of casserole
- 1 cup of soup
- 1/4 serving of a medium french fry
Some foods are so low in carbohydrates that you may not have to count them unless you eat large amounts. For example, most nonstarchy vegetables are low in carbohydrates. A 1/2-cup serving of cooked nonstarchy vegetables or a cup of raw vegetables has only about 5 grams of carbohydrate.
As you become familiar with which foods contain carbohydrates and how many grams of carbohydrate are in food you eat, carbohydrate counting will be easier.
Can you use carbohydrate counting if you are pregnant ?
You can use carbohydrate counting to help control your blood glucose levels when you are pregnant. Meeting your blood glucose targets during pregnancy is important for your and your baby’s health. High blood glucose during pregnancy can harm the baby and increase the baby’s chances of having type 2 diabetes later in life.
Women diagnosed with gestational diabetes—a type of diabetes that develops only during pregnancy—can also use carbohydrate counting to help control their blood glucose levels.
Talk with your doctor about using carbohydrate counting to help meet your blood glucose targets during your pregnancy.
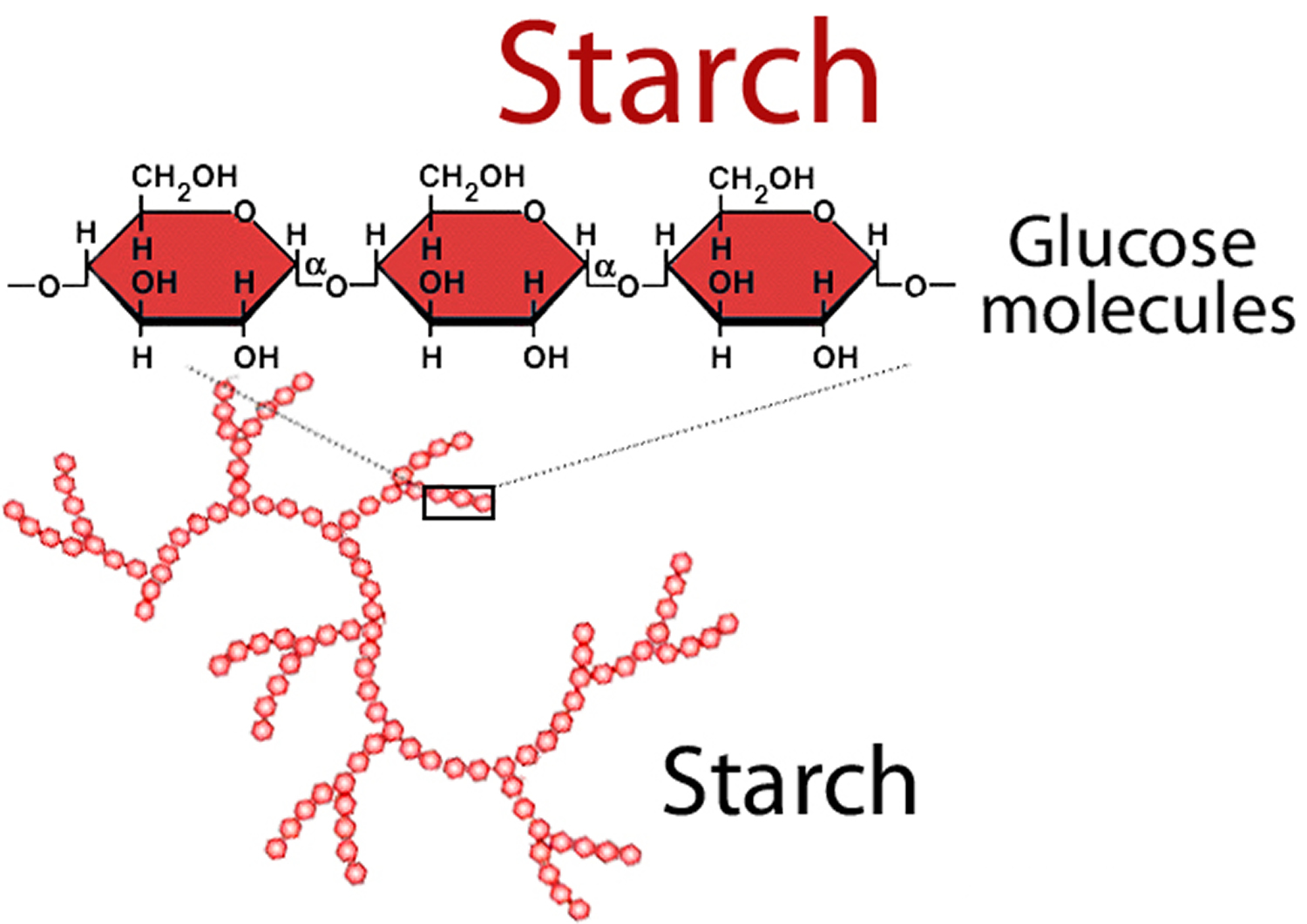
1) Starch (complex carbohydrates)
Foods high in starch include:
- Starchy vegetables like peas, corn, lima beans and potatoes
- Dried beans, lentils and peas such as pinto beans, kidney beans, black eyed peas and split peas
- Grains like oats, barley and rice. (The majority of grain products in the US are made from wheat flour. These include pasta, bread and crackers but the variety is expanding to include other grains as well.)
The grain group can be broken down even further into whole grain or refined grain.
A grain contains three parts:
- bran
- germ
- endosperm
The bran is the outer hard shell of the grain. It is the part of the grain that provides the most fiber and most of the B vitamins and minerals.
The germ is the next layer and is packed with nutrients including essential fatty acids and vitamin E.
The endosperm is the soft part in the center of the grain. It contains the starch. Whole grain means that the entire grain kernel is in the food.
If you eat a whole grain food, it contains the bran, germ, and endosperm so you get all of the nutrients that whole grains have to offer. If you eat a refined grain food, it contains only the endosperm or the starchy part so you miss out on a lot of vitamins and minerals. Because whole grains contain the entire grain, they are much more nutritious than refined grains.
2) Sugar (Sucrose)
Sucrose (table sugar) is made of two simpler sugars called glucose and fructose, is another type of carbohydrate. You may also hear sugar referred to as simple or fast-acting carbohydrate.
There are two main types of sugar:
- naturally occurring sugars such as those in milk or fruit
- added sugars such as those added during processing such as fruit canned in heavy syrup or sugar added to make a cookie
On the nutrition facts label, the number of sugar grams includes both added and natural sugars.
There are many different names for sugar. Examples of common names are table sugar, brown sugar, molasses, honey, beet sugar, cane sugar, confectioner’s sugar, powdered sugar, raw sugar, turbinado, maple syrup, high-fructose corn syrup, agave nectar and sugar cane syrup.
You may also see table sugar listed by its chemical name, sucrose. Fruit sugar is also known as fructose and the sugar in milk is called lactose. You can recognize other sugars on labels because their chemical names also end in “-ose.” For example glucose (also called dextrose), fructose (also called levulose), lactose and maltose.
If you are looking for information about artificial sweeteners, try this section.
What are added sugars ?
Added sugars are various forms of sugar added to foods or drinks during processing or preparation. Naturally occurring sugars such as those in milk and fruits are not added sugars but are carbohydrates. The most common sources of added sugars for Americans are
- sugar-sweetened soft drinks, fruit drinks, sports drinks, and energy drinks
- grain-based desserts, such as cakes, cookies, and doughnuts
- milk-based desserts and products, such as ice cream, sweetened yogurt, and sweetened milk
- candy
Reading the list of ingredients for foods and drinks can help you find added sugars, such as
- sugar, raw sugar, brown sugar, and invert sugar—a mixture of fructose and glucose :
- corn syrup and malt syrup
- high-fructose corn syrup, often used in soft drinks and juices
- honey, molasses, and agave nectar
- dextrose, fructose, glucose, lactose, and sucrose
For a healthier eating plan, limit foods and drinks with added sugars.
Current Guidelines for Sugar Intake
| US Department of Agriculture and US Department of Health and Human Services (2015-2020) | Limit consumption of added sugars to <10% of calories per day |
| World Health Organization (March 2015) | Restrict added sugar consumption to <10% of daily calories |
| American Heart Association (2009) | Limit added sugars to 5% of daily calories (for women, 100 calories/day; for men, 150 calories/day) |
The American Heart Association 14 recommends no more than half of your daily discretionary calorie allowance come from added sugars. Your daily discretionary calorie allowance consists of calories available after meeting nutrient needs. This is no more than 100 calories per day for most American women and no more than 150 per day for men (or about 6 teaspoons a day for women and 9 teaspoons a day for men).
The United Kingdom’s Scientific Advisory Committee on Nutrition published its final report “Carbohydrates and Health” in July 2015 15. It included recommendations that the average population intake of sugar should not exceed 5% of total dietary energy for the population aged two years upwards (halving the previous recommendation, in line with the American Heart Association and 50% less than the <10% recommended by the US Department of Agriculture and US Department of Health and Human ) and that consumption of sugar sweetened drinks should be minimised by both adults and children. These recommendations are based on United Kingdom Scientific Advisory Committee on Nutrition’s review of the evidence, which demonstrated that:
- in adults, when consuming an unrestricted daily diet, increasing the percentage of total energy from sugar leads to increases in energy intake
- greater consumption of sugar sweetened drinks is associated with increased risk of type 2 diabetes
- consumption of sugar sweetened drinks, compared to non-sugar sweetened drinks, results in greater weight gain and increases in body mass index in children and adolescents due to increased energy consumption
- higher consumption of sugar and sugar containing foods and drinks is associated with a greater risk of dental caries.
The evidence from the United Kingdom’s Scientific Advisory Committee on Nutrition published ‘Carbohydrates and Health’ report clearly and robustly presents the case for action to reduce sugar intakes to the new 5% recommendation and also to minimise consumption of sugar sweetened drinks.
This is the first time the United Kingdom’s Scientific Advisory Committee on Nutrition has made a recommendation to minimise consumption of a specific food and its importance must not be underestimated.
The report also includes recommendations for increasing fibre intake and confirmation that carbohydrates should provide around 50% of energy intakes (which also includes the new maximum sugar recommendation).
Sugar by Any Other Name
You don’t always see the word “sugar” on a food label. It sometimes goes by another name, like these:
- White sugar
- Brown sugar
- Raw sugar
- Agave nectar
- Brown rice syrup
- Corn syrup
- Corn syrup solids
- High-fructose corn syrup
- Invert sugar
- Dextrose
- Anhydrous dextrose
- Crystal dextrose
- Dextrin
- Evaporated cane juice
- Fructose sweetener
- Liquid fructose
- Glucose
- Lactose
- Honey
- Malt syrup
- Maple syrup
- Molasses
- Pancake syrup
- Sucrose
- Trehalose
- turbinado sugar
3) Fiber
Fiber comes from plant foods so there is no fiber in animal products such as milk, eggs, meat, poultry, and fish.
Fiber is the indigestible part of plant foods, including fruits, vegetables, whole grains, nuts and legumes. When you consume dietary fiber, most of it passes through the intestines and is not digested.
For good health, adults need to try to eat 25 to 30 grams of fiber each day. Most Americans do not consume nearly enough fiber in their diet, so while it is wise to aim for this goal, any increase in fiber in your diet can be beneficial. Most of us only get about half of what is recommended.
Fiber contributes to digestive health, helps to keep you regular, and helps to make you feel full and satisfied after eating.
Additional health benefits, of a diet high in fiber — such as a reduction in cholesterol levels — have been suggested by some so may be an additional benefit.
Good sources of dietary fiber include:
- Beans and legumes. Think black beans, kidney beans, pintos, chick peas (garbanzos), white beans, and lentils.
- Fruits and vegetables, especially those with edible skin (for example, apples, corn and beans) and those with edible seeds (for example, berries).
- Whole grains such as:
- Whole wheat pasta
- Whole grain cereals (Look for those with three grams of dietary fiber or more per serving, including those made from whole wheat, wheat bran, and oats.)
- Whole grain breads (To be a good source of fiber, one slice of bread should have at least three grams of fiber. Another good indication: look for breads where the first ingredient is a whole grain. For example, whole wheat or oats.) Many grain products now have “double fiber” with extra fiber added.
- Nuts — try different kinds. Peanuts, walnuts and almonds are a good source of fiber and healthy fat, but watch portion sizes, because they also contain a lot of calories in a small amount.
In general, an excellent source of fiber contains five grams or more per serving, while a good source of fiber contains 2.5 – 4.9 grams per serving.
It is best to get your fiber from food rather than taking a supplement. In addition to the fiber, these foods have a wealth of nutrition, containing many important vitamins and minerals. In fact, they may contain nutrients that haven’t even been discovered yet!
It is also important that you increase your fiber intake gradually, to prevent stomach irritation, and that you increase your intake of water and other liquids, to prevent constipation.
What is Carbohydrate Counting ?
Carbohydrate counting, also called carb counting, is a meal planning tool for people with type 1 or type 2 diabetes. Carbohydrate counting involves keeping track of the amount of carbohydrate in the foods you eat each day 16.
Carbohydrates are one of the main nutrients found in food and drinks. Protein and fat are the other main nutrients. Carbohydrates include sugars, starches, and fiber. Carbohydrate counting can help you control your blood glucose, also called blood sugar, levels because carbohydrates affect your blood glucose more than other nutrients.
Healthy carbohydrates
- such as whole grains, fruits, and vegetables, are an important part of a healthy eating plan because they can provide both energy and nutrients, such as vitamins and minerals, and fiber. Fiber can help you prevent constipation, lower your cholesterol levels, and control your weight.
Unhealthy carbohydrates
- are often food and drinks with added sugars. Although unhealthy carbohydrates can also provide energy, they have little to no nutrients. More information about which carbohydrates provide nutrients for good health and which carbohydrates do not is provided in the NIDDK health topic, Diabetes Diet and Eating.
The amount of carbohydrate in foods is measured in grams. To count grams of carbohydrate in foods you eat, you’ll need to
- know which foods contain carbohydrates
- learn to estimate the number of grams of carbohydrate in the foods you eat
- add up the number of grams of carbohydrate from each food you eat to get your total for the day
Your doctor can refer you to a dietitian or diabetes educator who can help you develop a healthy eating plan based on carbohydrate counting.
Which foods contain carbohydrates ?
Foods that contain carbohydrates include:
- grains, such as bread, noodles, pasta, crackers, cereals, and rice
- fruits, such as apples, bananas, berries, mangoes, melons, and oranges
- dairy products, such as milk and yogurt
- legumes, including dried beans, lentils, and peas
- snack foods and sweets, such as cakes, cookies, candy, and other desserts
- juices, soft drinks, fruit drinks, sports drinks, and energy drinks that contain sugars
- vegetables, especially “starchy” vegetables such as potatoes, corn, and peas
Potatoes, peas, and corn are called starchy vegetables because they are high in starch. These vegetables have more carbohydrates per serving than nonstarchy vegetables.
Examples of nonstarchy vegetables are asparagus, broccoli, carrots, celery, green beans, lettuce and other salad greens, peppers, spinach, tomatoes, and zucchini.
Foods that do not contain carbohydrates include meat, fish, and poultry; most types of cheese; nuts; and oils and other fats.
How can carbohydrate counting help you ?
Carbohydrate counting can help keep your blood glucose levels close to normal. Keeping your blood glucose levels as close to normal as possible may help you
- stay healthy longer
- prevent or delay diabetes problems such as kidney disease, blindness, nerve damage, and blood vessel disease that can lead to heart attacks, strokes, and amputations—surgery to remove a body part
- feel better and more energetic
You may also need to take diabetes medicines or have insulin shots to control your blood glucose levels. Discuss your blood glucose targets with your doctor. Targets are numbers you aim for. To meet your targets, you will need to balance your carbohydrate intake with physical activity and diabetes medicines or insulin shots.
Hormone contraceptives and how the body uses carbohydrates in women without diabetes
Hormone contraceptives may change how the body handles carbohydrates (starches and sugars). Changes may include lower ability to use sugar from food and more problems with the body’s insulin. Insulin is a hormone that helps the body use sugar. Problems with blood sugar can increase risk for diabetes and heart disease. These issues have been raised mainly with birth control methods that contain the hormone estrogen.
In April 2014, Cochrane Review researchers 17 looked for randomized trials of how the body handles carbohydrates when using birth control methods with hormones. Outcomes were blood glucose or insulin levels. Birth control methods could contain estrogen and progestin or just progestin. The type could be pills, shots (injections), implants (matchstick‐size rods put under the skin), the vaginal ring, or an intrauterine device (IUD). The studies had to compare two types of birth control or one type versus a placebo or ‘dummy’ method.
The review concluded that in women without diabetes, hormone contraceptives have little effect on the body’s carbohydrate use. Few studies compared the same types of birth control. Therefore, we cannot make strong statements. Many trials had small numbers of women, and many women dropped out. Older trials often did not report all the study methods. Many trials did not include overweight women.
Carbohydrate Metabolism Disorders
Metabolism is the process your body uses to make energy from the food you eat. Food is made up of proteins, carbohydrates, and fats. Chemicals in your digestive system (enzymes) break the food parts down into sugars and acids, your body’s fuel. Your body can use this fuel right away, or it can store the energy in your body tissues. If you have a metabolic disorder, something goes wrong with this process.
Sucrose (table sugar) is made of two simpler sugars called glucose and fructose. Lactose (milk sugar) is made of glucose and galactose. Both sucrose and lactose must be broken down into their component sugars by enzymes before the body can absorb and use them. The carbohydrates in bread, pasta, rice, and other carbohydrate-containing foods are long chains of simple sugar molecules. These longer molecules must also be broken down by the body. If an enzyme needed to process a certain sugar is missing, the sugar can accumulate in the body, causing problems.
Carbohydrate metabolism disorders are a group of metabolic disorders. Normally your enzymes break carbohydrates down into glucose (a type of sugar). If you have one of these disorders, you may not have enough enzymes to break down the carbohydrates. Or the enzymes may not work properly. This causes a harmful amount of sugar to build up in your body. That can lead to health problems, some of which can be serious. Some of the disorders are fatal 18.
These disorders are inherited 18. Newborn babies get screened for many of them, using blood tests. If there is a family history of one of these disorders, parents can get genetic testing to see whether they carry the gene. Other genetic tests can tell whether the fetus has the disorder or carries the gene for the disorder.
Treatments may include special diets, supplements, and medicines. Some babies may also need additional treatments, if there are complications. For some disorders, there is no cure, but treatments may help with symptoms 18.
Hereditary Fructose Intolerance
Hereditary fructose intolerance is caused by lack of the enzyme needed to metabolize fructose. Very small amounts of fructose cause low blood sugar levels and can lead to kidney and liver damage 19.
In this disorder, the body is missing an enzyme that allows it to use fructose, a sugar present in table sugar (sucrose) and many fruits. As a result, a by-product of fructose accumulates in the body, blocking the formation of glycogen and its conversion to glucose for use as energy. Ingesting more than tiny amounts of fructose or sucrose causes low blood sugar levels (hypoglycemia), with sweating, confusion, and sometimes seizures and coma. Children who continue to eat foods containing fructose develop kidney and liver damage, resulting in jaundice, vomiting, mental deterioration, seizures, and death. Chronic symptoms include poor eating, failure to thrive, digestive symptoms, liver failure, and kidney damage. For most types of this disorder, early diagnosis and dietary restrictions started early in infancy can help prevent these more serious problems.
The diagnosis is made when a chemical examination of a sample of liver tissue determines that the enzyme is missing. Treatment involves excluding fructose (generally present in sweet fruits), sucrose, and sorbitol (a sugar substitute) from the diet. Severe attacks of hypoglycemia respond to glucose given by vein. Milder attacks are treated with glucose tablets, which should be carried by anyone who has hereditary fructose intolerance 19.
Mucopolysaccharidoses
Complex sugar molecules called mucopolysaccharides are essential parts of many body tissues. In mucopolysaccharidoses, the body lacks enzymes needed to break down and store mucopolysaccharides. As a result, excess mucopolysaccharides enter the blood and are deposited in abnormal locations throughout the body.
Mucopolysaccharidoses are a group of inherited metabolic diseases, classified as lysosomal storage diseases, in which a defective or missing enzyme causes large amounts of complex sugar molecules, called lysosomes accumulate, to accumulate in harmful amounts in the body’s cells and tissues. This accumulation causes permanent, progressive cellular damage that affects appearance, physical abilities, organ and system functioning, and, in most cases, mental development. Depending on the type of mucopolysaccharidosis, affected individuals may have normal intellect or may be profoundly impaired, may experience developmental delay, or have severe behavioral problems. Physical symptoms generally include coarse or rough facial features, thick lips, an enlarged mouth and tongue, short stature with a disproportionately short trunk (dwarfism), abnormal bone size or shape (and other skeletal irregularities), thickened skin, enlarged organs such as the liver or spleen, hernias, and excessive body hair growth 20, 19.
- Mucopolysaccharidoses occur when the body lacks enzymes needed to break down and store complex sugar molecules (mucopolysaccharides).
- Typically, symptoms include short stature, hairiness, stiff finger joints, and coarseness of the face.
- The diagnosis is based on symptoms and a physical examination.
- Although a normal life span is possible, some types cause premature death.
- A bone marrow transplant may help.
During infancy and childhood, short stature, hairiness, and abnormal development become noticeable. The face may appear coarse. Some types of mucopolysaccharidoses cause intellectual disability to develop over several years. In some types, vision or hearing may become impaired. The arteries or heart valves can be affected. Finger joints are often stiff.
A doctor usually bases the diagnosis on the symptoms and a physical examination. The presence of a mucopolysaccharidosis in other family members also suggests the diagnosis. Urine tests may help but are sometimes inaccurate. X-rays may show characteristic bone abnormalities. Mucopolysaccharidoses can be diagnosed before birth by using amniocentesis or chorionic villus sampling.
Who is at risk of Mucopolysaccharidoses ?
It is estimated that one in every 25,000 babies born in the United States will have some form of the mucopolysaccharidoses. They are autosomal recessive disorders, meaning that only individuals inheriting the defective gene from both parents are affected. (The exception is MPS II, or Hunter syndrome, in which the mother alone passes along the defective gene to a son.) When both people in a couple have the defective gene, each pregnancy carries with it a one in four chance that the child will be affected. The parents and siblings of an affected child may have no sign of the disorder. Unaffected siblings and select relatives of a child with one of the mucopolysaccharidoses may carry the recessive gene and could pass it to their own children 20.
In general, the following factors may increase the chance of getting or passing on a genetic disease:
- A family history of a genetic disease.
- Parents who are closely related or part of a distinct ethnic or geographic circle.
- Parents who do not show disease symptoms but carry a disease gene.
What are the different types of the mucopolysaccharidoses ?
Seven distinct clinical types and numerous subtypes of the mucopolysaccharidoses have been identified. Although each mucopolysaccharidosis (MPS) differs clinically, most patients generally experience a period of normal development followed by a decline in physical and/or mental function 20.
- MPS I has historically been divided into three broad groups based on severity of symptoms–Hurler, Hurler-Scheie, and Scheie (in decreasing order of severity). It is now more appropriate to view MPS I as a continuous spectrum of disease, with the most severely affected individuals on one, the less severely affected (attenuated) on the other end, and a wide range of different severities in between. All individuals with MPS I have an absence of, or insufficient levels of, the enzyme alpha-L-iduronidase. Children with MPS I inherit a defective gene from both their mother and father.
In the more severe form of MPS I, developmental delay is evident by the end of the first year, and patients usually stop developing between ages 2 and 4. This is followed by progressive mental decline and loss of physical skills. Language may be limited due to hearing loss and an enlarged tongue. In time, the clear layers of the cornea become clouded and retinas may begin to degenerate. Carpal tunnel syndrome (or similar compression of nerves elsewhere in the body) and restricted joint movement are common.
Affected children may be quite large at birth and appear normal but may have inguinal (in the groin) or umbilical (where the umbilical cord passes through the abdomen) hernias. Growth in height may be faster than normal but begins to slow before the end of the first year and often ends around age 3. Many children develop a short body trunk and a maximum stature of less than 4 feet. Distinct facial features (including flat face, depressed nasal bridge, and bulging forehead) become more evident in the second year. By age 2, the ribs have widened and are oar-shaped. The liver, spleen, and heart are often enlarged. Children may experience noisy breathing and recurring upper respiratory tract and ear infections. Feeding may be difficult for some children, and many experience periodic bowel problems. Children with severe MPS I often die before age 10 from obstructive airway disease, respiratory infections, or cardiac complications.
Although symptoms generally begin to appear after age 5 in children with attenuated MPS I, the diagnosis is most commonly made after age 10. Children with attenuated MPS I have normal intelligence or may have mild learning disabilities; some may have psychiatric problems. Glaucoma, retinal degeneration, and clouded corneas may significantly impair vision. Other problems include carpal tunnel syndrome or other nerve compression, stiff joints, claw hands and deformed feet, a short neck, and aortic valve disease. Some affected individuals also have obstructive airway disease and sleep apnea. Persons with attenuated MPS I can live into adulthood.
Within the MPS I disease spectrum are children whose symptoms generally begin between ages 3 and 8. Children may have moderate mental impairment and learning difficulties. Skeletal and systemic irregularities include short stature, marked smallness in the jaws, progressive joint stiffness, compressed spinal cord, clouded corneas, hearing loss, heart disease, coarse facial features, and umbilical hernia. Respiratory problems, sleep apnea, and heart disease may develop in adolescence. Some persons need continuous positive airway pressure during sleep to ease breathing. Life expectancy is generally into the late teens or early twenties.
Although no studies have been done to determine the frequency of MPS I in the United States, studies in British Columbia estimate that one in 100,000 babies born has severe MPS I. The estimate for attenuated MPS I is one in 500,000 births and one in 115,000 births for individuals whose disease symptoms fall between severe and attenuated.
- MPS II, Hunter syndrome, is caused by lack of the enzyme iduronate sulfatase. Although it was once divided into two groups based on the severity of symptoms, MPS II is now considered a continuous spectrum of disease. MPS II is the only one of the mucopolysaccharidoses in which the mother alone can pass the defective gene to a son. The incidence of MPS II syndrome is estimated to be one in every 100,000 to 150,000 male births.
Children with the more severe form of MPS II share many of the same clinical features associated with severe MPS I but with milder symptoms. Onset of the disease is usually between ages 2 and 4. Developmental decline is usually noticed between the ages of 18 and 36 months, followed by progressive loss of skills. Other clinical features include coarse facial features, skeletal irregularities, obstructive airway and respiratory complications, short stature, joint stiffness, retinal degeneration (but no corneal clouding), communicating hydrocephalus, chronic diarrhea, enlarged liver and spleen, and progressive hearing loss. Whitish skin lesions may be found on the upper arms, back, and upper legs. Death from upper airway disease or cardiovascular failure usually occurs by age 15.
Physical characteristics of children with a less severe form of MPS II are less obvious and progress at a much slower rate. Diagnosis is often made in the second decade of life. Intellect and social development are not affected. Skeletal problems may be less severe, but carpal tunnel syndrome and joint stiffness can restrict movement and height is somewhat less than normal. Other clinical symptoms include hearing loss, poor peripheral vision, diarrhea, and sleep apnea, although respiratory and cardiac complications can contribute to premature death. Persons with attenuated MPS II may live into their 50s or beyond.
- MPS III, Sanfilippo syndrome, is marked by severe neurological symptoms. These include progressive dementia, aggressive behavior, hyperactivity, seizures, some deafness and loss of vision, and an inability to sleep for more than a few hours at a time. MPS III affects children differently, and its progress will be faster in some than in others. Early mental and motor skill development may be somewhat delayed. Affected children show a marked decline in learning between ages 2 and 6, followed by eventual loss of language skills and loss of some or all hearing. Some children may never learn to speak. Aggressive behavior, hyperactivity, profound dementia, and irregular sleep may make children difficult to manage, particularly those who retain normal physical strength. As the disease progresses, children become increasingly unsteady on their feet and most are unable to walk by age 10.
Thickened skin and mild changes in facial features, bone, and skeletal structures become noticeable with age. Growth in height usually stops by age 10. Other problems may include narrowing of the airway passage in the throat and enlargement of the tonsils and adenoids, making it difficult to eat or swallow. Recurring respiratory infections are common.
There are four distinct types of MPS III, each caused by alteration of a different enzyme needed to completely break down the heparan sulfate sugar chain. Little clinical difference exists between these four types but symptoms appear most severe and seem to progress more quickly in children with type A. Life expectancy in MPS III is extremely varied. Most persons with MPS III live into their teenage years, and some live longer, into their twenties or thirties.
- MPS IIIA is caused by the missing or altered enzyme heparan N-sulfatase.
- MPS IIIB is caused by the missing or deficient enzyme alpha-N-acetylglucosaminidase.
- MPS IIIC results from the missing or altered enzyme acetyl-CoAlpha-glucosaminide acetyltransferase.
- MPS IIID is caused by the missing or deficient enzyme N-acetylglucosamine 6-sulfatase.
The incidence of MPS III (for all four types combined) is about one in 70,000 births.
- MPS IV, Morquio syndrome, is estimated to occur in one of every 200,000 births. Its two subtypes result from the missing or deficient enzymes N-acetylgalactosamine 6-sulfatase (Type A) or beta-galactosidase (Type B) needed to break down the keratan sulfate sugar chain. Clinical features are similar in both types but appear milder in MPS IVB. Onset is between ages 1 and 3. Neurological complications include spinal nerve and nerve root compression resulting from extreme, progressive skeletal changes, particularly in the ribs and chest; conductive and/or neurosensitive loss of hearing (see “What are the signs and symptoms?”); and clouded corneas. Intelligence is normal unless hydrocephalus develops and is not treated.
Physical growth slows and often stops around age 8. Skeletal abnormalities include a bell-shaped chest, a flattening or curvature of the spine, shortened long bones, and dysplasia of the hips, knees, ankles, and wrists. The bones that stabilize the connection between the head and neck can be malformed (odontoid hypoplasia); in these cases, a surgical procedure called spinal cervical bone fusion can be lifesaving. Restricted breathing, joint stiffness, and heart disease are also common. Children with the more severe form of MPS IV may not live beyond their twenties or thirties.
- Children with MPS VI, Maroteaux-Lamy syndrome, usually have normal intellectual development but share many of the physical symptoms found in severe MPS I. Caused by the deficient enzyme N-acetylgalactosamine 4-sulfatase, MPS VI has a variable spectrum of severe symptoms. Neurological complications include clouded corneas, deafness, thickening of the dura (the membrane that surrounds and protects the brain and spinal cord), and pain caused by compressed or traumatized nerves and nerve roots.
Growth is normal at first but stops suddenly around age 8. By age 10 children have developed a shortened trunk, crouched stance, and restricted joint movement. In more severe cases, children also develop a protruding abdomen and forward-curving spine. Skeletal changes (particularly in the pelvic region) are progressive and limit movement. Many children also have umbilical or inguinal hernias. Nearly all children have some form of heart disease, usually involving valve dysfunction.
- MPS VII, Sly syndrome, one of the least common forms of the mucopolysaccharidoses, is estimated to occur in fewer than one in 250,000 births. The disorder is caused by deficiency of the enzyme beta-glucuronidase. In its rarest form, MPS VII causes children to be born with hydrops fetalis, in which extreme amounts of fluid are retained in the body. Survival is usually a few months or less. Most children with MPS VII are less severely affected. Neurological symptoms may include mild to moderate intellectual impairment by age 3, communicating hydrocephalus, nerve entrapment, corneal clouding, and some loss of peripheral and night vision. Other symptoms include short stature, some skeletal irregularities, joint stiffness and restricted movement, and umbilical and/or inguinal hernias. Some patients may have repeated bouts of pneumonia during their first years of life. Most children with MPS VII live into the teenage or young adult years.
- As of 2001, only one case of MPS IX had been reported. The disorder results from hyaluronidase deficiency. Symptoms included nodular soft-tissue masses located around joints, with episodes of painful swelling of the masses and pain that ended spontaneously within 3 days. Pelvic radiography showed multiple soft-tissue masses and some bone erosion. Other traits included mild facial changes, acquired short stature as seen in other MPS disorders, and normal joint movement and intelligence.
What are the signs and symptoms of mucopolysaccharidoses ?
The mucopolysaccharidoses share many clinical features but have varying degrees of severity. These features may not be apparent at birth but progress as storage of glycosaminoglycans affects bone, skeletal structure, connective tissues, and organs. Neurological complications may include damage to neurons (which send and receive signals throughout the body) as well as pain and impaired motor function. This results from compression of nerves or nerve roots in the spinal cord or in the peripheral nervous system, the part of the nervous system that connects the brain and spinal cord to sensory organs such as the eyes and to other organs, muscles, and tissues throughout the body.
Depending on the mucopolysaccharidoses subtype, affected individuals may have normal intellect or may be profoundly impaired, may experience developmental delay, or may have severe behavioral problems. Many individuals have hearing loss, either conductive (in which pressure behind the ear drum causes fluid from the lining of the middle ear to build up and eventually congeal), neurosensitive (in which tiny hair cells in the inner ear are damaged), or both. Communicating hydrocephalus, in which the normal circulation of cerebrospinal fluid becomes blocked over time and causes increased pressure inside the head, is common in some of the mucopolysaccharidoses. Surgically inserting a shunt into the brain can drain fluid. The eye’s cornea often becomes cloudy from intracellular storage, and degeneration of the retina and glaucoma also may affect the patient’s vision.
Physical symptoms generally include coarse or rough facial features (including a flat nasal bridge, thick lips, and enlarged mouth and tongue), short stature with disproportionately short trunk (dwarfism), dysplasia (abnormal bone size and/or shape) and other skeletal irregularities, thickened skin, enlarged organs such as liver or spleen, hernias, and excessive body hair growth. Short and often claw-like hands, progressive joint stiffness, and carpal tunnel syndrome can restrict hand mobility and function. Recurring respiratory infections are common, as are obstructive airway disease and obstructive sleep apnea. Many affected individuals also have heart disease, often involving enlarged or diseased heart valves.
Another lysosomal storage disease often confused with the mucopolysaccharidoses is mucolipidosis. In this disorder, excessive amounts of fatty materials known as lipids (another principal component of living cells) are stored, in addition to sugars. Persons with mucolipidosis may share some of the clinical features associated with the mucopolysaccharidoses (certain facial features, bony structure abnormalities, and damage to the brain), and increased amounts of the enzymes needed to break down the lipids are found in the blood.
How are the mucopolysaccharidoses diagnosed ?
Clinical examination and urine tests (excess mucopolysaccharides are excreted in the urine) are the first steps in the diagnosis of an MPS disease. Enzyme assays (testing a variety of cells or blood in culture for enzyme deficiency) are also used to provide definitive diagnosis of one of the mucopolysaccharidoses. Prenatal diagnosis using amniocentesis and chorionic villus sampling can verify if a fetus is affected with the disorder. Genetic counseling can help parents who have a family history of the mucopolysaccharidoses determine if they are carrying the mutated gene that causes the disorders.
Prognosis and Treatment
Currently there is no cure for these disease syndromes. The prognosis depends on the type of mucopolysaccharidosis. A normal life span is possible. Some types, usually those that affect the heart, cause premature death.
In one type of mucopolysaccharidosis, attempts at replacing the abnormal enzyme have had limited, temporary success. Bone marrow transplantation and umbilical cord blood transplantation have had limited success in treating the mucopolysaccharidoses in some people. Bone marrow transplantation and umbilical cord blood transplantation are high-risk procedures and are usually performed only after family members receive extensive evaluation and counseling 20. However, death or disability often results, and this treatment remains controversial 19.
Disorders of Pyruvate Metabolism
Pyruvate is a substance that is formed in the processing of carbohydrates and proteins and that serves as an energy source for cells. Problems with pyruvate metabolism can limit a cell’s ability to produce energy and allow a buildup of lactic acid, a waste product. Many enzymes are involved in pyruvate metabolism. A hereditary deficiency in any one of these enzymes results in one of a variety of disorders, depending on which enzyme is missing.
Pyruvate metabolism disorders are caused by a lack of the ability to metabolize a substance called pyruvate. These disorders cause a buildup of lactic acid and a variety of neurologic abnormalities 19.
- A deficiency in any one of the enzymes involved in pyruvate metabolism leads to one of many disorders.
- Symptoms include seizures, intellectual disability, muscle weakness, and coordination problems.
- Some of these disorders are fatal.
- Some children are helped by diets that are either high in fat and low in carbohydrates or high in carbohydrates and low in protein.
Symptoms may develop any time between early infancy and late adulthood. Exercise and infections can worsen symptoms, leading to severe lactic acidosis. These disorders are diagnosed by measuring enzyme activity in cells from the liver or skin.
Pyruvate Dehydrogenase Complex Deficiency
This disorder is caused by a lack of a group of enzymes needed to process pyruvate. This deficiency results in a variety of symptoms, ranging from mild to severe. Some newborns with this deficiency have brain malformations. Other children appear normal at birth but develop symptoms, including weak muscles, seizures, poor coordination, and a severe balance problem, later in infancy or childhood. Intellectual disability is common.
This disorder cannot be cured, but some children are helped by a diet that is high in fat and low in carbohydrates 19.
Absence of Pyruvate Carboxylase
Pyruvate carboxylase is an enzyme. A lack of this enzyme causes a very rare condition that interferes with or blocks the production of glucose from pyruvate in the body. Lactic acid and ketones build up in the blood. Often, this disease is fatal. Children who survive have seizures and severe intellectual disability, although there are recent reports of children with milder symptoms. There is no cure, but some children are helped by eating frequent carbohydrate-rich meals and restricting dietary protein 19.
Glycogen Storage Diseases
Glycogen is made of many glucose molecules linked together. The sugar glucose is the body’s main source of energy for the muscles (including the heart) and brain. Any glucose that is not used immediately for energy is held in reserve in the liver, muscles, and kidneys in the form of glycogen and is released when needed by the body.
Glycogen storage diseases occur when there is a defect in the enzymes that are involved in the metabolism of glycogen, resulting in growth abnormalities, weakness, and confusion 19.
- Glycogen storage diseases are caused by lack of an enzyme needed to change glucose into glycogen and break down glycogen into glucose.
- Typical symptoms include weakness, sweating, confusion, kidney stones, and stunted growth.
- The diagnosis is made by examining a piece of tissue under a microscope (biopsy).
Treatment depends on the type of glycogen storage disease and usually involves regulating the intake of carbohydrates.
There are many different glycogen storage diseases (also called glycogenoses), each identified by a roman numeral. These diseases are caused by a hereditary lack of one of the enzymes that is essential to the process of forming glucose into glycogen and breaking down glycogen into glucose. About 1 in 20,000 infants has some form of glycogen storage disease.
Types and Characteristics of Glycogen Storage Diseases
Name | Affected Organs, Tissues, or Cells | Symptoms |
Name | Affected Organs, Tissues, or Cells | Symptoms |
| Type O | Liver or muscle | Episodes of low blood sugar levels (hypoglycemia) during fasting if the liver is affected |
| von Gierke’s disease (type IA) | Liver and kidney | Enlarged liver and kidney, slowed growth, very low blood sugar levels, and abnormally high levels of acid, fats, and uric acid in blood |
| Type IB | Liver and white blood cells | Same as in von Gierke’s disease but may be less severe
Low white blood cell count, recurring infections, and inflammatory bowel disease |
| Pompe’s disease (type II) | All organs | Enlarged liver and heart and muscle weakness |
| Forbes’ disease (type III) | Liver, muscle, and heart | Enlarged liver or cirrhosis, low blood sugar levels, muscle damage, heart damage, and weak bones in some people |
| Andersen’s disease (type IV) | Liver, muscle, and most tissues | Cirrhosis, muscle damage, and delayed growth and development |
| McArdle disease (type V) | Muscle | Muscle cramps or weakness during physical activity |
| Hers’ disease (type VI) | Liver | Enlarged liver
Episodes of low blood sugar during fasting
Often no symptoms |
| Tarui’s disease (type VII) | Skeletal muscle and red blood cells | Muscle cramps during physical activity and red blood cell destruction (hemolysis) |
(Source 19)
Symptoms
Some of these diseases cause few symptoms. Others are fatal. The specific symptoms, age at which symptoms start, and their severity vary considerably among these diseases. For types II, V, and VII, the main symptom is usually weakness. For types I, III, and VI, symptoms are low levels of sugar in the blood and protrusion of the abdomen (because excess or abnormal glycogen may enlarge the liver). Low levels of sugar in the blood cause weakness, sweating, confusion, and sometimes seizures and coma. Other consequences for children may include stunted growth, frequent infections, and sores in the mouth and intestines.
Glycogen storage diseases tend to cause uric acid (a waste product) to accumulate in the joints, which can cause gout, and in the kidneys, which can cause kidney stones. In type I glycogen storage disease, kidney failure is common in the second decade of life or later.
Diagnosis and Treatment
The specific type of glycogen storage disease is diagnosed by examining a piece of muscle or liver tissue under a microscope (biopsy).
Treatment depends on the type of glycogen storage disease. For most types, eating many small carbohydrate-rich meals every day helps prevent blood sugar levels from dropping. For people who have glycogen storage diseases that cause low blood sugar levels, levels are maintained by giving uncooked cornstarch every 4 to 6 hours around the clock. For others, it is sometimes necessary to give carbohydrate solutions through a stomach tube all night to prevent low blood sugar levels from occurring at night.
Galactosemia
Galactose is a sugar that is present in milk and in some fruits and vegetables. A deficient enzyme or liver dysfunction can alter the metabolism, which can lead to high levels of galactose in the blood (galactosemia). There are different forms of galactosemia, but the most common and the most severe form is referred to as classic galactosemia.
Galactosemia (a high blood level of galactose) is caused by lack of one of the enzymes necessary for metabolizing galactose, a sugar present in lactose (milk sugar). A metabolite that is toxic to the liver and kidneys builds up. The metabolite also damages the lens of the eye, causing cataracts 19.
- Galactosemia is caused by lack of one of the enzymes needed to metabolize the sugar in milk.
- Symptoms include vomiting, jaundice, diarrhea, and abnormal growth.
- The diagnosis is based on a blood test.
- Even with adequate treatment, affected children still develop mental and physical problems.
- Treatment involves completely eliminating milk and milk products from the diet.
What causes galactosemia ?
Classic galactosemia is a rare genetic metabolic disorder. The gene defect for Galactosemia is a recessive genetic trait. This faulty gene only emerges when two carriers have children together and pass it to their offspring. For each pregnancy of two such carriers, there is a 25% chance that the child will be born with the disease and a 50% chance that the child will be a carrier for the gene defect. A child born with classic galactosemia inherits a gene for galactosemia from both parents, who are carriers. A child with Duarte galactosemia inherits a gene for classic galactosemia from one parent and a Duarte variant gene from the other parent 21.
Symptoms
Newborns with galactosemia seem normal at first but, within a few days or weeks, lose their appetite, vomit, become jaundiced, have diarrhea, and stop growing normally. White blood cell function is affected, and serious infections can develop. If treatment is delayed, affected children remain short and become intellectually disabled or may die.
Diagnosis
Galactosemia is detectable with a blood test. This test is done as a routine screening test for newborns in all states in the United States. Before conception, adults with a sibling or child known to have the disorder can be tested to find out whether they carry the gene that causes the disease. If two carriers conceive a child, that child has a 1 in 4 chance of being born with the disease.
Prognosis
If galactosemia is recognized at birth and adequately treated, liver and kidney problems do not develop, and initial mental development is normal. However, even with adequate treatment, children with galactosemia may have a lower intelligence quotient (IQ) than their siblings, and they often have speech problems. Girls often have ovaries that do not function, and only a few are able to conceive naturally. Boys, however, have normal testicular function.
Treatment
Galactosemia is treated by completely eliminating milk and milk products—the source of galactose—from an affected child’s diet. Galactose is also present in some fruits, vegetables, and sea products, such as seaweed. Doctors are not sure whether the small amounts in these foods cause problems in the long term. People who have the disorder must restrict galactose intake throughout life.
References- Wade LG., Jr . Organic Chemistry. Prentice-Hall, Inc; 1999.
- Use of lectins as diagnostic and therapeutic tools for cancer. Mody R, Joshi S, Chaney W. J Pharmacol Toxicol Methods. 1995 Feb; 33(1):1-10. https://www.ncbi.nlm.nih.gov/pubmed/7727802/
- On the role of cell surface carbohydrates and their binding proteins (lectins) in tumor metastasis. Gorelik E, Galili U, Raz A. Cancer Metastasis Rev. 2001; 20(3-4):245-77. https://www.ncbi.nlm.nih.gov/pubmed/12085965/
- Lectins as cell recognition molecules. Sharon N, Lis H. Science. 1989 Oct 13; 246(4927):227-34. https://www.ncbi.nlm.nih.gov/pubmed/2552581/
- Carbohydrates in cell recognition. Sharon N, Lis H. Sci Am. 1993 Jan; 268(1):82-9. https://www.ncbi.nlm.nih.gov/pubmed/7678182/
- Carbohydrates and glycoconjugates. Glycomics: the new era of carbohydrate biology. Feizi T, Mulloy B. Curr Opin Struct Biol. 2003 Oct; 13(5):602-4. https://www.ncbi.nlm.nih.gov/pubmed/14568615/
- Concepts and principles of glycobiology. Opdenakker G, Rudd PM, Ponting CP, Dwek RA. FASEB J. 1993 Nov; 7(14):1330-7. https://www.ncbi.nlm.nih.gov/pubmed/8224606/
- Regulatory role of GM3 ganglioside in alpha 5 beta 1 integrin receptor for fibronectin-mediated adhesion of FUA169 cells. Zheng M, Fang H, Tsuruoka T, Tsuji T, Sasaki T, Hakomori S. J Biol Chem. 1993 Jan 25; 268(3):2217-22. https://www.ncbi.nlm.nih.gov/pubmed/8420989/
- Taylor ME, Drickamer K. Introduction to Glycobiology. Second. Oxford University Press; 2006.
- American Diabetes Association. Glycemic Index and Diabetes. http://www.diabetes.org/food-and-fitness/food/what-can-i-eat/understanding-carbohydrates/glycemic-index-and-diabetes.html
- American Diabetes Association. Carbohydrate Counting. http://www.diabetes.org/food-and-fitness/food/what-can-i-eat/understanding-carbohydrates/carbohydrate-counting.html
- U.S. Department of Health and Human Services and U.S. Department of Agriculture. 2015–2020 Dietary Guidelines for Americans. 8th Edition. December 2015. https://health.gov/dietaryguidelines/2015/
- Cell Metab. 2015 Sep 1;22(3):427-36. doi: 10.1016/j.cmet.2015.07.021. Epub 2015 Aug 13. Calorie for Calorie, Dietary Fat Restriction Results in More Body Fat Loss than Carbohydrate Restriction in People with Obesity. https://www.ncbi.nlm.nih.gov/pubmed/26278052
- American Heart Association – Sugar Intake – http://www.heart.org/HEARTORG/Encyclopedia/Heart-and-Stroke-Encyclopedia_UCM_445084_ContentIndex.jsp?title=sugar%20intake
- The Scientific Advisory Committee on Nutrition. (2015) Carbohydrates and Health. Online. Available from: https://www.gov.uk/government/groups/scientific-advisory-committee-on-nutrition
- National Institute of Diabetes and Digestive and Kidney Diseases. Carbohydrate Counting & Diabetes. https://www.niddk.nih.gov/health-information/diabetes/overview/diet-eating-physical-activity/carbohydrate-counting
- Cochrane Review 30 April 2014. Steroidal contraceptives: effect on carbohydrate metabolism in women without diabetes mellitus. http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD006133.pub5/abstract;jsessionid=DEC8C81D2DDFF87C4197FF95D69BE4EE.f04t03
- U.S. National Library of Medicine. Medline Plus. Carbohydrate Metabolism Disorders. https://medlineplus.gov/carbohydratemetabolismdisorders.html
- Merck Sharp & Dohme Corp. Merck Manual. Disorders of Carbohydrate Metabolism. https://www.merckmanuals.com/home/children-s-health-issues/hereditary-metabolic-disorders/disorders-of-carbohydrate-metabolism
- National Institute of Neurological Disorders and Stroke. Mucopolysaccharidoses Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Mucopolysaccharidoses-Information-Page
- American Liver Foundation. Galactosemia. http://www.liverfoundation.org/abouttheliver/info/galactosemia/



{kind=link}