
What is Usher syndrome
Usher syndrome is a rare genetic disorder characterized by partial or total hearing loss and vision loss that worsens over time and sometimes it also affects balance 1. The hearing loss is classified as sensorineural hearing loss, which means that it is caused by abnormalities of the inner ear. The loss of vision is caused by an eye disease called retinitis pigmentosa, which affects the layer of light-sensitive tissue at the back of the eye (the retina). Vision loss occurs as the light-sensing cells of the retina gradually deteriorate. Night vision loss begins first, followed by blind spots that develop in the side (peripheral) vision. Over time, these blind spots enlarge and merge to produce tunnel vision (loss of all peripheral vision). In some cases, vision is further impaired by clouding of the lens of the eye (cataracts). However, many people with retinitis pigmentosa retain some central vision throughout their lives.
Usher syndrome is the most common genetic cause of combined deafness and blindness. Usher syndrome is the most common genetic disorder involving both hearing and vision abnormalities. Usher syndrome types 1 and 2 account for approximately 10 percent of all cases of moderate to profound deafness in children. More than 400,000 people are affected by Usher syndrome worldwide. Higher than average numbers of people with Usher syndrome have been found among Jewish people in Israel, Berlin, Germany; French Canadians of Louisiana; Argentineans of Spanish descent; and Nigerian Africans. USH3, the rarest form in most populations, comprises about 40% of Usher patients in Finland.
There is currently no cure for Usher syndrome 2.
Researchers have identified three major types of Usher syndrome, designated as types 1, 2, and 3. The different types are distinguished by the severity of hearing loss, the presence or absence of balance problems, and the age at which signs and symptoms appear. The types are further divided into subtypes based on their genetic cause. All three types of Usher syndrome are inherited in an autosomal recessive manner 1. Treatment for the hearing loss may include hearing aids or surgery for a cochlear implant. Vitamin A palmitate is useful for treating the vision loss in people with Usher syndrome type 2 3.
Most individuals with Usher syndrome type 1 are born with severe to profound hearing loss. Progressive vision loss caused by retinitis pigmentosa becomes apparent in childhood. This type of Usher syndrome also causes abnormalities of the vestibular system, which is the part of the inner ear that helps maintain the body’s balance and orientation in space. As a result of the vestibular abnormalities, children with the condition have trouble with balance. They begin sitting independently and walking later than usual, and they may have difficulty riding a bicycle and playing certain sports.
Usher syndrome type 2 is characterized by hearing loss from birth and progressive vision loss that begins in adolescence or adulthood. The hearing loss associated with this form of Usher syndrome ranges from mild to severe and mainly affects the ability to hear high-frequency sounds. For example, it is difficult for affected individuals to hear high, soft speech sounds, such as those of the letters d and t. The degree of hearing loss varies within and among families with this condition, and it may become more severe over time. Unlike the other forms of Usher syndrome, type II is not associated with vestibular abnormalities that cause difficulties with balance.
People with Usher syndrome type 3 experience hearing loss and vision loss beginning somewhat later in life. Unlike the other forms of Usher syndrome type 3 is usually associated with normal hearing at birth. Hearing loss typically begins during late childhood or adolescence, after the development of speech, and becomes more severe over time. By middle age, most affected individuals have profound hearing loss. Vision loss caused by retinitis pigmentosa also develops in late childhood or adolescence. Some people with Usher syndrome type 3 develop vestibular abnormalities that cause problems with balance.
Usher syndrome types 1 and 2 are the most common forms of Usher syndrome in most countries. Certain genetic mutations resulting in type 1 Usher syndrome are more common among people of Ashkenazi (eastern and central European) Jewish or French Acadian heritage than in the general population.
Usher syndrome type 3 represents only about 2 percent of all Usher syndrome cases overall. However, Usher syndrome type 3 occurs more frequently in the Finnish population, where it accounts for about 40 percent of cases, and among people of Ashkenazi Jewish heritage.
Why is early diagnosis of Usher syndrome important?
Early diagnosis is critical for children with Usher syndrome. While there is no definitive cure for Usher syndrome, there are a lot of treatments available. There are treatments for hearing loss, treatments for balance issues, and, yes, treatments for vision loss. And many of these treatments are most successful when begun very early in life.
In short, there is something that can be done. Lots of somethings. But you need a definitive diagnosis and you need it as early as possible. Here are just some of the reasons why early diagnosis is a good thing for a family:
Communication Skills
Kids with Usher syndrome have hearing loss. Regardless of the communication method a family chooses, be it sign language or oral communication, early detection is the key. Most language is developed in the first five years of life. You need to get started right away and knowing whether you’re dealing with Usher needs to be part of the decision process.
Safety Concerns
A child with hearing loss does not need mobility training, but a child with Usher syndrome might. Many parents of children with Usher report suspecting that their child had problems seeing at night, but they often only recognize it after an accident has occurred.
Balance is also a concern in families with Usher. Riding a bike, ice skating or simply hiking a steep trail requires different preparation, skills and awareness for kids with Usher. Parents and kids can get creative and plan ahead when they have a diagnosis.
Educational Support Planning
Families that have a child with undiagnosed Usher syndrome may only consider supports for hearing loss as part of the Individualized Education Plan (IEP). Early diagnosis of Usher can change that before visual struggles start to appear. Vision loss changes the way a child learns. For example, a child with night vision problems might need a high contrast or tactile diagram of the night sky for an astronomy class. Those types of visual accommodations need to be part of a child’s Individualized Education Plan.
Future Clinical Trials
There are treatments nearing or in clinical trial that hold the hope of slowing, stopping, or even reversing the vision loss associated with Usher syndrome. But almost all of these treatments are diagnosis specific. In other words, it’s not enough to suspect Usher syndrome. You have to know definitively and you have to know the specific genetic cause.
Family Planning
Many families of children with hearing loss ask the question “will my next child have hearing loss, too?” Usher syndrome is an autosomal recessive genetic disorder. Each child born to carrier parents has a 25% chance of having Usher syndrome. A definitive diagnosis of Usher syndrome will assist parents who are planning to expand their family to make an informed decision.
Satisfies the need to know
Knowing removes the doubt. There are thousands of adults with Usher syndrome living happy, productive lives. Knowing this gives families a chance to support their child to take control of their own lives, to find positive adult role models with Usher syndrome, and to envision a future that is vibrant and bright.
Usher syndrome types
There are 3 types of Usher syndrome:
| Type 1 | Type 2 | Type 3 | |
|---|---|---|---|
| Hearing | Profound hearing loss or deafness at birth. | Moderate to severe hearing loss at birth. | Progressive hearing loss in childhood or early teens. |
| Vision | Decreased night vision by age 10, progressing to severe vision loss by midlife. | Decreased night vision by adolescence, progressing to severe vision loss by midlife. | Varies in severity and age of onset; night vision problems often begin in teens and progress to severe vision loss by midlife. |
| Balance (vestibular function) | Balance problems from birth. | Normal balance. | Normal to near-normal balance in childhood; chance of later problems. |
Usher syndrome type 1
People with Usher syndrome type 1 are deaf from birth and have severe balance problems from a young age. Vision problems usually start by age 10 and lead to blindness.
Usher syndrome type 1 is a recessive genetic disease. This means that a child receives two copies of the same Usher 1 gene, one from each parent.
Children with Usher type 1 are usually born profoundly deaf and experience progressive vision loss due to a retinal disease called retinitis pigmentosa (retinitis pigmentosa). The symptoms of retinitis pigmentosa first manifest as difficulty seeing in dimly lit areas – or night blindness – and a gradual loss of peripheral vision (tunnel vision). These symptoms will generally be noticed before the age of 10 and continue through adulthood.
Children and adults with Usher type 1 usually have vestibular issues which affects balance. Balance issues manifest early in children with Usher type 1, and they often learn to sit up and walk later than their peers. Balance problems can become more pronounced as visual fields decrease.
Today, Usher syndrome is usually diagnosed before adulthood, but many older people report that the diagnosis wasn’t made until later in life when the vision loss from retinitis pigmentosa became severe enough to interfere with their mobility. Genetic testing – usually through a simple blood test – is the only way to definitively diagnose Usher syndrome.
Hearing
Hearing loss in children with Usher syndrome type 1 is usually severe to profound and present at birth.
Vision
A person with Usher syndrome type 1 may become legally blind as a young adult primarily because of severe tunnel vision. Central vision is usually retained into adulthood, allowing individuals to continue to read print and use a computer. Some adults with Usher syndrome type 1 will lose central vision more quickly, retaining only light perception.
Balance
Individuals with Usher syndrome type 1 often have limited vestibular functionality. This can result in severe balance issues. Children with Usher type I are often late sitters or late walkers. It is not uncommon for children with type I Usher to begin walking at 24-36 months. These balance issues are present throughout life and often become more pronounced in low light or as the vision degrades.
Physical and occupational therapy can help individuals with type I with their balance issues. Core strengthening and hippotherapy can improve a person’s ability to compensate for the decreased vestibular functionality. Mobility training can also be also helpful.
Genes involved in Usher syndrome type 1
Usher syndrome type I is the most severe form of Usher syndrome and is characterized by congenital, profound, sensorineural hearing loss; vestibular dysfunction, often manifested as delayed walking (>18 months); and the onset of retinitis pigmentosa by the age of ten 4. Usher syndrome type 1 is further subdivided into 5 types. Mutations in the MYO7A, USH1C, CDH23, PCDH15, and USH1G (SANS) genes cause Usher syndrome type 1B, type 1C, type 1D, type 1F and type 1G respectively 4.
Mutations in these genes account for most cases of Usher syndrome Type 1. Mutations in MYO7A are the most common accounting for 39-55% of cases. An Ashkenazi Jewish founder mutation, R245X, has been identified the PCDH15 gene which has a carrier frequency of up to 2.5% in this population. MYO7A, USH1C, CDH23, and PCDH15 mutations have also been reported in several families with recessive nonsyndromic hearing loss. In addition, a few families with autosomal dominant nonsyndromic hearing loss (DFNA11) have also been described with mutations in MYO7A.
Although these are the most common characteristics of Usher syndrome due to mutations in these genes, there can be variations. Overlapping and atypical presentations have been described for all three types of Usher syndrome. For example some individuals with mutations in type I genes may have a milder presentation (moderate hearing loss and/or a normal vestibular system). In addition, certain genes (MYO7A, USH1C, CDH23, PCDH15, and DFNB31) can cause isolated hearing loss without developing retinitis pigmentosa.
Identified disease causing mutations in all of these genes include missense, nonsense, frameshift, splice-site as well as deletions distributed across nearly all exons.
Usher syndrome type 1 subtypes
Although all individuals with Usher type 1 demonstrate similar symptoms, the genetic causes differ. Some are more common than others. Usher subtype 1b is by far the most common form of Usher type 1 and accounts for over 40% of all cases. Subtype 1d appears to be next most common and is responsible for about 25% of all cases. Usher Ic is not very common in the general USA population, but it is common among French Acadians in Louisiana. Usher 1f and 1g are uncommon and so far only a few cases have been reported. There is a form of Usher 1f that is common among people with Ashkenazi Jewish ancestry. Usher 1a does not exist and 1e is quite rare. There is evidence that there are more Usher 1 subtypes that have not yet been identified.
Usher syndrome type 2
People with Usher syndrome type 2 have moderate to severe hearing loss and normal balance. Vision problems start in the early teens and get worse more slowly than in type 1.
Individuals with Usher syndrome type 2 are born hard-of-hearing and gradually lose their vision due to retinitis pigmentosa. Hearing tests show a sloping hearing loss that is mild to moderate in the low frequencies and severe to profound in the high frequencies. Vestibular problems are absent in Usher syndrome type 2, which distinguishes it from Usher syndrome type 1. Retinitis pigmentosa generally manifests in the teen years and progresses throughout life.
Today, most diagnoses are made before adulthood, but older people still report that the diagnosis wasn’t made until later in life when the retinitis pigmentosa became severe enough to interfere with their mobility.
Hearing
People with Usher syndrome type 2 are born with a moderate to severe hearing loss. The hearing loss is milder in the low frequencies and more severe in the higher one. Since most speech involves the lower frequencies, this means that adults and children with Usher syndrome type 2 will usually have good oral communication skills.
Vision
A person with Usher syndrome 2 may become legally blind as a young adult primarily because of severe tunnel vision. While most adults retain good central vision for most of their lives, some with Usher syndrome 2 will lose their functional vision as they age.
Balance
Individuals with Usher syndrome type 2 rarely experience balance issues.
Genes involved in Usher syndrome type 2
There are three subtypes of Usher syndrome type 2 which have known gene locations: USH2A, GPR98 (VLGR1)and DFNB31 (WHRN).
USH2A accounts for approximately 80%, GPR98 (VLGR1) accounts for approximately 15%, and DFNB31 accounts for approximately 5% of Usher syndrome type 2 cases. DFNB31 mutations have also been reported in several families with recessive nonsyndromic hearing loss. Identified disease-causing mutations in all of these genes include missense, nonsense, frameshift, splice-site as well as deletions distributed across nearly all exons.
Some mutations in USH2A can cause retinitis pigmentosa without hearing loss.
Usher syndrome type 2 subtypes
Subtype 2c appears to be uncommon. Because the genes for 2b and 2d have not been identified, we don’t have any idea about their frequencies, but we believe them to be uncommon. There is evidence that there are more Usher 2 subtypes that have not been recognized.
Usher syndrome type 3
People with Usher syndrome type 3 are born with normal hearing and near-normal balance but develop vision problems and then hearing loss.
Usher syndrome type 3 is the rarest form of Usher syndrome, characterized by later onset hearing loss, retinitis pigmentosa that manifests between the second and fourth decades of life and variable vestibular dysfunction. Forty-two percent of those with Usher syndrome in Finland are thought to have USH3. It is also found in individuals of Ashkenazi Jewish heritage.
Hearing
Individuals are born with normal hearing. Hearing loss begins during lat childhood or adolescence and progresses to profound hearing loss.
Vision
Children born with Usher type 3 have retinitis pigmentosa that manifests in later childhood or early adolescence.
Vestibular Function
Vestibular (balance) function is typical at birth and may become affected in some with Usher syndrome type 3 in adult years.
Genes involved Usher syndrome type 3
To date, only mutations in the CLRN1 (USH3A) and HARS (USH3B) genes are known to cause Usher syndrome type 3. Identified disease causing mutations in these genes include missense, nonsense, frameshift, splice-site as well as deletions distributed across nearly all exons.
What is retinitis pigmentosa?
Retinitis pigmentosa refers to a group of rare inherited diseases causing retinal degeneration — that involve a breakdown and loss of cells in the retina. The retina is a thin piece of tissue lining the back of the eye. It converts light into electrical signals that the brain interprets as vision. People with retinitis pigmentosa experience a gradual decline in their vision, because photoreceptor cells in the retina degenerate. Common symptoms include difficulty seeing at night and a loss of side (peripheral) vision.
An estimated 100,000 people in the U.S. have retinitis pigmentosa, mainly caused by gene mutations (variations) inherited from one or both parents. Forms of retinitis pigmentosa and related diseases include Usher syndrome, Leber congenital amaurosis, and Bardet-Biedl syndrome, among others.
Retinitis pigmentosa is characterized by diffuse progressive dysfunction of predominantly rod photoreceptors, with subsequent degeneration of cone photoreceptors, and retinal pigment epithelium (RPE) 5. In the early stages of retinitis pigmentosa, rods are more severely affected than cones. As the rods die, people experience night blindness and a progressive loss of the visual field, the area of space that is visible at a given instant without moving the eyes. The loss of rods eventually leads to a breakdown and loss of cones. In the late stages of retinitis pigmentosa, as cones die, people tend to lose more of the visual field, developing ìtunnel vision. They may have difficulty performing essential tasks of daily living such as reading, driving, walking without assistance, or recognizing faces and objects.
Pathologic findings of an enucleated eye in a patient with autosomal recessive retinitis pigmentosa showed that the rod and cone outer segments were shortened and disorganized in the patient’s best field of vision, while in the area of visual loss; there was total loss of outer segments and a decrease in photoreceptors number 6. Two types of pigmented cells were found invading the retina: typical retinal pigment epithelium (RPE) cells that were migrating away from the retinal pigment epithelial layer, and macrophage-like cells that contained melanin. These changes were thought to be a reactive response to photoreceptor damage, since the retinal pigment epithelium (RPE) appeared relatively normal morphologically in areas of early photoreceptor involvement. A recent review described histopathologic findings in 10 patients with autosomal dominant retinitis pigmentosa, including poorly organized, shortened or absent outer segments with shortened inner segments. Inclusion bodies and/or perinuclear cytoplasmic membranous swirls were found in three cases 7. Visual impairment usually manifest as night blindness and progressive visual field loss. Retinitis pigmentosa prevalence is 1:3000 to 1:5000 8. Retinitis pigmentosa may be seen in isolation (typical retinitis pigmentosa) or in association with systemic disease.
Figure 1. Retinitis Pigmentosa
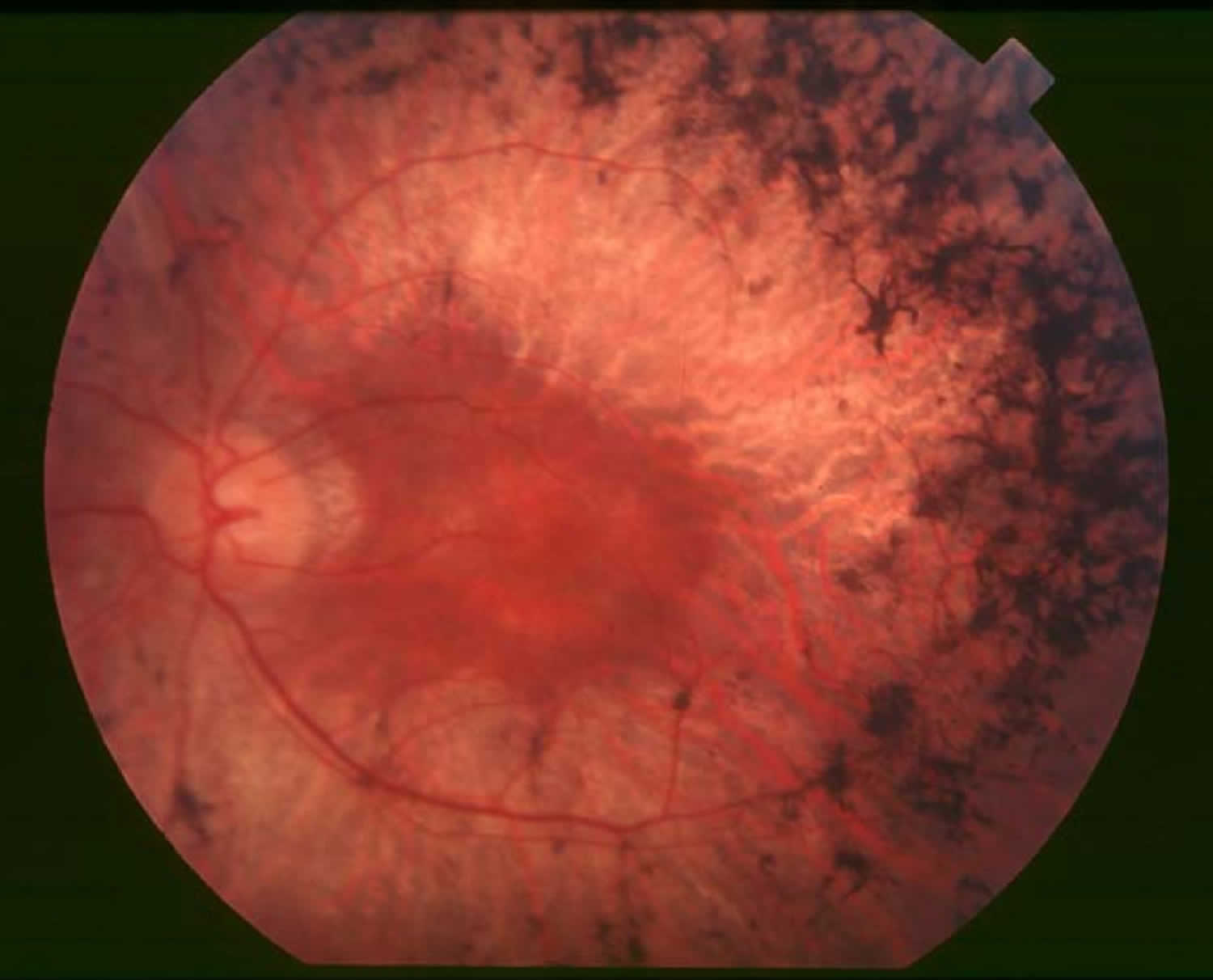
Retinitis pigmentosa symptoms
Symptoms depend on whether rods or cones are initially involved. In most forms of retinitis pigmentosa, rods are affected first. Because rods are concentrated in the outer portions of the retina and are triggered by dim light, their degeneration affects peripheral and night vision. When the disease progresses and cones become affected, visual acuity, color perception, and central vision are diminished.
Night blindness is one of the earliest and most frequent symptoms of retinitis pigmentosa. People with mainly cone degeneration, however, first experience decreased central vision and reduced ability to discriminate colors and perceive details.
Patients typically presents with night vision problems (unable to see in the dark or slow to adjusting to dark), progressive peripheral vision restriction, and tunnel vision at later stage of the disease.
Retinitis pigmentosa is typically diagnosed in adolescents and young adults. It is a progressive disorder. The rate of progression and degree of visual loss varies from person to person. Most people with retinitis pigmentosa are legally blind by age 40, with a central visual field of less than 20 degrees in diameter. It is a genetic disorder and, therefore, is almost always inherited.
It is rare for patients to lose all vision in both eyes. In a large study involving close to 1,000 patients with retinitis pigmentosa and Usher Syndrome at age 45 or older, one fourth of the patients had a visual acuity of 20/200 or worse in both eyes, and more than half had a visual acuity of 20/40 or better in at least one eye. Only 0.5% of patients were completely blind in both eyes 9. In one study, about 50% of retinitis pigmentosa patients reported having headaches, and 35% of retinitis pigmentosa patients reported light flashes 10.
How does retinitis pigmentosa progress?
The symptoms of retinitis pigmentosa typically appear in childhood. Children often have difficulty getting around in the dark. It can also take abnormally long periods of time to adjust to changes in lighting. As their visual field becomes restricted, patients often trip over things and appear clumsy. People with retinitis pigmentosa often find bright lights uncomfortable, a condition known as photophobia. Because there are many gene mutations that cause the disorder, its progression can differ greatly from person to person. Some people retain central vision and a restricted visual field into their 50s, while others experience significant vision loss in early adulthood. Eventually, most individuals with retinitis pigmentosa will lose most of their sight.
How is retinitis pigmentosa diagnosed?
Retinitis pigmentosa is diagnosed in part through an examination of the retina. An eye care professional will use an ophthalmoscope, a tool that allows for a wider, clear view of the retina. This typically reveals abnormal, dark pigment deposits that streak the retina. These pigment deposits are in part why the disorder was named retinitis pigmentosa. Other tests for retinitis pigmentosa include:
Diagnostic procedures
- Full-Field Electroretinogram (ERG): Electroretinogram measures the electrical potential generated by rods and cones after a light stimulus and is essential in the diagnosis of retinitis pigmentosa. The most important parameters being measured include a- and b-wave amplitudes and implicit times. In early stages of the disease, there is reduction in a- and b-wave amplitudes but implicit time can be prolonged or normal. Patients with advanced stages have non-detectable electroretinogram.
- Dark adaptometry (DA): Visual threshold is the minimum intensity of light that will stimulate the rods or cones to elicit a subjective response. Dark adaptometry measures the absolute threshold of rods at given time intervals as the retina adapts to the dark. In retinitis pigmentosa, there is increased absolute rod threshold and dark adaptation is usually prolonged. This test maybe useful in detecting early cases 11.
- Visual field: Kinetic perimetry with Goldmann perimeter characteristically shows a ring scotoma in the mid-periphery of the visual field. They usually start as a group of isolated scotomas around 20 degrees from fixation, and gradually coalesce to form a partial followed by a complete ring. The outer edge of the ring expands relatively quickly to the periphery, while the inner edge constricts slowly toward fixation. Patients often have good central vision from a small central island (“tunnel vision”) until their 50’s or 60’s 12. Visual field testing is useful in monitoring the progression of disease and document the status of legal blindness.
- Electrooculogram (EOG) is a measurement of standing potential between the cornea and the retina and is a measurement of function of the retinitis pigment epitehlium and photoreceptors. It is usually abnormal in retinitis pigmentosa. However, electroretinogram is considered a more sensitive test for detection of photoreceptor function and consequently electrooculogram is not routinely done.
- Optical coherence tomography (OCT): Optical coherence tomography is a quick, inexpensive, and widely available tool to detect cystic macular lesions, epiretinal membrane, and vitreomaular traction syndrome observed in some retinitis pigmentosa patients with decreased central vision. One study also showed mild inner retinal layer thinning and severe outer retinal layer thinning using spectral domain optical coherence tomography 13
- Fluorescein angiography: Fluorescein angiography may have a role in documenting early deterioration of the retinal pigment epithelium and especially in female carriers of X-linked retinitis pigmentosa. It has a role in patients with cystic macular lesions and exudative vasculopathy.
Laboratory test
- Genetic testing can be helpful in confirming the diagnosis. It also helps assess the risk of passing retinitis pigmentosa from parent to offspring.
- Other laboratory testings that can be helpful in differentiating atypical cases of retinitis pigmentosa from other ocular disorders include serologies for syphilis, serum ornithine level or ornithine-lysine ratio (for gyrate atrophy of the retina and choroid), and serum phytanic acid level (for Refsum disease).
Retinitis pigmentosa treatment
Many treatments have been explored without proven benefit for the isolated forms of retinitis pigmentosa 14. These include various vitamins and minerals, vasodilators, tissue therapy with placental extract, cortisone, cervical sympathectomy, injections of a hydrolysate of yeast RNA, ultrasound, transfer factor, dimethyl sulfoxide, ozone, muscle transplants, and subretinal injections of fetal retinal cells. None of the above treatments were conducted in randomized, controlled clinical trials. It is important to note that anecdotal treatment with subjective improvement of visual function should be interpreted with caution due to fluctuation in visual acuity and visual fields in this disease. ERG (electroretinogram) is a better objective measure of remaining retinal function. Any potential therapy will likely require several years of follow-up to assess efficacy due to the nature of slow progression of this disease.
Medical therapy
Controversies exist regarding the use of high dose vitamin A, docosahexaenoic acid (DHA), and lutein to slow the progression of retinitis pigmentosa. Berson et al. 15 conducted three large randomized, controlled, double-masked trials. In the first study, 601 adult patients were randomized to one of four treatment groups: vitamin A, 15,000 IU/day plus vitamin E 3 IU/day; vitamin A 75 IU/day plus vitamin E, 3 IU/day; vitamin A, 15,000 IU/day plus vitamin E, 400 IU/day; and vitamin A, 75 IU/day plus vitamin E, 400 IU/day. The main outcome variable was the 30-Hz cone flicker ERG (electroretinogram). In summary, patients who are on the higher dose of vitamin A had the slowest annual rate of decline in remaining ERG (electroretinogram) amplitude (8.3% of decline per year) while those on high dose vitamin E had the fastest (11.8%). The results were more significant in the cohort with higher amplitudes to start with (i.e., > 0.68 μV).
In the second study, patients who were given vitamin A palmitate 15,000 IU/day were randomized to either docosahexaenoic acid (DHA) capsules (1200 mg/day) or control fatty acid capsules. The main outcome variable was the total point score of the 30-2 Humphrey visual field. Overall, DHA supplementation by capsules did not slow the course of retinitis pigmentosa over a 4-year interval. However, for those who are taking vitamin A for the first time, a subgroup analysis concluded DHA supplement slowed the rate of visual field loss and log ERG amplitude loss in years 1 and 2, but not in years 3 and 4 after the start of treatment.
In the third study, they evaluated the supplemental effects of lutein 12 mg/day combined with high dose vitamin A and high dietary intake of DHA on the rate of retinitis pigmentosa visual field loss. The investigators reported no difference between groups in the rate of decline in the total point score for the HFA 30-2 program (primary outcome measure, p=0.66), nor loss of HFA 30-2 plus 60-4 total point score, logERG amplitude, and logMAR visual acuity (secondary outcomes). However, they did report a significant effect of treatment on the rate of decline for the HFA 60-4 total point score.
Based on these studies, the authors 15 concluded that patients with retinitis pigmentosa would benefit from taking 12 mg of lutein per day in addition to 15,000 IU/d of vitamin A and weekly meals of oily fish, of which DHA is a major component. However, there were some debates regarding these recommendations 16. For example, members of the Data and Safety Monitoring Committee from the first study reported that much of the originally reported significant difference was a consequence of pooling the data and could be attributed to early and consistently large differences between the vitamin E group and all of the other groups 17. In the 2nd and 3rd study, conclusions were drawn based on secondary outcomes and subgroup analyses, rather than primary outcome 16. Therefore, the use of high dose vitamin A and other supplements must be weighed against their potential side effects.
The precise mechanism by which vitamin A supplementation provides its benefit is not known. It has been speculated that vitamin A rescues remaining cones, thereby explaining how one supplement may help a group of patients with different rod-specific gene defects. Vitamin E may lead to an adverse effect on the course of retinitis pigmentosa by inhibiting the absorption or transport of vitamin A. DHA is thought to facilitate the release of vitamin A from its carrier protein (interphotoreceptor retinoid binding protein) in the subretinal space.
Other treatment considerations
Patients who develop cystic macular lesions (about 30%) may benefit from oral acetazolamide 18, topical dorzolamide drops 19, and intravitreal steroids in some cases. Anti-VEGF intravitreal injection has also been shown to be effective in a small case series 20. The long-term efficacy of topical dorzolamide in improving the macular cystic lesions in patients with retinitis pigmentosa and Usher syndrome has been been demonstrated in a retrospective series with a mean follow-up of 39 months 21.
Although light deprivation has not been shown to be of benefit in altering the course of retinal degeneration 22, it is generally advisable for patients to use ultraviolet and short-wavelength (blue) blocking sunglasses for outdoor activities. Audiology consults should be considered for patients with possible or known diagnosis of Usher syndrome. Low vision services are designed to benefit those whose ability to function is compromised by visual impairment. A low vision examination may be useful to help optimize the use of remaining visual function. Genetic counseling can provide patients and families with information on the inheritance and implications of their genetic disorders and can help them make informed medical and personal decisions.
Medical follow up
Annual ocular examinations usually are sufficient to measure visual acuity and Goldmann visual field. If medical treatment is initiated, more frequent visits and laboratory blood work may be indicated. For example, patients with red blood cell docosahexaenoic acid (DHA) level of at least 4% of total red blood cell fatty acids has been reported to have, on average, a slower rate of decline of visual field sensitivity than those with lower levels 23. Vitamin A levels and liver function tests should also be done annually if treatment has been initiated.
Treatment of rare forms of retinitis pigmentosa
In patients with hereditary abetalipoproteinemia (Bassen–Kornzweig syndrome), mutations in the gene encoding a microsomal triglyceride transfer protein lead to depletion of vitamin A in the liver and the retina 24. A low-fat diet and supplementation of fat soluable vitamins A, E, and K are recommended. In patients with Refsum disease, they inherit a defective enzyme that can lead to accumulation of excess phytanic acid 25. Findings other than retinopathy include peripheral neuropathy and ataxia. Treatment consists of restricting food items that contain phytanic acid (including animal fats, milk products, and green leafy vegetables containing phytol) while maintaining body weight.
Surgery
There is now an FDA approved Humanitarian Device, called the ARGUS II implant, which may help patients with end-stage retinitis pigmentosa. It is approved for use in patients with bare light to no light perception. It consists of 3 parts: a video recorder, a transmitter and the implant itself. The implant is an epiretinal electrode chip coated in silicone that stimulates the retina electrically. It is connected to a silicone strip that carries the electrodes from the receiver. This strip encircles the eyeball and is surgically sewn onto the sclera. The wireless receiver receives electrical signals from a video recorder which is mounted to glasses on the patient’s face. The video unit converts the video images into electrical impulses which are transmitted to the receiver. The retinal stimulation results in the patient seeing lines or dots of light that indicate edges or objects in the patient’s field of vision. The patient does not see in color and the resolution does not allow for “seeing faces or small details.” Prior research on the ARGUS II showed that patients are better able to find doors, walk along a path and identify the location and movement of objects with the device turned “on” than without the device 26.
In patients with another form of retinitis pigmentosa, Leber’s variant, gene therapy for retinitis pigmentosa epithelium 65 is being performed. This technique requires the injection of the gene into the eye, specifically into the space under the center of the retina (macular subretinal space). Replacement of the gene in younger patients (versus adults) has allowed patients to gain vision. This treatment is produced by Spark Therapeutics.
If the patient develops a cataract, it is generally advisable to defer surgical removal until the patient can no longer read with the better eye. In one study of 30 patients with retinitis pigmentosa, 83% improved by 2 lines on the Snellen visual acuity chart with cataract surgery 27.
Retinitis pigmentosa treatment complications
In general, toxicity from vitamin A treatment is rare. As a safety measure, patients should have a pretreatment assessment of fasting serum vitamin A levels and liver function and annually thereafter. Because of the potential for birth defects, women who are pregnant or planning to conceive are advised not to take high doses of vitamin A (15,000 IU/day). In older adults, long-term vitamin A supplementation has been associated with a decrease in bone density and up to a 1% increased risk of hip fractures 28. Therefore, postmenopausal women and men over the age of 49 who are taking vitamin A should consult with their primary care physician regarding their bone health. Patients with renal failure or renal transplant should not take vitamin A due to excessive renal re-absorption. Finally, vitamin A should not be given to patients on chronic doxycycline because the combination can lead to increased intracranial pressure.
The 5 year study of the ARGUS II Implant supports the long term safety and benefit of the implant for those blind from retinitis pigmentosa 29. A collaborative has also recently published their recommendations to optimize patient outcomes 30. Most common complications are conjunctival erosion and hypotony. It is rare that the implant would require removal.
Retinitis pigmentosa prognosis
Some studies suggest that the rate of progression, age of onset, and eventual visual loss are related to the mode of inheritance. Autosomal dominant retinitis pigmentosa has the best prognosis, with the majority of patients under 30 years having visual acuity of 20/30 or better. X-linked is the most severe form with appreciable impairment of central visual acuity to 20/200 or less by the fifth decade of life. Autosomal recessive and sporadic cases were intermediate in severity 31. In terms of visual field loss, a study of 104 patients with autosomal dominant retinitis pigmentosa shows 93% of patients under age 20, 89% of those from 20-40, and 60% over the age of 40 had a central visual field radius of 10 degrees or greater with the IV4e test object 32.
Living with vision loss
A number of services and devices are available to help people with vision loss carry out daily activities and maintain their independence. In addition to an eye care professional, it’s important to have help from a team of experts, which may include occupational therapists, orientation and mobility specialists, certified low vision therapists, and others.
Children with retinitis pigmentosa may benefit from low vision aids that maximize existing vision. For example, there are special lenses that magnify central vision to expand visual field and eliminate glare. Computer programs that read text are readily available. Closed circuit televisions with a camera can adjust text to suit oneís vision. Portable lighting devices can adjust a dark or dim environment. Mobility training can teach people to use a cane or a guide dog, and eye scanning techniques can help people to optimize remaining vision. Once a child is diagnosed, he or she will be referred to a low vision specialist for a comprehensive evaluation. Parents may also want to meet with the child’s school administrators and teachers to make sure that necessary accommodations are put in place.
For parents of children with retinitis pigmentosa, one challenge is to determine when a child might need to learn to use a cane or a guide dog. Having regular eye examinations to measure the progress of the disorder will help parents make informed decisions regarding low vision services and rehabilitation.
Does Usher syndrome always result in blindness?
Very few people with Usher syndrome will become totally blind – that is, have no light perception 33. Some adults maintain a small degree of central vision throughout their lives while others lose all usable vision. Natural history studies are currently collecting data to map the “typical” progression of vision, hearing and vestibular loss for the various types of Usher syndrome.
Children will begin to develop night blindness in dimly lit areas. Progressive loss of visual fields will affect their ability to see to their right, left, above and below. Typically, central vision for reading and fine detail is functional into the adult years, as long as good lighting is available. It may take a minute or so for individuals with RP to adjust to glare and dramatic changes in lighting. As visual fields narrow, individuals with RP will often stop and scan the environment before entering a building or room. Objects and people at a distance will be easier to discern than those that are near to them.
Sometimes children and adults with Usher appear to be clumsy because of vestibular/balance issues. This can be even more noticeable as visual fields decrease, or in dimly lit areas. Depth perception may be affected, as well as the ability to see monotones such as grey, white and black.
Low vision specialists can suggest optical aids and devices that maximize remaining vision. Orientation and mobility instructors can provide training in safe travel.
Usher syndrome symptoms
Usher syndrome is characterized by deafness due to an impaired ability of the inner ear and auditory nerves to transmit sensory (sound) input to the brain (sensorineural hearing loss) as well as abnormal accumulation of colored (pigmented) material on the nerve-rich membrane (the retina) lining the eyes (retinitis pigmentosa or RP). RP eventually causes retinal degeneration leading to progressive loss of vision and legal blindness. Sensorineural nerve deafness may be profound or mild, and may be progressive. The vision loss caused by RP may begin during childhood or later during life, and often first presents with difficulty seeing at night or in low light (“night blindness”). Studies show that clear central vision may be maintained for many years even while side (peripheral) vision decreases. These narrowed visual fields are also referred to as “tunnel vision.” Issues with balance are seen in individuals with Usher syndrome types 1 and 3.
Usher syndrome type 1 is characterized by profound hearing loss in both ears at birth (congenital deafness) and balance problems. In many cases, affected children do not learn to walk until 18 months of age or later. Vision problems usually begin at approximately the age of ten years to early teens, although some parents report onset in children younger than 10. Usher syndrome type 2 is characterized by moderate to severe hearing loss in both ears at birth. In some cases, hearing loss may worsen over time. Onset of night blindness occurs during the late teens or early twenties. Peripheral vision loss is ongoing, but central vision is usually retained into adulthood. Visual problems associated with Usher syndrome type 2 tend to progress more slowly than those associated with type 1.
Usher syndrome type 3 is characterized by later onset hearing loss, variable balance (vestibular) dysfunction and RP that can present between the second and fourth decade of life. Balance issues occur in approximately 50% of individuals with Usher syndrome type 3.
What causes Usher syndrome
Usher syndrome can be caused by mutations in several different genes. So far, Usher syndrome has been associated with mutations in at least ten genes:
- Usher syndrome type 1: MYO7A (USH1B), USH1C, CDH23, PCDH15 (USH1F), SANS (USH1G), and possibly CIB2
- Usher syndrome type 2: USH2A, ADGRV1 (previously called VLGR1) WHRN (DFNB31)
- Usher syndrome type 3: USH3A (CLRN1), HARS
Mutations in at least six genes can cause Usher syndrome type 1. The most common of these are MYO7A gene mutations, followed by mutations in the CDH23 gene 1. Usher syndrome type 2 can result from mutations in three genes; USH2A gene mutations account for most cases of type 2. Usher syndrome type 3 is most often caused by mutations in the CLRN1 gene 1.
The genes associated with Usher syndrome provide instructions for making proteins involved in normal hearing, balance, and vision. In the inner ear, these proteins are involved in the development and function of specialized cells called hair cells, which help to transmit sound and signals from the inner ear to the brain. In the retina, the proteins contribute to the maintenance of light-sensing cells called rod photoreceptors (which provide vision in low light) and cone photoreceptors (which provide color vision and vision in bright light). For some of the proteins related to Usher syndrome, their exact role in hearing, balance, and vision is unknown.
Most of the gene mutations responsible for Usher syndrome lead to a loss of hair cells in the inner ear and a gradual loss of rods and cones in the retina. Degeneration of these sensory cells causes the hearing loss, balance problems, and vision loss that occur with Usher syndrome.
In some people with Usher syndrome, the genetic cause of the condition has not been identified. Researchers suspect that several additional genes are probably associated with this disorder.
Usher syndrome inheritance pattern
All of the types of Usher syndrome are inherited in an autosomal recessive pattern, which means both copies of a gene in each cell have a mutation. The parents of an individual with Usher syndrome each carry one copy of the mutated gene, but they do not have any signs and symptoms of the condition.
Many people with Usher syndrome inherit a faulty gene from both their parents. This means they have 2 copies of the faulty gene.
The parents themselves might only have 1 copy of the faulty gene. This is known as being a “carrier”. They do not have any signs and symptoms of the condition.
If you have Usher syndrome, any children you have will be carriers of the faulty gene so are unlikely to have problems. There’s only a risk they could be born with the condition if your partner is a carrier.
In autosomal recessive inheritance, it takes two copies of the mutant gene to give rise to the disorder. An individual with a recessive gene mutation is known as a carrier. When two carriers have a child, there is a:
- 1 in 4 chance the child will have the disorder
- 1 in 2 chance the child will be a carrier
- 1 in 4 chance the child will neither have the disorder nor be a carrier
Genetic counseling may help you understand the risks of passing Usher syndrome on to any children you have.
- To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. Online directories are provided by the American College of Medical Genetics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) and the National Society of Genetic Counselors (https://www.findageneticcounselor.com/).
- USH Yellow Book: Here you will find researchers and support providers who are all familiar with Usher syndrome, as well as resources for genetic testing (https://www.usher-syndrome.org/what-is-usher-syndrome/find-an-expert.html)
Figure 2. Usher syndrome autosomal recessive inheritance pattern
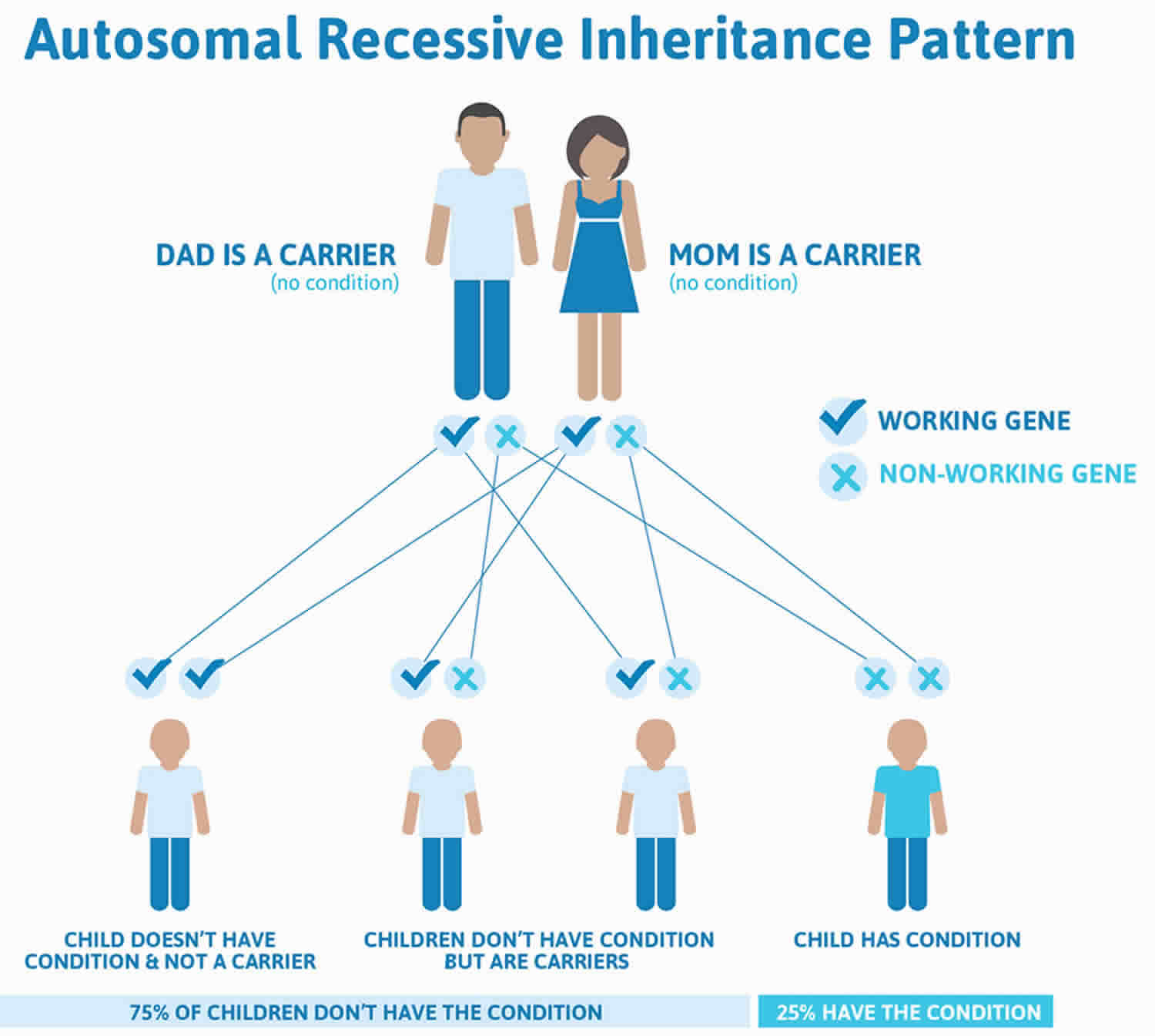
Footnote: Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and, therefore, have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Carriers of Usher syndrome type 1
Usher syndrome type 1 occurs when an individual inherits two copies of the same type of Usher 1 gene – one from each parent. If an individual has one Usher syndrome type 1 gene and one gene that is not Usher, they are considered to be a “carrier” of the Usher gene. Carriers have typical vision, hearing and balance. If two carriers of the same gene have a child, there is a 25% chance in each pregnancy that their child will inherit two Usher genes – one from each parent – and that child will have Usher syndrome. If a child inherits one Usher 1 gene and one non-Usher 1 gene, that child will be a carrier, like their parents. He or she will have typical vision, hearing and balance. It is also possible that a child of 2 carriers will inherit 2 non-Usher syndrome genes, which means that they will not have Usher syndrome and they will not be a carrier.
Carriers of Usher syndrome type 2
When an individual has one Usher syndrome type 2 gene (carrier), half the normal amount of the protein usherin is produced. Scientists believe that this is sufficient for normal vision and hearing, therefore no observable symptoms result. There are theoretical reasons to think that there may be very mild hearing and vision problems in older adults who are carriers, but this has never been studied. It may be that these genes are partly responsible for some of the hearing and vision losses that all of us have as we grow older.
How much does a genetic test cost?
The cost and turn-around-time of a genetic test may vary depending on the lab and the methods used for testing. Some Usher syndrome screens cost around $500. More accurate and detailed testing may cost between $2000-$5000. A typical range of time to get the results might be 8-12 weeks. Insurance companies will often pay for genetic tests, but you should check with your company before your doctor orders the test.
Usher syndrome diagnosis
Usher syndrome is diagnosed by hearing, balance and vision examinations. A hearing (audiologic) exam measures the frequency and loudness of sounds that a person can hear. An electroretinogram measures the electrical response to the light-sensitive cells in the retina of the eyes. A retinal exam is done to observe the retina and other structures in the back of the eye. Vestibular (balance) function can be assessed by a variety of tests that evaluate different parts of the balance system. Genetic testing is clinically available for most of the genes associated with Usher syndrome.
Early diagnosis is important, as it improves treatment success. An eye care specialist can use dilating drops to examine the retina for signs of retinitis pigmentosa. Visual field testing measures peripheral vision. An electroretinogram measures the electrical response of the eye’s light-sensitive cells in the retina. Optical coherence tomography may be helpful to assess for macular cystic changes. Videonystagmography measures involuntary eye movements that could signify a balance problem. Audiology testing determines hearing sensitivity at a range of frequencies.
Genetic testing may help in diagnosing Usher syndrome. So far, researchers have found nine genes that cause Usher syndrome. Genetic testing is available for all of them:
- Type 1 Usher syndrome: MY07A, USH1C, CDH23, PCHD15, USH1G
- Type 2 Usher syndrome: USH2A, GPR98, DFNB31
- Type 3 Usher syndrome: CLRN1
Usher syndrome treatment
Presently, there is no cure for Usher syndrome. Treatment involves managing hearing, vision, and balance problems. Early diagnosis helps tailor educational programs that consider the severity of hearing and vision loss and a child’s age and ability. Treatment and communication services may include hearing aids, assistive listening devices, cochlear implants, auditory (hearing) training, and/or learning American Sign Language. Independent-living training may include orientation and mobility training for balance problems, Braille instruction, and low-vision services.
Vitamin A may slow the progression of retinitis pigmentosa, according to results from a long-term clinical trial supported by the National Eye Institute and the Foundation Fighting Blindness 34. Based on the study, adults with a common form of retinitis pigmentosa may benefit from a daily supplement of 15,000 IU (international units) of the palmitate form of vitamin A. Patients should discuss this treatment option with their health care provider before proceeding. Because people with type 1 Usher syndrome did not take part in the study, high-dose vitamin A is not recommended for these patients.
General precautions for vitamin A supplementation:
- Do not substitute vitamin A palmitate with a beta-carotene supplement.
- Do not take vitamin A supplements greater than the recommended dose of 15,000 IU or modify your diet to select foods with high levels of vitamin A.
- Pregnant women should not take high-dose vitamin A supplements due to the increased risk of birth defects. Women considering pregnancy should stop taking high-dose vitamin A supplements for six months before trying to conceive.
Because long-term high-dose vitamin A supplementation (e.g., exceeding 25,000 IU) may cause certain adverse effects, such as liver disease, patients should be regularly monitored by their doctors when taking such supplementation. (The body’s reserves of vitamin A are primarily stored in the liver.) It is essential that any patients with retinitis pigmentosa considering such supplementation consult with their doctors for necessary evaluation to determine whether it is appropriate or inadvisable in their particular case.
Individuals with retinitis pigmentosa in association with Usher syndrome may find low-vision aids to be helpful. Other treatment for Usher syndrome is symptomatic and supportive. Agencies that provide services to individuals with hearing and visual loss can be helpful.
Genetic counseling is recommended for affected individuals and their families.
References- Usher syndrome. https://ghr.nlm.nih.gov/condition/usher-syndrome
- Usher Syndrome Coalition. https://www.usher-syndrome.org/what-is-usher-syndrome/
- Lentz J, Keats B. Usher Syndrome Type II. 1999 Dec 10 [Updated 2016 Jul 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1341
- https://www.usher-syndrome.org/what-is-usher-syndrome/frequent-questions.html#content_75481e1df0cc116b11edc26c9b9df51c_item_7501419
- Mizuno K, Nishida S: Electron microscopic studies of human retinitis pigmentosa. Part I. Two cases of advanced retinitis pigmentosa. Am J Ophthalmol. 1967;63(4):791-803.
- Bunt-Milam AH, Kalina RE, Pagon RA. Clinical-ultrastructural study of a retinal dystrophy. Invest Ophthalmol Vis Sci. 1983;24(4):458-469.
- Ben-Arie-Weintrob Y, Berson EL, Dryja TP. Histopathologic-genotypic correlations in retinitis pigmentosa and allied diseases. Ophthalmic Genet. 2005;26(2):91-100.
- Weleber RG, Gregory-Evans K. Retinitis pigmentosa and allied disorders. In: Ryan SJ, ed. Retina, 4th edn. Philadelphia, PA: Elsevier; 2006:394-485.
- Grover S, Fishman GA, Anderson RJ, et al. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology. 1999;106(9):1780-1785.
- Heckenlively JR, Yoser SL, Friedman LH et al. Clinical findings and common symptoms in retinitis pigmentosa. Am J Ophthalmol. 1988; 105(5):504-511.
- Alexander KR, Fishman GA. Prolonged rod dark adaptation in retinitis pigmentosa. Br J Ophthalmol. 1984;68(8):561-569.
- Grover S, Fishman GA, Brown J Jr. Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology. 1998;105(6):1069-1075.
- Lim JI, Tan O, Fawzi AA, et al: A pilot study of Fourier-domain optical coherence tomography of retinal dystrophy patients. Am J Ophthalmol. 2008;146(3):417-426.
- Berson EL. Retinitis Pigmentosa and Allied Diseases. In: Albert D, Miller J, Azar D, Blodi B, eds. Albert and Jakobiec, 3rd edn. Philadelphia, PA: Elsevier; 2008:Ch. 177.
- Berson EL, Rosner B, Sandberg MA, et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol. 2010;128(4):403-411.
- Massof RW, Fishman GA. How strong is the evidence that nutritional supplements slow the progression of retinitis pigmentosa? Arch Ophthalmol. 2010;128(4):493-495.
- Norton EWD. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa [letter to the editor]. Arch Ophthalmol. 1993;111(11):1460.
- Cox SN, Hay E, Bird AC: Treatment of chronic macular edema with acetazolamide. Arch Ophthalmol. 1988; 106(9):1190-1195.
- Grover S, Apushkin MA, Fishman GA: Topical Dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa. Am J Ophthalmol. 2006; 141(5):850-858.
- Yuzbasioglu E, Artunay O, Rasier R, et al. Intravitreal bevacizumab (Avastin) injection in retinitis pigmentosa. Curr Eye Res. 2009;34(3):231-237.
- Genead MA, Fishman GA. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with retinitis pigmentosa and Usher syndrome. Arch Ophthalmol. 2010;128(9):1146-1150.
- Berson EL: Light deprivation and retinitis pigmentosa. Vision Res. 1980;20(12):1179-1184.
- Berson EL, Rosner B, Sandberg MA, et al: Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Arch Ophthalmol. 2004; 122(9):1306-1314.
- Narcisi TME, Shoulders CC, Chester SA, et al: Mutations of the microsomal triglyceride-transfer-protein gene in abetalipoproteinemia. Am J Hum Genet. 1995;57(6):1298-1310.
- Refsum S: Heredopathia atactica polyneuritiformis: A familial syndrome not hitherto described. Acta Psychiatr Neurol Scand. 1946;38(Suppl):1
- Luo YH, Zhong JJ, da Cruz L. The use of Argus® II retinal prosthesis by blind subjects to achieve localisation and prehension of objects in 3-dimensional space. Graefes Arch Clin Exp Ophthalmol. 2014 Dec 31.
- Stronks HC, Dagnelie G. The functional performance of the Argus II retinal prosthesis. Expert Rev Med Devices. 2014 Jan;11(1):23-30.
- Michaelsson K, Lithell H, Vessby B, Melhus H: Serum retinol levels and the risk of fracture. N Engl J Med. 2003;348(4):287-294.
- deCruz et al. Five-Year Safety and Performance Results from the Argus II Retinal Prosthesis. Ophthalmology. 2016 Oct;123(10):2248-54.
- Ghodasra DH, et al. Worldwide Argus II implantation: recommendations to optimize patient outcomes. BMC Ophthalmol. 2016 May 6;16:52. doi: 10.1186/s12886-016-0225-1.
- Fishman GA, Farber MD, Derlacki DJ. X-linked retinitis pigmentosa. Profile of clinical findings. Arch Ophthalmol. 1988;106(3):369-375.
- Lyness AL, Ernst W, Quinlan MP, et al. A clinical, psychophysical, and electroretinographic survey of patients with autosomal dominant retinitis pigmentosa. Br J Ophthalmol. 1985;69(5):326-339.
- https://www.usher-syndrome.org/what-is-usher-syndrome/frequent-questions.html#content_75481e1df0cc116b11edc26c9b9df51c_item_7501420
- Berson, E.L. (1998). Treatment of retinitis pigmentosa with vitamin A (link is external). Digital Journal of Ophthalmology, 4(7).





{kind=link}