WAGR syndrome
WAGR syndrome also called 11p deletion syndrome, is a rare genetic syndrome that affects many body systems and is named for its main features: Wilms tumor, Anirida, Genitourinary anomalies such as undescended testicles or hypospadias in males, or internal genital or urinary anomalies in females, and Range of developmental delays such as intellectual disability (mental retardation) 1. A combination of two or more of these conditions is usually present in most individuals with WAGR syndrome 2. WAGR syndrome is caused by a deletion of genetic material on the short (p) arm of chromosome 11 3. In most cases, this genetic change occurs spontaneously during early embryonic development (de novo) for unknown reasons (sporadic). Only rarely is the mutation inherited.
People with WAGR syndrome have a 45 to 60 percent chance of developing Wilms tumor, a rare form of kidney cancer. This type of cancer is most often diagnosed in children but is sometimes seen in adults.
Most people with WAGR syndrome have aniridia, an absence of the colored part of the eye (the iris). This can cause reduction in the sharpness of vision (visual acuity) and increased sensitivity to light (photophobia). Aniridia is typically the first noticeable sign of WAGR syndrome. Other eye problems may also develop, such as clouding of the lens of the eyes (cataracts), increased pressure in the eyes (glaucoma), and involuntary eye movements (nystagmus).
Abnormalities of the genitalia and urinary tract (genitourinary anomalies) are seen more frequently in males with WAGR syndrome than in affected females. The most common genitourinary anomaly in affected males is undescended testes (cryptorchidism). Females may not have functional ovaries and instead have undeveloped clumps of tissue called streak gonads. Females may also have a heart-shaped (bicornate) uterus, which makes it difficult to carry a pregnancy to term.
Another common feature of WAGR syndrome is intellectual disability. Affected individuals often have difficulty processing, learning, and properly responding to information. Some individuals with WAGR syndrome also have psychiatric or behavioral problems including depression, anxiety, attention-deficit/hyperactivity disorder (ADHD), obsessive-compulsive disorder (OCD), or a developmental disorder called autism spectrum disorder that affects communication and social interaction.
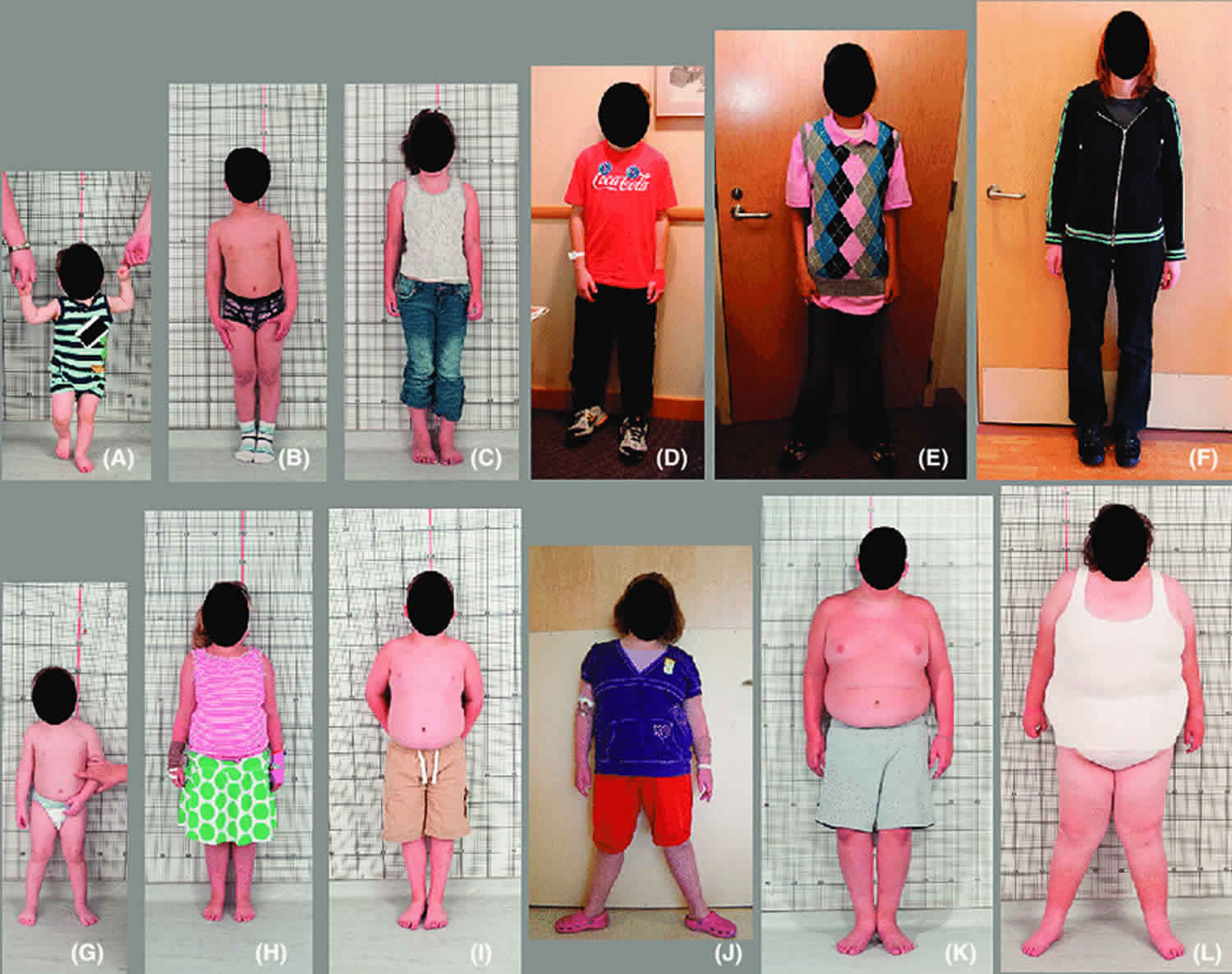
Other signs and symptoms of WAGR syndrome can include childhood-onset obesity, inflammation of the pancreas (pancreatitis), and kidney failure. When WAGR syndrome includes childhood-onset obesity, it is often referred to as WAGRO syndrome.
The prevalence of WAGR syndrome ranges from 1 in 500,000 to one million individuals. It is estimated that one-third of people with aniridia actually have WAGR syndrome. Approximately 7 in 1,000 cases of Wilms tumor can be attributed to WAGR syndrome 1.
Treatment of WAGR syndrome is aimed at addressing the specific symptoms that are present in each individual.
WAGR syndrome causes
WAGR syndrome is caused by a deletion of genetic material on the short (p) arm of chromosome 11. The size of the deletion varies among affected individuals.
The signs and symptoms of WAGR syndrome are related to the loss of multiple genes on the short arm of chromosome 11. WAGR syndrome is often described as a contiguous gene deletion syndrome because it results from the loss of several neighboring genes. The PAX6 and WT1 genes are always deleted in people with the typical signs and symptoms of this disorder. Because changes in the PAX6 gene can affect eye development, researchers think that the loss of the PAX6 gene is responsible for the characteristic eye features of WAGR syndrome. The PAX6 gene may also affect brain development. Wilms tumor and genitourinary abnormalities are often the result of mutations in the WT1 gene, so deletion of the WT1 gene is very likely the cause of these features in WAGR syndrome.
In people with WAGRO syndrome, the chromosome 11 deletion includes an additional gene, BDNF. This gene is active (expressed) in the brain and plays a role in the survival of nerve cells (neurons). The protein produced from the BDNF gene is thought to be involved in the management of eating, drinking, and body weight. Loss of the BDNF gene is likely responsible for childhood-onset obesity in people with WAGRO syndrome. People with WAGRO syndrome may be at greater risk of neurological problems such as intellectual disability and autism than those with WAGR syndrome. It is unclear whether this increased risk is due to the loss of the BDNF gene or other nearby genes.
Research is ongoing to identify additional genes deleted in people with WAGR syndrome and to determine how their loss leads to the other features of the disorder.
WAGR syndrome inheritance pattern
Most cases of WAGR syndrome are not inherited. They result from a chromosomal deletion that occurs as a random event during the formation of reproductive cells (eggs or sperm) or in early fetal development. Affected people typically have no history of the disorder in their family.
Some affected individuals inherit a chromosome 11 with a deleted segment from an unaffected parent. In these cases, the parent carries a chromosomal rearrangement called a balanced translocation, in which no genetic material is gained or lost. Balanced translocations usually do not cause any health problems; however, they can become unbalanced as they are passed to the next generation. Children who inherit an unbalanced translocation can have a chromosomal rearrangement with extra or missing genetic material. Individuals with WAGR syndrome who inherit an unbalanced translocation are missing genetic material from the short arm of chromosome 11, which results in an increased risk of Wilms tumor, aniridia, genitourinary anomalies, and intellectual disability.
WAGR syndrome symptoms
WAGR syndrome (11p deletion syndrome) is defined as a genetic syndrome in which there is a predisposition to Wilms’ tumor; aniridia; abnormalities of the reproductive and urinary tracts (genitourinary); and intellectual disability. The specific symptoms that occur depend upon the combination of disorders present.
Wilms’ tumor (nephroblastoma) is the most common form of kidney cancer in early childhood. It occurs in approximately one half of all cases of WAGR syndrome (11p deletion syndrome). In the early stages of Wilms’ tumor, there are usually no symptoms. The first signs of the disease may include blood in the urine (hematuria), low-grade fever, loss of appetite, paleness, weight loss, fatigue and lack of energy (lethargy), and swelling of the abdomen. In the later stages, slight pain may occur at intervals (intermittent), or pain may be sudden and sharp. (For more information on this disorder, choose “Wilms’ tumor” as your search term in the Rare Disease Database.) Abnormally persistent clusters of embryonal cells within the kidneys (nephrogenic rests) are not uncommon in children with WAGR syndrome (11p deletion syndrome). This tissue is noticeable on ultrasound examination, and is sometimes difficult to distinguish from Wilms’ tumor. It is thought that nephrogenic rests, or clusters of rests (nephroblastomatosis), may give rise to Wilms’ tumor in some cases.
In infants with aniridia that is associated with WAGR syndrome (11p deletion syndrome), the irises fail to develop normally before birth (prenatally). This results in the partial or complete absence of the round, colored (pigmented) portion of the eye (iris). Aniridia is almost always present in individuals with WAGR syndrome (11p deletion syndrome); however, at least four cases of WAGR syndrome (11p deletion syndrome) have been confirmed without aniridia. In almost all cases, aniridia occurs in combination with other disorders of the eye. These accompanying disorders may include clouding of the lens of the eye (cataract); rapid, involuntary movements of the eye (nystagmus); and/or partial or complete loss of vision due to abnormally high pressure of the fluid in the eye (glaucoma). Progressive scarring and opacity of the cornea (aniridic keratopathy, also called corneal pannus) is common in adolescents and adults with aniridia, but may also occur in children. (For more information on these disorders, choose “Aniridia” and “Cataracts” as your search terms in the Rare Disease Database.)
In the medical literature, the “G” in the acronym WAGR refers to “Genitourinary abnormalities,” “ambiguous Genitalia,” or “Gonadoblastoma.” Gonadoblastoma, a cancer of the cells that form the testes in males or the ovaries in females (gonads), occurs exclusively in people with defective development of the gonads (gonadal dysgenesis), as is the case in some infants with WAGR syndrome (11p deletion syndrome). Although gonadoblastoma is not always manifested in WAGR syndrome (11p deletion syndrome), it is important to be aware of the genetic predisposition for this potentially serious disorder so that appropriate steps can be taken. (See the “Standard Therapies” section of this report for more information.)
Other abnormalities of the reproductive and urinary tracts (genitourinary) may be present in many children with WAGR syndrome (11p deletion syndrome). In males, these may include the failure of one or both testes to descend into the scrotum (cryptorchidism) and placement of the urinary opening (meatus) on the underside of the penis (hypospadias). In females, these abnormalities may include underdeveloped (streak) ovaries, and malformations of the uterus, fallopian tubes, or vagina. These abnormalities may also include duplicate ureters or horseshoe kidney.
In addition, individuals with WAGR syndrome (11p deletion syndrome) may have the gonads of one sex and external genitalia resembling that of the opposite sex (ambiguous genitalia), making their sexual assignment (i.e., male or female) uncertain (pseudohermaphroditism).
Intellectual disability is common in children with WAGR syndrome (11p deletion syndrome). However, the severity of the impairment varies greatly from case to case, ranging from severe to mild intellectual disability (IQ of 20 to 70) ). Some children with WAGR syndrome (11p deletion syndrome) may have normal intelligence (IQ at or above 100).
A variety of behavioral and psychiatric disorders have been reported in WAGR syndrome (11p deletion syndrome). These include autism spectrum disorders, attention-deficit disorder (with or without hyperactivity), obsessive-compulsive disorder, other anxiety disorders, and depression.
Although hearing is usually normal in people with WAGR syndrome (11p deletion syndrome), many individuals have difficulty with the way the brain processes auditory information, particularly with recognizing and interpreting the sounds involved with speech (auditory processing disorder).
Metabolic abnormalities present in some individuals with WAGR syndrome (11p deletion syndrome) may include early-onset overweight (obesity), and high serum cholesterol (hyperlipidemia). Some individuals with WAGR syndrome (11p deletion syndrome) have a combination of conditions including insulin resistance, high blood pressure, and high serum cholesterol which can increase the risk for coronary artery disease, stroke, and type 2 diabetes (metabolic syndrome).
Chronic kidney failure occurs in approximately 60% of individuals with WAGR syndrome (11p deletion syndrome), most often after age 12. This failure is usually the result of Focal Segmental Glomerulosclerosis (FSGS) a disorder which results in scarring of the filtering tubes of the kidneys. Chronic kidney failure may occur in an individual with WAGR syndrome (11p deletion syndrome) regardless of their history of Wilms tumor.
Frequent, recurrent upper respiratory infections, ear and sinus infections, asthma, and pneumonia are common in WAGR syndrome (11p deletion syndrome), particularly in young children with the disorder. Delayed loss of primary teeth and severely crowded or uneven teeth (dental malocclusion) are also common. A temporary suspension of breathing occurring repeatedly during sleep (sleep apnea) may occur in both children and adults with WAGR syndrome (11p deletion syndrome).
Abnormalities of muscle tone or strength (hypertonia/hypotonia) are common in WAGR syndrome (11p deletion syndrome), particularly during infancy and early childhood. Seizure disorder (epilepsy) has been reported frequently and chronic inflammation of the pancreas (pancreatitis) has also been reported.
In rare cases, other symptoms of WAGR syndrome (11p deletion syndrome) which may be present at birth (congenital) may include: defects of the heart or kidneys, partial or complete absence of the structure which connects the two hemispheres of the brain (agenesis of the corpus callosum) a weak area of the abdomen which allows part of the intestines to push through (inguinal hernia) an abnormal opening in the diaphragm which allows part of the abdominal organs to migrate into the chest cavity (diaphragmatic hernia) extra fingers or toes (polydactyly) webbing or fusing of fingers or toes (syndactyly) absence or closure of ducts which drain bile from the liver (biliary atresia) weakness or floppiness of the walls of the windpipe (tracheomalacia) and hearing impairment.
In rare cases, other symptoms of WAGR syndrome (11p deletion syndrome) which may develop or be diagnosed after birth may include: an abnormal enlargement of a part of the body (hemihypertrophy), growth retardation, communication disorders, inability of the brain to integrate information received from the body’s five sensory systems (sensory integration disorder) feeding/swallowing disorders, gastroesophageal reflux, multiple bony lumps or spurs on the bones (multiple exostoses) and curvature of the spine (scoliosis).
The disorders most commonly associated with WAGR syndrome (11p deletion syndrome) (Wilms’ tumor, aniridia, genitourinary abnormalities, and intellectual disability) as well as those listed above may appear together or in a variety of combinations. In the medical literature, these various groupings (disorder subdivisions) have been referred to as distinct disorders including “Aniridia-Wilms’ Tumor Association” (AWTA); “Aniridia-ambiguous Genitalia-mental Retardation” (AGR triad); or “Aniridia- Wilms’ Tumor-Gonadoblastoma.” While all individuals with WAGR syndrome (11p deletion syndrome) will be found to have deletions in chromosome 11p13, great variation in the size and nature of these deletions is possible among individuals. These variations in missing genetic material are thought to account for the differences in symptoms and manifestations of the disorder.
WAGR syndrome diagnosis
WAGR syndrome (11p deletion syndrome) can be diagnosed at birth, based upon a clinical evaluation, characteristic physical findings, and chromosomal studies (high-resolution karyotyping and molecular cytogenetic tests). In many cases, the partial or complete absence of the iris of the eye (aniridia) may be the only physical feature associated with WAGR Syndrome/11p deletion syndrome that is obvious at birth. In other cases, genitourinary abnormalities associated with the syndrome may also be apparent.
If a child has aniridia, chromosomal studies are necessary to determine whether the child has a genetic predisposition for the disorders associated with WAGR syndrome (11p deletion syndrome).
WAGR syndrome treatment
Treatment of WAGR syndrome (11p deletion syndrome) is directed toward the specific symptoms that are apparent in each individual.
Wilms’ Tumor
Wilms’ tumor will occur in approximately 50% of children with WAGR syndrome (11p deletion syndrome) whose deletion encompasses the WT1 gene. The onset of Wilms’ tumor in these children is most often between the ages of 1 and 3. The majority of cases of Wilms’ tumor have been detected by age 8, but rare cases have occurred in individuals with WAGR syndrome (11p deletion syndrome) as late as age 25. Surveillance for the development of Wilms’. tumor in children with WAGR syndrome (11p deletion syndrome) should begin at birth or as soon as WAGR syndrome (11p deletion syndrome) is suspected. This surveillance consists of abdominal ultrasound every 3 months until at least age 8. In addition, feeling the abdomen for signs of swelling or masses (palpation) may be done by both the pediatrician and, with instruction, the parents.
Wilms’ tumor can often be treated successfully depending on the stage of the tumor at detection. Treatment programs may combine surgical techniques (including kidney removal), radiation therapy, and chemotherapy. The chemotherapy regimen currently preferred to treat Wilms’ tumor consists of the drugs dactinomycin and vincristine, which may be combined with doxorubicin. Cyclophosphamide may also be used with this drug regimen to treat tumors that have not responded to the first line of chemotherapy. Other regimens sometimes used to treat Wilms’ tumor include a combination of the drugs cisplatin and etoposide or a regimen that combines ifosfamide and mesna. Abnormally persistent clusters of embryonal cells within the kidneys (nephrogenic rests or nephroblastomatosis) are thought to be precursors of Wilms’ tumor in some cases. Serial abdominal ultrasound may be used to monitor this tissue for changes which indicate malignancy (cancer), and surgery or chemotherapy is sometimes indicated if changes are noted.
Aniridia
The treatment of aniridia is usually directed at preserving vision. Drugs or surgery may be helpful for glaucoma and/or cataracts. Individuals with aniridia often lack limbal cells, which serve to regenerate the cornea. For this reason, care should be taken to prevent injury to the corneas. Contact lenses should be avoided if possible, and when needed, preservative-free ocular lubricants and antibiotics should be used. In the past, cornea transplants typically failed in persons with aniridia, but simultaneous treatment with corneal limbal stem cells has been found to significantly increase the success rate.. An artificial cornea (Boston Keratoprosthesis) and several types of artificial iris implants are currently in clinical trials.
Gonadoblastoma and genitourinary abnormalities
Children with WAGR syndrome (11p deletion syndrome) should be regularly evaluated to detect abnormal development of the ovaries (streak gonads) or testes. Surgical removal of abnormal gonads (gonadectomy) may be indicated to prevent the occurrence of gonadoblastoma. In cases when gonadoblastoma is actually present, surgery to remove the gonad(s) with the tumor is performed. If one gonad is cancer-free, it still may be removed since it may be at risk for developing gonadoblastoma. Individuals who have had both gonads removed are given hormone treatment to help them develop sexual characteristics that are usually manifested during puberty. Because hormone therapy may cause secondary uterine cancer to develop, the uterus may be surgically removed (if one is present) when the gonadectomy is initially performed.
In males with WAGR syndrome (11p deletion syndrome), one or both testes may fail to descend into the scrotum (cryptorchidism). If a testis does not properly descend into the scrotum on its own before the child is 1 year of age, hormone treatment may be given. If this treatment is not successful, surgery may be performed to move the undescended testis into the scrotum and attach it in a fixed position so it will not retract (orchiopexy).
Males with cryptorchidism may also have the urinary opening on the underside of the penis (hypospadias). When hypospadias is identified in infants, routine removal of the penis’s foreskin (circumcision) soon after birth should not be performed. The foreskin can be essential in aiding surgical repair later in life. Surgical correction is performed as necessary for cosmetic, reproductive, and/or psychological purposes and/or to correct problems with urination. Surgical correction is usually performed before the child is 1 year of age.
In females with WAGR syndrome (11p deletion syndrome), the ovaries may be small, or may not have developed properly (streak ovaries). Abnormal ovaries may or may not function well enough to produce adequate levels of hormones for the development of puberty and for menstruation. If abnormal ovaries are detected, surveillance for the development of gonadoblastoma is necessary. Pelvic ultrasound or MRI (magnetic resonance imaging) may be used.
Individuals with WAGR syndrome (11p deletion syndrome) may have the gonads of one sex and external genitalia resembling that of the opposite sex (ambiguous genitalia), making their sexual assignment (i.e., male or female) uncertain (pseudohermaphroditism). Surgery may be performed to correct some abnormalities of the external genitalia, and hormone treatment may be instituted.
Intellectual disability
The intellectual capacity of individuals with WAGR syndrome (11p deletion syndrome) may range from severe impairment (IQ of 35 or lower) to mild impairment (IQ of 70 ) to intelligence that is within normal limits (IQ of approximately 100). Most individuals with WAGR syndrome (11p deletion syndrome) fall into the range of mild to moderate impairment (IQ of 35 to 70) Children with WAGR syndrome (11p deletion syndrome) should be referred for Early Intervention services soon after birth or diagnosis. Physical, Occupational, and Speech therapies, as well as Special Education services can maximize normal development and can ensure that appropriate steps are taken to help affected individuals reach their potential.
Kidney failure
The renal failure associated with WAGR syndrome (11p deletion syndrome) often causes high blood pressure (hypertension) high blood cholesterol (hyperlipidemia) and the leakage of protein from the blood into the urine (proteinuria). Treatment consists of medications called “ACE” (angiotensin converting enzyme) inhibitors, or “ARBs” (angiotensin II receptor blockers). If given early in the course of kidney failure, these drugs may help maintain blood pressure within a normal range, reduce the loss of protein through the blood, and may prolong the function of the kidney(s). Some individuals with WAGR syndrome (11p deletion syndrome) and renal failure may require kidney transplant.
Genetic counseling is important for individuals with WAGR syndrome (11p deletion syndrome) and for their families. Chromosomal studies are necessary to determine parents’ risk for WAGR syndrome (11p deletion syndrome) in subsequent children. Ongoing genetic counseling will also allow for up-to-date genetic testing and for the provision of information regarding new treatments and therapies.
WAGR syndrome prognosis
Patients with WAGR syndrome have an excellent prognosis for long-term survival. Morbidity and mortality associated with late development of renal failure may be more significant than Wilms tumor.
Life-limiting abnormalities include genitourinary anomalies in the first year of life. Lifelong disabilities may include vision loss and mental retardation.
In the approximately 30% of patients with WAGR syndrome in whom a Wilms tumor develops, the prognosis is related to the histologic features and the stage of the tumor.
Early detection seems to improve the outcome.
The overall survival rate of patients with Wilms tumor is excellent and is related to the histologic features of the tumor (favorable vs unfavorable) and the stage of the disease, as follows:
- In stage 1, the disease is localized to the kidney.
- In stage 2, the disease extends through the capsule of the kidney.
- In stage 3, the disease extends to ipsilateral structures or beyond the line connecting the poles.
- In stage 4, the distinct metastases are present.
- In stage 5, bilateral kidney involvement is present.
In the third National Wilms Tumor Study, the survival rate ranged from 95% for stage I to almost 80% for stage 4 4. Patients with stage 5 tumors, some of whom had WAGR syndrome, had an overall survival rate of approximately 87%.
References- WAGR syndrome. https://ghr.nlm.nih.gov/condition/wagr-syndrome
- WAGR Syndrome/11p Deletion Syndrome. https://rarediseases.org/rare-diseases/wagr-syndrome11p-deletion-syndrome
- WAGR syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=893
- D’Angio GJ, Breslow N, Beckwith JB, et al. Treatment of Wilms’ tumor. Results of the Third National Wilms’ Tumor Study. Cancer. 1989 Jul 15. 64(2):349-60.





{kind=link}