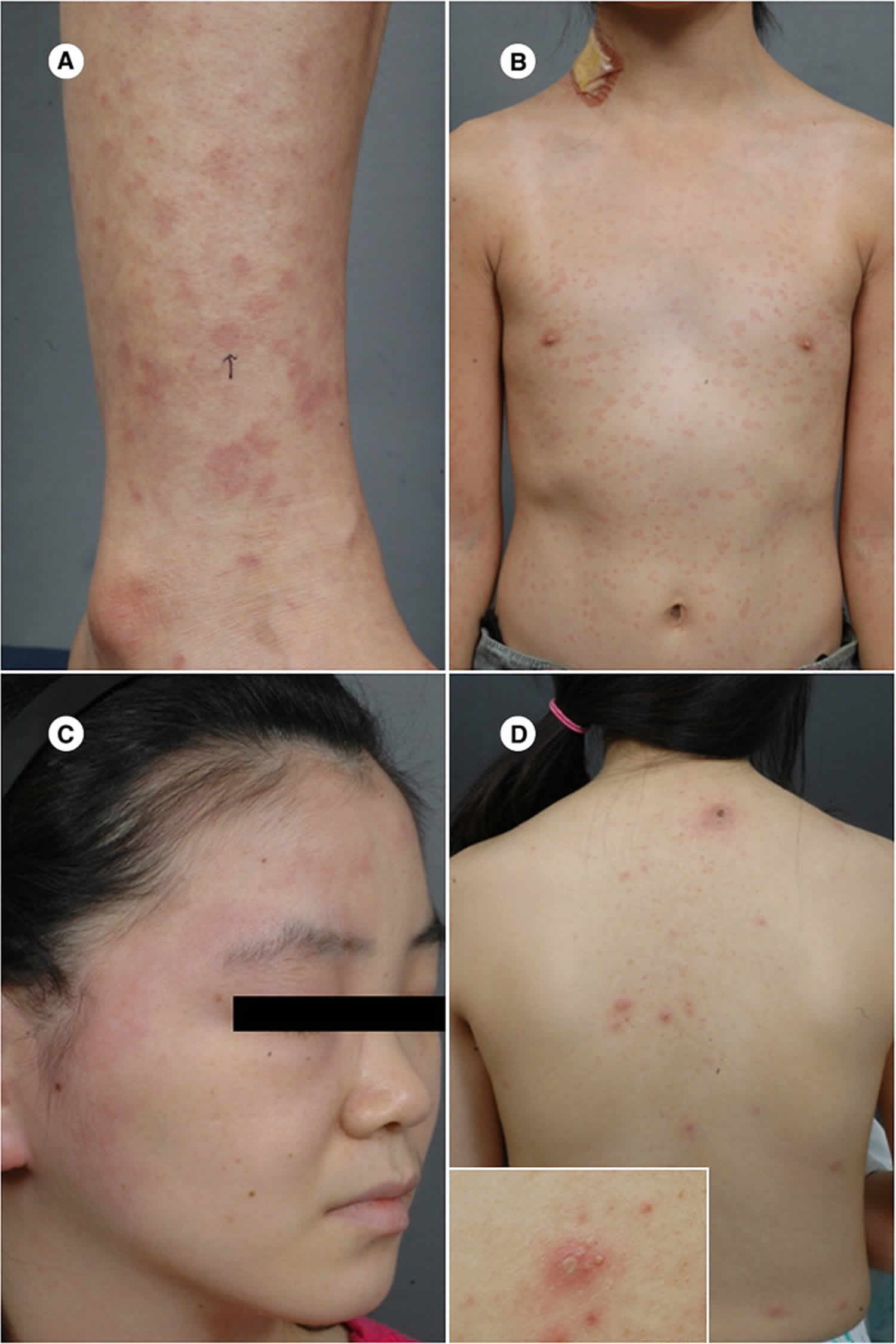
What is Kikuchi disease
Kikuchi-Fujimoto disease also known as histiocytic necrotizing lymphadenitis, is an extremely rare benign (non-cancerous) condition of the lymph nodes 1. Kikuchi-Fujimoto disease was first described in Japan in 1972, by Japanese Pathologists Kikuchi and Fujimoto, is benign and self-limiting disease that mainly affects young women of Asian origin. Kikuchi-Fujimoto disease largely affects young adults (young than 30 years) and is most frequently found in East Asian and Japanese populations but rare in the United States and Europe. This geographic association can be related to certain HLA alleles such as HLA class II alleles, HLA-DPA1, and HLA-DPB1, which are more prevalent in Asian Kikuchi-Fujimoto disease patients and rare in Caucasians 2. Kikuchi-Fujimoto disease can affect both males and females. The ratio of affected males to females in three series was 1:4, 1:1.6, and 1:1.26, respectively. In a Korean report of 20 individuals with Kikuchi-Fujimoto disease that were younger than 18 years, the gender distribution was equal. Most patients are younger than 40 years of age, but this condition has been reported in patients ranging in age from six to 80 years, most of whom were previously well. The mean age at presentation in a United States series was 30 years. Although initially described in two separate cases from Japan, Kikuchi-Fujimoto disease has since been found in all racial and ethnic groups and many countries, including the United States. In a study of 88 patients with Kikuchi-Fujimoto disease in the United States, 75 percent were Caucasian. The frequency of this condition varies widely in different groups, but it has been most frequently reported from Asia.
The main symptoms of Kikuchi disease include swollen lymph nodes in the neck, mild fever, and night sweats. Less common symptoms include weight loss, nausea, vomiting, and sore throat. While the exact cause of Kikuchi-Fujimoto disease is unknown, infectious and autoimmune causes have been suggested. Kikuchi disease usually gets better (resolves) on its own within one to four months (although it may take up to a year), with or without treatment. However, treatments are available to relieve some of the associated signs and symptoms.
Is Kikuchi disease contagious?
The cause of Kikuchi-Fujimoto disease is unknown 1. Several infectious agents have been suggested as possible causative agents, for example, herpes simplex virus type 1 and 2, varicella-zoster virus, cytomegalovirus, Epstein-Barr virus (EBV), human herpesvirus (HHV 6, 7, 8), parvovirus B19, human papillomavirus, hepatitis B virus, human T-lymphotropic virus 1, Brucella, toxoplasma, and Yersinia. However, none of them have been proven to have any significant role in the causation of Kikuchi-Fujimoto disease 1.
What causes Kikuchi disease?
Kikuchi-Fujimoto disease is a disease of unknown cause. Several infectious agents have been suggested as possible causative agents, for example, herpes simplex virus type 1 and 2, varicella-zoster virus, cytomegalovirus, Epstein-Barr virus (EBV), human herpesvirus (HHV 6, 7, 8), parvovirus B19, human papillomavirus, hepatitis B virus, human T-lymphotropic virus 1, Brucella, toxoplasma, and Yersinia. However, none of them have been proven to have any significant role in the causation of Kikuchi-Fujimoto disease. Epstein-Barr virus has been studied extensively in Kikuchi-Fujimoto disease, but no causative link has been demonstrated. On the other hand, some authors have hypothesized that Kikuchi-Fujimoto disease has an autoimmune etiology induced by virus-infected transformed lymphocytes and is also associated with systemic lupus erythematosus. This is because histopathological findings in the lymphocytes and the endothelial cells in patients with systemic lupus erythematosus are similar to those found in patients with Kikuchi-Fujimoto disease. Kikuchi-Fujimoto disease can mimic systemic lupus erythematosus. However, the association of Kikuchi-Fujimoto disease with systemic lupus erythematosus remains unclear. There have been some patients with Kikuchi-Fujimoto disease that have developed systemic lupus erythematosus. So it is important that the patients with Kikuchi-Fujimoto disease should have a regular follow up with rheumatology for a detailed autoimmune work up.
Kikuchi disease pathophysiology
While the pathogenesis of Kikuchi-Fujimoto disease is unknown, the clinical presentation, course, and histologic changes suggest an immune response of T cells and histiocytes to an infectious agent. An observation that is consistent with a viral etiology is increased levels of interferon-alpha and some other proteins stimulated by interferon-alpha including 2′,5′-oligoadenylate synthetase and tubuloreticular structures in the cytoplasm of stimulated lymphocytes, histiocytes, and vascular endothelium.
A possible role for interferon-gamma and interleukin (IL)-6 in the pathogenesis of this syndrome is suggested by one study of four men with biopsy-proven Kikuchi-Fujimoto disease. During the acute phase of illness, these patients had elevated serum levels of interferon-gamma and IL-6 but not interferon-alpha, tumor necrosis factor, or IL-2. The interferon-gamma and IL-6 levels returned to normal during convalescence.
Kikuchi disease symptoms
Kikuchi Fujimoto disease is a benign disease of the lymph nodes. Kikuchi-Fujimoto disease presents classically as swollen lymph nodes in the neck (cervical lymphadenopathy) predominantly unilateral and posterior cervical lymph nodes with or without fever in young women, which tend to develop suddenly. Cervical lymph nodes are most commonly involved (70% to 98%) followed by axillary (14%) and supraclavicular lymph nodes (12%). Fever is the first symptom in 30% to 50% of the cases. Some patients have non-specific presentation including weight loss, nausea, vomiting, night sweats, generalized lymphadenopathy, and hepatosplenomegaly. Cutaneous involvement involves maculopapular, morbilliform, urticarial rashes, or a disseminated erythema. Also, there has been one case report of a patient presenting with eyelid edema revealing Kikuchi-Fujimoto disease. Clinically, Kikuchi-Fujimoto disease resembles a lot of other diseases, and the differential is broad including reactive lymphadenitis, viral infections (HIV, Epstein-Barr, herpes simplex, cytomegalovirus), tuberculosis, systemic lupus erythematosus, lymphoma, and metastasis. Kikuchi-Fujimoto disease is also underdiagnosed in a lot of cases that get labeled with a presumptive diagnosis of viral infection or reactive lymphadenitis.
Lymphadenopathy features are as follows:
- Cervical nodes are affected in about 70% to 98% of cases
- Posterior cervical nodes are frequently involved (65-70% of cases)
- Lymphadenopathy is isolated to a single location in 83% of cases, but multiple chains may be involved
- Cases of generalized adenopathy involving axillary, inguinal, and mesenteric nodes are unusual
Characteristics of lymphadenopathy are as follows:
- Lymphadenopathy is isolated to a single location in 83% of patients, although multiple nodal chains may be involved
- Cervical lymph nodes are affected in 80% of patients; of these, 65-70% involve posterior triangle cervical nodes
- Less commonly affected nodes include those in axillary, mediastinal, celiac, abdominal, and inguinal locations
- The lymph nodes are usually described as painless or mildly tender
- The lymph nodes tend to be 2-3 cm in diameter, although masses of multiple nodes may reach 6 cm
- The lymph nodes are usually firm and mobile, but they are not fluctuant or draining
Other signs and symptoms may include a flulike prodrome with fever is present in 50% of cases 3:
- Fever.
- Headache
- Nausea and vomiting
- Malaise, fatigue
- Joint pain (arthralgia)
- Muscle pain (myalgia)
- Night sweats
- Rash (up to 30%)
- Abdominal pain
- Weight loss
- Thoracic pain
Extranodal findings are as follows:
- Skin: The incidence of skin involvement varies from 5-30%; findings are varied and nonspecific and include maculopapular lesions, morbilliform rash, nodules, urticaria, and malar rash, which may resemble that of systemic lupus erythematosus (SLE); skin lesions resolve in a few weeks to months 4
- Hepatosplenomegaly (enlarged spleen and liver): This finding is not uncommon; monitor lactate dehydrogenase (LDH) levels
- Neurologic involvement: Neurologic involvement is rare but has included conditions such as aseptic meningitis, acute cerebellar ataxia, and encephalitis 5; patients with aseptic meningitis may report headache, but they do not exhibit nuchal rigidity or positive Kernig or Brudzinski signs; cerebrospinal fluid (CSF) findings are similar to those noted in patients with aseptic meningitis of viral etiology
- Rarely involved extranodal sites include the bone marrow, myocardium, uvea, and thyroid and parotid glands
- Arthritic involvement: Asymmetric polyarthritis, enthesitis, and dactylitis of the toes was reported in the case of a 14-year-old boy 6
- Widespread involvement of multiple organ systems in Kikuchi disease has been described in solid-organ transplant patients
Kikuchi disease diagnosis
Routine laboratory investigations do not help much in making a diagnosis of Kikuchi-Fujimoto disease. The only significant lab abnormalities are leukopenia, elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). Around 30% of the patients also have atypical lymphocytosis. Some patients with Kikuchi-Fujimoto disease have elevated aminotransferases and lactate dehydrogenase (LDH) levels. Antibodies against nuclear antigens (ANA), double-stranded DNA (dsDNA), Rheumatoid factor (RF) and ANCA are also negative even though systemic lupus erythematosus and several other autoimmune diseases have been described in patients with Kikuchi-Fujimoto disease. CT and MRI scans are not diagnostic for Kikuchi-Fujimoto disease as there are no characteristic radiological features. In fact, imaging is not able to differentiate Kikuchi-Fujimoto disease from other nodal diseases including lymphoma, metastasis or Tuberculosis. Kwon et al. did a retrospective analysis of CT scans of 96 patients with confirmed Kikuchi-Fujimoto disease and found out that 94% of the enlarged lymph nodes had a size of less than 2.5 cm. This can somehow help in differentiating Kikuchi-Fujimoto disease from lymphoma that typically produces larger lymphadenopathy 7.
The gold standard diagnostic test for Kikuchi-Fujimoto disease is the histopathological analysis of excised lymph node biopsy specimen. The histopathological features can be classified into three stages: (1) proliferative stage expressing various histiocytes, plasmacytoid monocytes, lymphoid cells containing karyorrhectic nuclear fragments, and eosinophilic apoptotic debris; (2) necrotizing stage showing a degree of coagulative necrosis; and (3) xanthomatous stage predominantly containing foamy histiocytes. The absence of granulocytes is also an important feature. Histologically it can mimic lymphoma. However, the absence of monoclonal lymphocyte receptors on the histologic analysis rules out a lymphoma. Other histological differential diagnoses of Kikuchi-Fujimoto disease include reactive lymphadenitis associated with systemic lupus erythematosus, infectious mononucleosis, tuberculosis, and metastatic cancer.
Kikuchi disease treatment
Because kikuchi-fujimoto disease is rare and seen in Japanese individuals, it is best managed by a multidisciplinary team that includes an infectious disease expert, internist, pathologist, and a pharmacist. There is no specific treatment available for Kikuchi-Fujimoto disease. It is typically a self-limited disease, and spontaneous resolution occurs in one to four months. However, the possible recurrence rate of 3% to 4% has been reported that can occur even eight to nine years later. Management is symptomatic including analgesic (e.g., NSAIDs) and antipyretic. Some authors have seen a positive clinical response to glucocorticoids in Kikuchi-Fujimoto disease patients with a severe or relapsing clinical course. Takada et al. reported a case of Kikuchi-Fujimoto disease that dramatically resolved with oral minocycline suggesting that the causative agent of Kikuchi-Fujimoto disease might be especially sensitive to this antibiotic. There have been some case reports demonstrating hydroxychloroquine and intravenous immunoglobulins as successful alternative treatment options for Kikuchi-Fujimoto disease 8.
Kikuchi disease prognosis
The course of Kikuchi disease is generally benign and self-limited. Lymphadenopathy most often resolves over several weeks to 6 months, although the disease occasionally persists longer. The disease recurs in about 3% of cases. Four deaths have been reported, from hemophagocytic syndrome and severe infection, pulmonary hemorrhage, acute heart failure, and multiorgan failure and disseminated intravascular coagulation 9.
- Masab M, Farooq H. Kikuchi Disease. [Updated 2019 Mar 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430830[↩][↩][↩]
- Al-Allaf AW, Yahia YM. Kikuchi-Fujimoto Disease Associated with Sjögren’s Syndrome: A Case Report. Eur J Case Rep Intern Med. 2018;5(5):000856.[↩]
- Kikuchi disease. https://emedicine.medscape.com/article/210752-overview[↩]
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008 Jul. 59(1):130-6.[↩]
- Khishfe BF, Krass LM, Nordquist EK. Kikuchi disease presenting with aseptic meningitis. Am J Emerg Med. 2014 Oct. 32 (10):1298.e1-2.[↩]
- Singh YP, Agarwal V, Krishnani N, Misra R. Enthesitis-related arthritis in Kikuchi-Fujimoto disease. Mod Rheumatol. 2008 May 10. epub ahead of print.[↩]
- Delplanque M, Chasset F, Hirsch P, Malard F, Ditchi Y, Fain O, Mekinian A. Cutaneous lupus with Kikuchi disease-like inflammatory pattern associated with myelodysplastic syndrome. Rheumatology (Oxford). 2018 Dec 19[↩]
- Zuckerman R, Damiani L, Ayyad HA, Alpert DR. Persistent cervical lymphadenitis in a patient with prior thyroid cancer attributed to Kikuchi-Fujimoto disease. BMJ Case Rep. 2018 Oct 21;2018[↩]
- Barbat B, Jhaj R, Khurram D. Fatality in Kikuchi-Fujimoto disease: A rare phenomenon. World J Clin Cases. 2017 Feb 16. 5 (2):35-39.[↩]

{kind=link}