Lysosomal storage disease
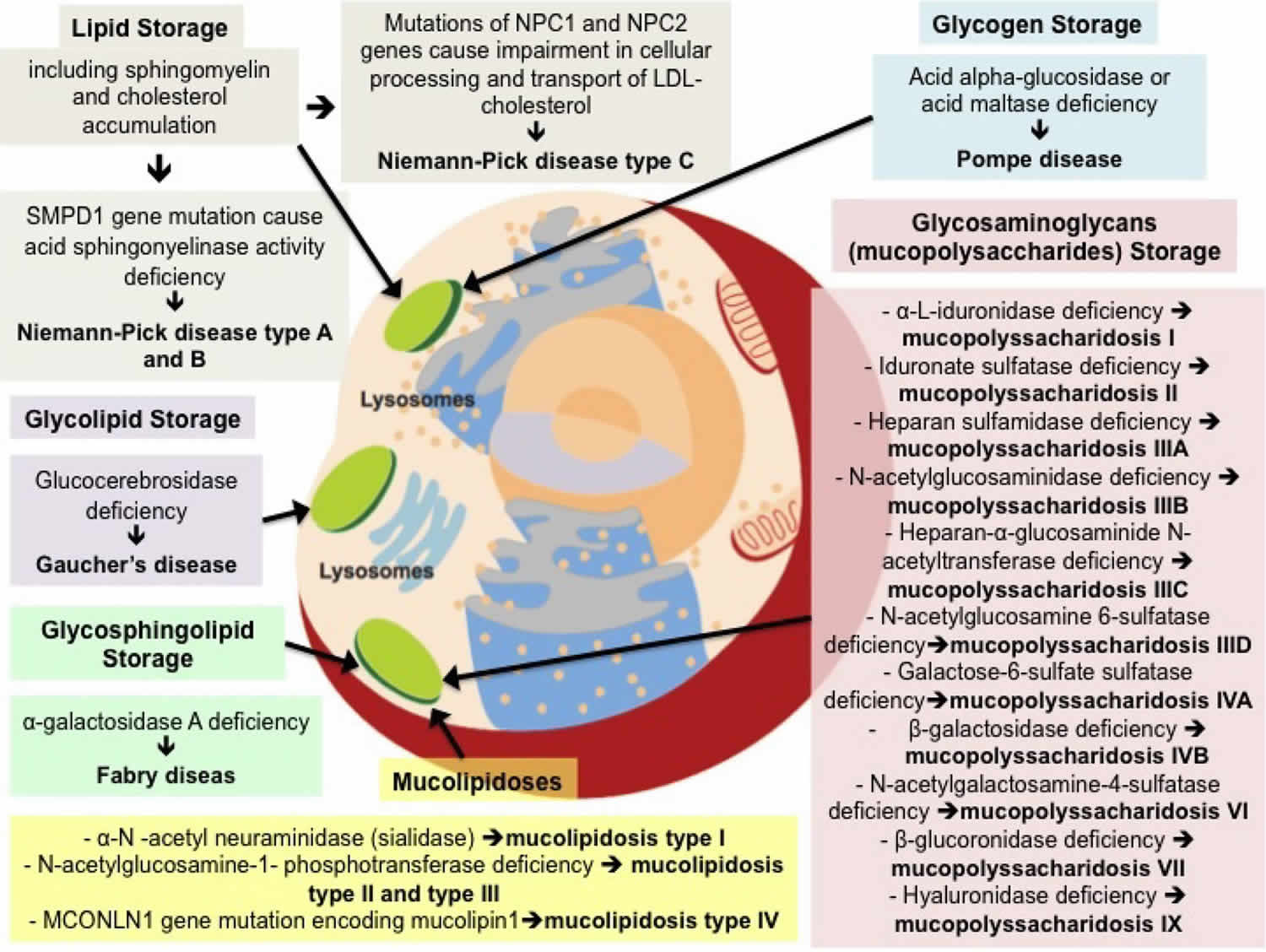
Lysosomal storage diseases comprise a group of over 70 inherited metabolic disorders caused by deficiency of certain enzymes in certain compartments of the cells that generally involve progressive neurological manifestations and that primarily affect children 1. Lysosomal storage diseases are individually rare but collectively affect 1 in 5,000 live births.
All lysosomal storage diseases share a common pathogenesis: a genetic defect in one or more specific lysosomal enzymes, activator protein or membrane protein, resulting in deficient enzymatic activity. In other words, when the lysosome does not function normally, excess products destined for breakdown and recycling is stored in the cell.
Lysosomal storage disease symptoms vary greatly among the specific disorders and other variables like the age of onset and can be mild or severe. Neurological signs and symptoms can include developmental delay, movement disorders and seizures, dementia, deafness and/or blindness. Other symptoms might affect the bones that grow abnormally, gastrointestinal, enlarged livers (hepatomegaly), enlarged spleens (splenomegaly), dermatological, pulmonary and cardiac problems and ophthalmologic systems. A patient with lysosomal storage disease not only develops physical deformation in cell structures throughout the body, but these cells often die resulting in a wide variety of clinical symptoms. If diagnosed late and/or left untreated patients are at risk of developing significant, irreversible damage and loss of body functions and life-threatening complications.
Although most of these lysosomal storage diseases were described in the 20th century, the cellular pathogenesis of these diseases is complex and is currently incompletely understood.
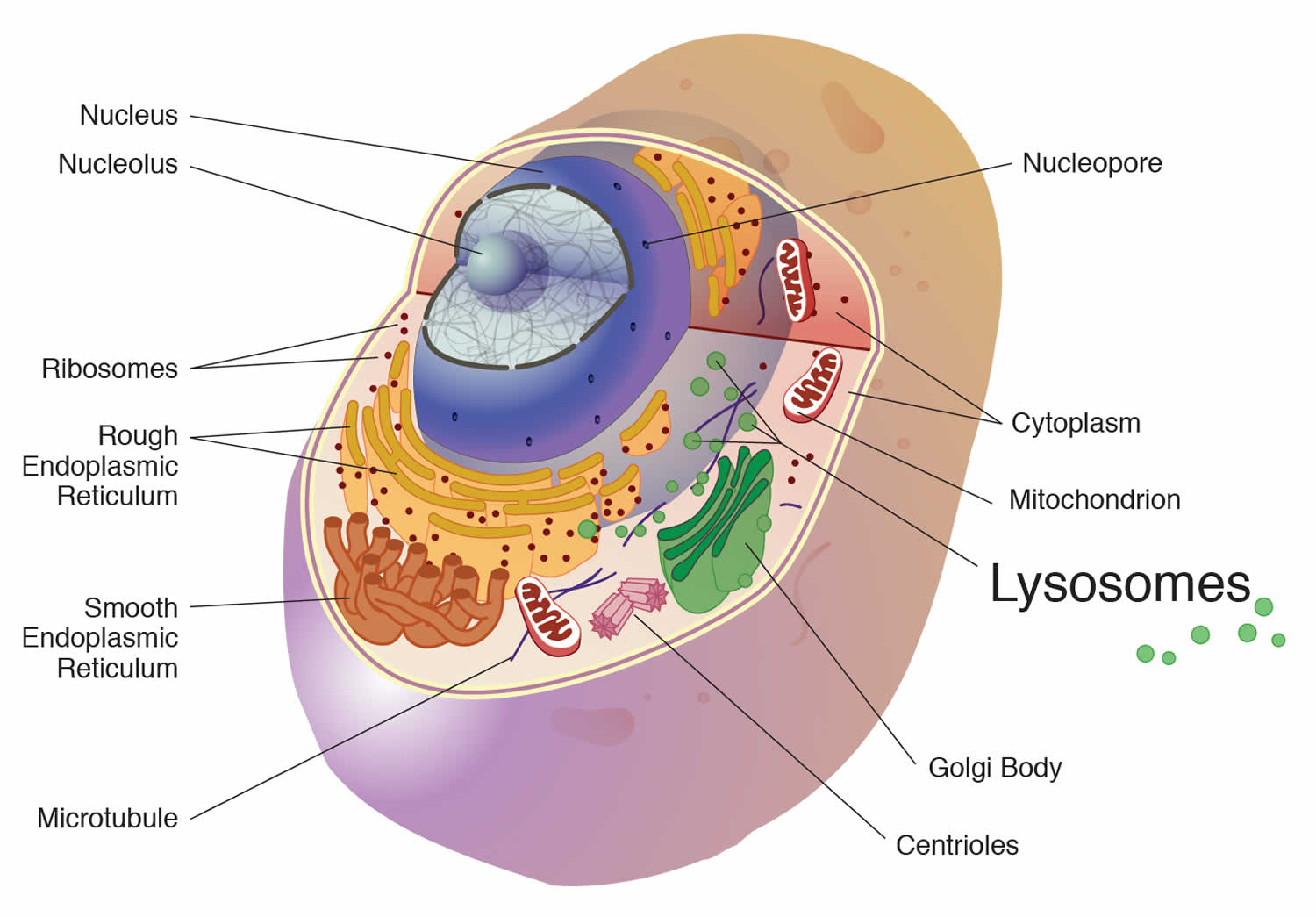
In all cells, including neurons, the primary degradative organelle is the lysosome. The lysosome is the key cellular hub for macromolecule catabolism, recycling and signalling. Lysosomes are subcellular organelles responsible for the physiologic turnover of cell constituents. Lysosomes contain catabolic enzymes, which require a low pH environment in order to function optimally. Lysosomes receive their ‘substrates’ through various pathways including endocytosis, phagocytosis, and autophagy. Substrates are degraded via a finely orchestrated network of membrane bound lysosomal proteins, soluble lysosomal hydrolases, lysosomal related organelles, and other cellular constituents. Defects that impair any of these functions cause the accumulation of undigested or partially digested macromolecules in lysosomes (that is, ‘storage’) or impair the transport of molecules, which can result in cellular damage. When lysosomal function is impaired, neuronal dysfunction and neurodegeneration can occur.
The lysosome itself was discovered in 1955 and by 1960 its role in cellular digestion was well understood. Pompe disease was the first to be formally diagnosed. By 1970s the scientific community had recognized many more lysosomal storage diseases and categorized them based on the type of enzymatic defect and/or stored substrate product.
What is lysosome?
A lysosome is a membrane-bound cell organelle that contains digestive enzymes called hydrolytic enzymes and they break down large molecules into small molecules. Lysosomes digest excess or worn out organelles, food particles, and engulfed viruses or bacteria. The membrane surrounding a lysosome prevents the digestive enzymes inside from destroying the cell. Lysosomes are involved with various cell processes. Lysosomes break down excess or worn-out cell parts. They may be used to destroy invading viruses and bacteria. If the cell is damaged beyond repair, lysosomes can help it to self-destruct in a process called programmed cell death or apoptosis.
Lysosomes also breakdown large proteins into amino acids or large carbohydrates into simple sugars or large lipids into single fatty acids. And when they do that, they provide for the rest of the cell the nutrients that it needs to survive. So, for example, if lysosomes can’t do that, they can’t break down large molecules into small molecules. You’ll have storage of those large molecules, and this is called lysosomal storage diseases. There’s also another type of lysosome storage disease in which the small molecules that are produced from those large molecules can’t get out of the lysosome. They’re stored there because the transporters for moving these small molecules out are missing genetically. And finally, one other function of the lysosome is to ingest bacteria so that the bacteria can be destroyed. So the lysosomes also provide a function against infection, and the cell will often engorge a bacterium and put it into its lysosome for destruction. So here’s an important organelle that has function against infection and function in a way in nutrition to break down large molecules into small molecules so that they can be reutilized.
Figure 1. Lysosome
Lysosomal storage disease list
More than 70 lysosomal storage diseases have been described 1. They are classified below 2.
Glycogen storage disease type 2
Glycogen storage disease type 2 or acid alpha-glucosidase (acid maltase) a lysosomal enzyme deficiency, is an inherited disorder of glycogen metabolism resulting from defective activity of the lysosomal enzyme alpha-glucosidase in tissues of affected individuals. In turn, this defect results in intralysosomal accumulation of glycogen of normal structure in numerous tissues.
Two types are as follows:
- Infantile-onset Pompe disease
- Late-onset Pompe disease
The mode of inheritance is autosomal recessive, and the gene encoding for acid alpha-glucosidase has been localized to arm 17q23.
The disorder is genetically heterogeneous with missense, nonsense, and frameshift mutations, as well as splice-site and partial deletions.
Phenotypic expression is variable, and the severity is probably correlated with residual acid alpha-glucosidase activity.
Mucopolysaccharidoses
Mucopolysaccharidoses (MPS) and mucolipidosis are genetic lysosomal storage diseases caused by the body’s inability to produce specific enzymes. The missing or insufficient enzyme prevents cells from recycling waste, resulting in the storage of materials in cells throughout the body. As the disease progresses, there is widespread damage throughout the body, including the heart, bones, joints, respiratory system and central nervous system, leading to a shortened lifespan.
Mucopolysaccharides are long chains of sugar molecule used in the building of connective tissues in the body.
- “muco” refers to the thick jelly-like consistency of the molecules
- “poly” means many
- “saccharide” is a general term for a sugar molecule (think of saccharin)
There is a continuous process in the body of replacing used materials and breaking them down for disposal.
Many mucopolysaccharidoses can be detected via urine glycosaminoglycan assay. Mucopolysaccharidoses Types 1-7 and type 9:
- Mucopolysaccharidosis type 1 H, Hurler syndrome (alpha-L-iduronidase deficiency)
- Mucopolysaccharidosis type 1 H/S, Hurler-Scheie syndrome
- Mucopolysaccharidosis type 1 S, Scheie syndrome
- Mucopolysaccharidosis type 2 A, Hunter syndrome, severe (iduronate sulfatase deficiency)
- Mucopolysaccharidosis type 2 B, Hunter syndrome, mild (iduronate sulfatase deficiency)
- Mucopolysaccharidosis type 3 A-D, Sanfilippo A, B, C and D syndrome (heparan N -sulfatase deficiency)
- Mucopolysaccharidosis type 4, Morquio A and B syndrome, classic (galactose 6-sulfatase deficiency)
- Mucopolysaccharidosis type 5, formerly Scheie Syndrome
- Mucopolysaccharidosis type 6, Maroteaux-Lamy syndrome (arylsulfatase B deficiency)
- Mucopolysaccharidosis type 7, Sly syndrome (beta-glucuronidase deficiency)
- Mucopolysaccharidosis type 9 (Hyaluronidase deficiency) is a condition that was first noted in 1996. It is caused by a deficiency of the enzyme, hyaluronidase. This enzyme is used in the lysosome to break down a complex string of sugars known as Glycosaminoglycans (GAGs), often referred to as mucopolysaccharides. In individuals inflicted with mucopolysaccharidosis type 9 , the GAG accumulates resulting in symptoms of this disorder.
Mucolipidoses
The mucolipidoses consist of the following:
- Mucolipidosis 1: This term that has been used to describe sialidosis.
- Mucolipidosis 2 (I-cell disease): Pseudo-Hurler Polydystrophy
- Mucolipidosis 3 (phosphotransferase deficiency): Pseudo-Hurler Polydystrophy
- Mucolipidosis 4 (mucolipidin 1 deficiency): Clinical features include psychomotor retardation, corneal clouding, and retinopathy. Screening may be accomplished by screening serum gastrin levels, which are typically significantly elevated.
Oligosaccharidoses
Many oligosaccharidoses can be detected by screening urine for oligosaccharide accumulation.
- Schindler disease/Kanzaki disease (alpha-N -acetylgalactosaminidase deficiency)
- Alpha-mannosidosis and beta-mannosidosis
- Alpha-fucosidosis: Clinical features include progressive neuromotor deterioration, seizures (including myoclonic seizures), coarse facial features, dysostosis multiplex, angiokeratoma corporis diffusum, hepatosplenomegaly, and growth retardation.
- Sialidosis (mucolipidosis I; alpha-N -acetyl neuraminidase [sialidase] deficiency)
- Aspartylglucosaminuria (aspartylglucosaminase deficiency): The progress of this disease is slower than many other lysosomal storage diseases. Patients appear healthy during infancy and generally live 25-45 years. Clinical features include intellectual disability, neurobehavioral symptoms, milder skeletal abnormalities, hepatosplenomegaly, and facial coarsening.
Lipidoses
The lipidoses include the following:
- Niemann-Pick disease types C and D 3
- Neuronal ceroid lipofuscinoses
- Wolman disease (acid lipase deficiency, mild form cholesterol ester storage disease) (See Wolman Disease and Cholesteryl Ester Storage Disease for detailed information.) 4
Sphingolipidoses
The sphingolipidoses are as follows:
- Niemann-Pick disease type A (sphingomyelinase deficiency) and Niemann-Pick disease type B (sphingomyelinase deficiency)
- Gaucher disease types I, II, and III (beta-glucosidase deficiency)
- Krabbe disease, infantile globoid-cell leukodystrophy (galactosylceramidase deficiency)
- Fabry disease (alpha-galactosidase A)
- GM1 gangliosidosis and Morquio B disease (beta-galactosidase deficiency)
- GM2 gangliosidoses: These include Tay-Sachs disease (hexosaminidase A deficiency) and Sandhoff disease (hexosaminidase A and B deficiency)
- Metachromatic leukodystrophy (arylsulfatase A deficiency)
- Farber disease, disseminated lipogranulomatosis (ceramidase deficiency): Farber disease starts to manifest in infancy as a hoarse cry or swollen tender joints followed by the development of subcutaneous nodules, flesh-colored papules, and periarticular tumors or nodules. Clinical features include coarsening facial features, osteopenia, and neurodegeneration. Histopathology shows foam cells and granulomatous infiltration. Ultramicroscopically, curvilinear tubular bodies are present as comma-shaped tubular structures consisting of 2 single membranes separated by a clear space in dermal fibroblasts. Banana bodies, variably membrane-bound structures that have a spindle and usually a curved shape, are found predominantly in Schwann cells of peripheral nerves.
- Multiple sulfatase deficiency (sulfatase-modifying factor-1 mutation): Mutation in SUMF1 leads to deficiency of 7 sulfatases. The resulting disorder combines features of metachromatic leukodystrophy and mucopolysaccharidosis. Clinical features include proptosis, ichthyosis, broad thumbs and index fingers, progressive leukoencephalopathy, and hepatosplenomegaly.
- Galactosialidosis (cathepsin A deficiency): Mutation in CTSA leads to a combined deficiency of lysosomal beta-galactosidase and neuraminidase as a result of a primary defect in the protective protein/cathepsin A (PPCA). Clinical features include coarse facial features, macular cherry-red spots, angiokeratomas, dysostosis multiplex, epilepsy, myoclonus, and ataxia.
Lysosomal transport diseases
The lysosomal transport diseases are as follows:
- Cystinosis (cystine transporter deficiency): Clinical features include nephropathy (most common inherited cause of renal Fanconi syndrome), short stature, myopathy, corneal crystals, and possibly neurodegeneration in adulthood. Screening can be performed via cystine level assay in leukocytes. It can be treated with cysteamine.
- Sialic acid storage disease (Salla disease; sialic acid transporter deficiency): Clinical features include hypotonia, spasticity, ataxia, intellectual disability, growth retardation, and epilepsy. It is more common in the Finnish population owing to a founder effect. It may be detected by assay of free sialic acid levels in urine.
Lysosomal storage disease symptoms
Lysosomal storage diseases typically present in infancy and childhood, although adult-onset forms also occur. Most lysosomal storage diseases have a progressive neurodegenerative clinical course, although symptoms in other organ systems are frequent.
Glycogen storage disease type 2 symptoms
Two major presentations are (1) infantile acid maltase disease, or Pompe disease, and (2) slowly progressive acid maltase disease.
Infantile acid maltase disease or Pompe disease, is rapidly progressive and usually has an onset in the first 6 months of life. Infants with Pompe disease typically experience rapidly progressive muscle weakness (myopathy) and poor muscle tone (hypotonia) as indicated by feeding and respiratory difficulties, an enlarged liver (hepatomegaly), and progressive cardiomegaly. Affected infants may also fail to gain weight and grow at the expected rate (failure to thrive) and have breathing problems. If untreated, this form of Pompe disease leads to death from heart failure in the first year of life. Death prior to age 2 years may be due to cardiorespiratory failure 5.
Slowly progressive acid maltase disease is characterized by an onset of symptoms in childhood or adult life. Affected individuals may have progressive proximal weakness with manifestations limited to the skeletal muscles. Respiratory dysfunction with early ventilatory insufficiency may be out of proportion to the degree of limb weakness.
The non-classic form of infantile-onset Pompe disease usually appears by age 1. It is characterized by delayed motor skills (such as rolling over and sitting) and progressive muscle weakness. The heart may be abnormally large (cardiomegaly), but affected individuals usually do not experience heart failure. The muscle weakness in this disorder leads to serious breathing problems, and most children with non-classic infantile-onset Pompe disease live only into early childhood.
The late-onset type of Pompe disease may not become apparent until later in childhood, adolescence, or adulthood. Late-onset Pompe disease is usually milder than the infantile-onset forms of this disorder and is less likely to involve the heart. Most individuals with late-onset Pompe disease experience progressive muscle weakness, especially in the legs and the trunk, including the muscles that control breathing. As the disorder progresses, breathing problems can lead to respiratory failure.
Mucopolysaccharidoses symptoms
Mucopolysaccharidosis type 1
Mucopolysaccharidosis type 1 (MPS I) comprises a wide spectrum of severity and clinical involvement. Children with the classic, severe form of Hurler disease have progressive developmental delay, severe progressive physical problems and early advancement of the disease. Children and adults with Scheie disease do not have progressive developmental delay and their physical problems advance more slowly. There are others whose disease pattern will fall between the two ends of the spectrum. It is important to remember that mucopolysaccharidosis type 1 is extremely varied in its effects.
Mucopolysaccharidosis type 1 (MPS I) is currently divided into the severe and attenuated types.
Children with MPS I often appear normal at birth, although some have a soft out-pouching around the belly-button (umbilical hernia) or lower abdomen (inguinal hernia). People with severe MPS I generally begin to show other signs and symptoms of the disorder within the first year of life, while those with the attenuated form have milder features that develop later in childhood.
Individuals with MPS I may have a large head (macrocephaly), a buildup of fluid in the brain (hydrocephalus), heart valve abnormalities, distinctive-looking facial features that are described as “coarse,” an enlarged liver and spleen (hepatosplenomegaly), and a large tongue (macroglossia). Vocal cords can also enlarge, resulting in a deep, hoarse voice. The airway may become narrow in some people with MPS I, causing frequent upper respiratory infections and short pauses in breathing during sleep (sleep apnea).
People with MPS I often develop clouding of the clear covering of the eye (cornea), which can cause significant vision loss. Affected individuals may also have hearing loss and recurrent ear infections.
Some individuals with MPS I have short stature and joint deformities (contractures) that affect mobility. Carpal tunnel syndrome develops in many children with this disorder and is characterized by numbness, tingling, and weakness in the hand and fingers. Narrowing of the spinal canal (spinal stenosis) in the neck can compress and damage the spinal cord.
While both forms of MPS I can affect many different organs and tissues, people with severe MPS I experience a decline in intellectual function and a more rapid disease progression. Developmental delay is usually present by age 1, and severely affected individuals eventually lose basic functional skills (developmentally regress). In this form of the disorder, death typically occurs by age 10. Individuals with attenuated MPS I typically live into adulthood and may or may not have a shortened lifespan. Some people with the attenuated type have learning disabilities, while others have no intellectual impairments. Heart disease and airway obstruction are major causes of death in people with both types of MPS I.
Mucopolysaccharidosis type 2
Mucopolysaccharidosis type 2 (MPS II) also known as Hunter disease comprises a wide spectrum of severity. Until recently mucopolysaccharidosis type 2 had been described as either mild or severe. However, based on current understanding of the enzyme and its gene, mucopolysaccharidosis type 2 comprises a wide spectrum of severity. Clinical manifestations include severe airway obstruction, skeletal deformities, cardiomyopathy and, in most patients, neurological decline. Death usually occurs in the second decade of life, although some patients with less severe disease have survived into their fifth or sixth decade. Some individuals will have progressive developmental delay and severe progressive physical problems. Others will have normal intelligence and progressive physical problems, some being more severely affected than others.
It is important to remember that mucopolysaccharidosis type 2 is extremely varied in its effects.
Mucopolysaccharidosis type 3
Mucopolysaccharidosis type 3 (MPS III) also known as Sanfilippo disease, is marked by severe neurological symptoms. These include progressive dementia, aggressive behavior, hyperactivity, seizures, some deafness and loss of vision, and an inability to sleep for more than a few hours at a time. This disorder tends to have three main stages. During the first stage, early mental and motor skill development may be somewhat delayed. Affected children show a marked decline in learning between ages 2 and 6, followed by eventual loss of language skills and loss of some or all hearing.
Some children may never learn to speak. In the syndrome’s second stage, aggressive behavior, hyperactivity, profound dementia, and irregular sleep may make children difficult to manage, particularly those who retain normal physical strength. In the syndrome’s last stage, children become increasingly unsteady on their feet and most are unable to walk by age 10.
Thickened skin and mild changes in facial features, bone, and skeletal structures become noticeable with age. Growth in height usually stops by age 10. Other problems may include narrowing of the airway passage in the throat and enlargement of the tonsils and adenoids, making it difficult to eat or swallow. Recurring respiratory infections are common.
To date, four different enzyme deficiencies have been found to cause MPS III and thus the condition is described as MPS III Type A, B, C or D. Type A is the most common form found in most populations.
- MPS III A is missing the enzyme heparan N sulphatase
- MPS III B is missing the enzyme alpha-Nacetylglucosaminidase
- MPS III C is missing acetyl-CoA:alpha-glucosaminide acetyltransferase
- MPS III D is missing N-acetylglucosamine-6-sulphatase
It is important to note that there are no significant, clinical, physical differences between the different subtypes of mucopolysaccharidosis type 3 disease, although there have been cases of late onset MPS III Type B where the individuals have remained relatively unaffected into adult life. The latest understanding is that some people seem to produce some enzyme activity which helps to slow down the progression of the disease whilst those with more severe symptoms appear to have no enzyme activity at all.
Mucopolysaccharidosis type 4
Mucopolysaccharidosis type 4 (MPS IV) also known as Morquio disease, is a progressive condition that mainly affects the skeleton. There are two types of Morquio disease which are referred to as MPS IVA and MPS IVB. The subtypes of this disease are as a result of the type of enzyme that is missing or malfunctioning within the cells of the body: A missing or malfunctioning enzyme known as ‘N-acetylgalactosamine 6-sulfatase’ results in mucopolysaccharidosis type 4A. In mucopolysaccharidosis type 4B the missing or malfunctioning enzyme is known as ‘beta-galactosidase’.
Morquio disease comprises a wide spectrum of severity and clinical involvement. Some individuals may not achieve a height of one meter by adulthood, whilst others grow significantly taller and have fewer problems related to severely restricted growth.
The first signs and symptoms of MPS IV usually become apparent during early childhood. Affected individuals develop various skeletal abnormalities, including short stature, knock knees, and abnormalities of the ribs, chest, spine, hips, and wrists. People with MPS IV often have joints that are loose and very flexible (hypermobile), but they may also have restricted movement in certain joints. A characteristic feature of this condition is underdevelopment (hypoplasia) of a peg-like bone in the neck called the odontoid process. The odontoid process helps stabilize the spinal bones in the neck (cervical vertebrae). Odontoid hypoplasia can lead to misalignment of the cervical vertebrae, which may compress and damage the spinal cord, resulting in paralysis or death.
In people with MPS IV, the clear covering of the eye (cornea) typically becomes cloudy, which can cause vision loss. Some affected individuals have recurrent ear infections and hearing loss. The airway may become narrow in some people with MPS IV, leading to frequent upper respiratory infections and short pauses in breathing during sleep (sleep apnea). Other common features of this condition include mildly “coarse” facial features, thin tooth enamel, multiple cavities, heart valve abnormalities, a mildly enlarged liver (hepatomegaly), and a soft out-pouching around the belly-button (umbilical hernia) or lower abdomen (inguinal hernia). Unlike some other types of mucopolysaccharidosis, MPS IV does not affect intelligence.
The life expectancy of individuals with MPS IV depends on the severity of symptoms. Severely affected individuals may survive only until late childhood or adolescence. Those with milder forms of the disorder usually live into adulthood, although their life expectancy may be reduced. Spinal cord compression and airway obstruction are major causes of death in people with MPS IV.
Only palliative measures are currently available for treatment of patients with Morquio syndrome (mucopolysaccharidosis type IV). Potential strategies for treatment of patients with the other mucopolysaccharidoses (MPSs), which are currently at different levels of development, include enzyme replacement therapy (ERT), gene therapy, and allogenic bone marrow transplantation in which engrafted cells provide the normal enzyme.
Mucopolysaccharidosis type 6
Mucopolysaccharidosis type 6 (MPS VI) also known as Maroteaux-Lamy disease. The disease varies enormously in the severity of the problems it causes. It is important to remember this if you are a parent of a newly diagnosed child.
Children with MPS VI, Maroteaux-Lamy syndrome, usually have normal intellectual development but share many of the physical symptoms found in Hurler syndrome. Caused by the deficient enzyme N-acetylgalactosamine 4-sulfatase, Maroteaux-Lamy syndrome has a variable spectrum of severe symptoms. Neurological complications include clouded corneas, deafness, thickening of the dura (the membrane that surrounds and protects the brain and spinal cord), and pain caused by compressed or traumatized nerves and nerve roots.
Growth is normal at first but stops suddenly around age 8. By age 10 children have developed a shortened trunk, crouched stance, and restricted joint movement. In more severe cases, children also develop a protruding abdomen and forward-curving spine. Skeletal changes (particularly in the pelvic region) are progressive and limit movement. Many children also have umbilical or inguinal hernias. Nearly all children have some form of heart disease, usually involving valve dysfunction.
Mucopolysaccharidosis type 7
Mucopolysaccharidosis type 7 (MPS VII) also known as Sly disease, one of the least common forms of the mucopolysaccharidoses, is estimated to occur in fewer than one in 250,000 births.
Mucopolysaccharidosis type 7 (Sly syndrome) causes children to be born with hydrops fetalis, in which extreme amounts of fluid are retained in the body.
Survival is usually a few months or less. Most children with Sly syndrome are less severely affected. Neurological symptoms may include mild to moderate mental retardation by age 3, communicating hydrocephalus, nerve entrapment, corneal clouding, and some loss of peripheral and night vision. Other symptoms include short stature, some skeletal irregularities, joint stiffness and restricted movement, and umbilical and/or inguinal hernias. Some patients may have repeated bouts of pneumonia during their first years of life. Most children with Sly syndrome live into the teenage or young adult years.
Mucopolysaccharidosis type 9
Mucopolysaccharidosis type 9 (MPS IX) also known as hyaluronidase deficiency that was first noted in 1996. It is caused by a deficiency of the enzyme, hyaluronidase. Hyaluronidase enzyme is used in the lysosome to break down a complex string of sugars known as Glycosaminoglycans (GAGs), often referred to as mucopolysaccharides. In individuals inflicted with MPS IX, the GAG accumulates resulting in symptoms of this disorder.
Symptoms include nodular soft-tissue masses located around joints, with periodic episodes of painful swelling of the masses. This pain spontaneously ends within three days.
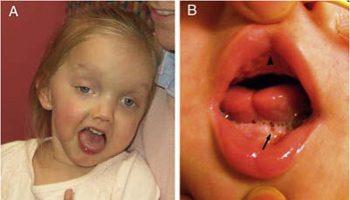
Other symptoms of this disease include a flattened nose bridge and a cleft palate. Short stature is also a noted symptom.
Gaucher’s disease
Gaucher’s disease is a rare, inherited disorder that causes too much of a substance called glucocerebroside to build up in your spleen, liver, lungs, bones and sometimes in your brain. The buildup prevents these organs from working properly.
There are three types:
- Type 1, the most common form, causes liver and spleen enlargement, bone pain and broken bones, and, sometimes, lung and kidney problems. It does not involve the brain. It can occur at any age.
- Type 2, which causes severe brain damage, appears in infants. Most children who have it die by age 2.
- In type 3, there may be liver and spleen enlargement, and signs of brain involvement appear gradually.
Fabry disease
Fabry disease is caused by the lack of or faulty enzyme needed to metabolize lipids, fat-like substances that include oils, waxes, and fatty acids. The enzyme is known as ceramide trihexosidase, also called alpha-galactosidase-A. A mutation in the gene that controls this enzyme causes insufficient breakdown of lipids, which build up to harmful levels in the eyes, kidneys, autonomic nervous system, and cardiovascular system. Since the gene that is altered is carried on a mother’s X chromosome, her sons have a 50 percent chance of inheriting the disorder and her daughters have a 50 percent chance of being a carrier.
Symptoms usually begin during childhood or adolescence and include:
- Burning sensations in the hands that gets worse with exercise and hot weather, and
- Small, raised reddish-purple blemishes on the skin.
Some boys will also have eye manifestations, especially cloudiness of the cornea. Lipid storage may lead to impaired arterial circulation and increased risk of heart attack or stroke. The heart may also become enlarged and the kidneys may become progressively involved.
Other symptoms include:
- Decreased sweating,
- Fever, and
- Gastrointestinal difficulties, particularly after eating.
- Hearing loss and tinnitus (ringing in the ears) may develop in adulthood.
Lysosomal storage disease diagnosis
The majority of the patients are initially screened by enzyme assay, which is the most efficient method to arrive at a definitive diagnosis. In some families where the disease-causing mutation(s) is known and in certain genetic isolates, mutation analysis may be performed. In addition, after a diagnosis is made by biochemical means, mutation analysis may be performed for certain disorders.
Lysosomal storage disease treatment
There are no cures for lysosomal storage diseases and treatment is mostly symptomatic, although bone marrow transplantation and enzyme replacement therapy (ERT) have been successful in some cases. Enzyme replacement therapy (ERT) appears safe and effective for peripheral manifestations in patients with Gaucher disease types I and III, Fabry disease, mucopolysaccharidosis I (Hurler, Hurler-Scheie, and Scheie syndromes), mucopolysaccharidosis II (Hunter syndrome), mucopolysaccharidosis VI (Maroteaux-Lamy syndrome), Pompe disease, and recently Batten disease (neuronal ceroid lipofuscinoses, CLN2). Efforts are underway to develop enzyme replacement options for several other disorders. Thus far, enzyme replacement therapy (ERT) has been largely unsuccessful in improving central nervous system manifestations of the lysosomal storage diseases, putatively due to difficulty in penetrating the blood-brain barrier. This has led to active clinical trials evaluating the safety and efficacy of intrathecal enzyme delivery in several lysosomal storage diseases.
In addition, umbilical cord blood transplantation is being performed at specialized centers for a number of these diseases. In addition, substrate reduction therapy, a method used to decrease the accumulation of storage material, is currently being evaluated for some of these diseases. Furthermore, chaperone therapy, a technique used to stabilize the defective enzymes produced by patients, is being examined for certain of these disorders. Gene therapy involves replacing the patient’s mutated gene with a normal copy to allow proper enzyme production. However, it is still in pre-clinical (animal studies) and much research is needed.
Accumulated data indicate that hematopoietic stem cell transplantation may be effective under optimal conditions in preventing the progression of central nervous system symptoms in neuronopathic forms of lysosomal storage diseases (such as Krabbe disease), including some of the mucopolysaccharidoses, oligosaccharidoses, sphingolipidoses, and lipidoses as well as peroxisome disorders such as X-linked adrenoleukodystrophy. Although longitudinal natural history data are limited, published guidelines are available to assist with decisions related to the pursuit of transplantation and whether to use bone marrow or umbilical cord blood–derived cells. In general, transplantation yields the best results when performed early in the course of the disease (ie, in an asymptomatic affected sibling of a child with a lysosomal storage disorder), in centers with experience in performing transplantations to treat inherited metabolic disorders, and in patients healthy enough to tolerate the conditioning and transplantation regimen.
Some evidence indicates that at least in certain disorders, combination ERT and hematopoietic stem cell transplantation together might be superior to hematopoietic stem cell transplantation alone in patients who are appropriate candidates.
The availability of both ERT and hematopoietic stem cell transplantation has prompted ongoing consideration of newborn screening efforts to diagnose lysosomal storage diseases. Although screening for these disorders has not been widely implemented, the potential to treat these disorders is likely to drive further efforts at development.
Gene therapy is experimental but in the future may help correct both somatic and neurologic abnormalities in a lysosomal storage disorder 6.
- Lysosomal storage disorders. Nature Reviews Disease Primersvolume 4, Article number: 28, 2018[↩][↩]
- Lysosomal Storage Disease. https://emedicine.medscape.com/article/1182830-overview[↩]
- Trilck M, Hübner R, Seibler P, Klein C, Rolfs A, Frech MJ. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J Rare Dis. 2013 Sep 18. 8(1):144.[↩]
- Reynolds T. Cholesteryl ester storage disease: a rare and possibly treatable cause of premature vascular disease and cirrhosis. J Clin Pathol. 2013 Sep 2.[↩]
- Bodamer OA, Dajnoki A. Diagnosing lysosomal storage disorders: Pompe disease. Curr Protoc Hum Genet. 2012 Oct. Chapter 17:Unit17.11.[↩]
- Tomatsu S, Azario I, Sawamoto K, Pievani AS, Biondi A, Serafini M. Neonatal cellular and gene therapies for mucopolysaccharidoses: the earlier the better?. J Inherit Metab Dis. 2015 Nov 17.[↩]

{kind=link}