
Congenital cystic adenomatoid malformation
Congenital cystic adenomatoid malformation also known as CCAM or congenital pulmonary airway malformation (CPAM), is a rare benign (non-cancerous) congenital lung cystic disease in which the adenomatoid proliferation of the bronchiolar epithelium results in the formation of multiple cysts in the lung’s lobes 1. CCAMs are considered part of the spectrum of bronchopulmonary foregut malformations. The routine use of prenatal ultrasonography has led to frequent prenatal diagnosis and has provided great insight into the natural history of cystic adenomatoid malformation. Improvements in surgical techniques (ie, both prenatal and postnatal) as well as greatly enhanced imaging modalities have altered the surgical approach to this lesion.
CCAM is the most common type of fetal lung lesion. CCAM account for ~25% of congenital lung lesions. The estimated incidence is approximately 1:1500-4000 live births and there is a male predominance. Congenital cystic adenomatoid malformation develops before a baby is born, and can vary in size and be either fluid-filled or solid. A large cyst is called a macrocystic lesion, and a small cyst or solid appearing lesion is called microcystic.
Approximately 80% of CCAMs are detected and diagnosed prenatally or during the neonatal period when patients present with dyspnea and cyanosis. Cystic adenomatoid malformations are also sometimes detected in infants and school-age children following respiratory infections, but they are rarely found in adults. In most cases, CCAM is in only one of the lungs. In 80 to 95 percent of cases, it is only present in one lobe of the affected lung. The right lung has three lobes, and the left lung has two lobes. CPAM can affect any of these lobes. Less than 2 percent are in both lungs.
Some CCAMs can be life-threatening if they are not treated, so early and accurate diagnosis is important. The vast majority of CCAM lesions are small enough that they will not cause any problems to the baby during pregnancy and the CCAM can be removed after birth. However, some large lesions can cause serious and even fatal complications, including fetal heart failure (also called fetal hydrops) or maternal mirror syndrome. These cases may require treatment before birth.
CCAM is often associated with other organ anomalies and aplasia, and these patients have poor prognoses.
Most series report a mortality rate of 25-30% of all children who present in the newborn period with CCAM; however, these figures do not include asymptomatic children who present later in life. Furthermore, the use of elective abortion may lead to an underestimation of perinatal mortality by preferentially terminating fetuses with a higher risk of mortality. The reported mortality rate of prenatally diagnosed CCAMs ranges from 9-49%. Reviews of children who are asymptomatic in the neonatal period with antenatally diagnosed lesions suggest that 3-10% will develop symptoms in the first year of life 2. A more recent study suggested that 18 of 21 patients developed symptoms at a median age of 2 years 3.
Risk factors for a poor outcome include hydrops fetalis 4. Other indicators of poor prognosis include the type of lesion; microcystic CCAM is associated with much poorer outcomes 4.
The overall size of the lesion has also been reported as being an important predictor of survival 5; however, this index may be compromised by the fact that CCAM may decrease in size or even resolve over time in utero 4. The major morbidity is related to pulmonary compromise. A large lesion may be associated with pulmonary hypoplasia 2. This can cause respiratory distress at birth.
Some authorities have suggested that the presence of bilateral lesions is associated with a worse outcome. More controversially, left-sided lesions may be associated with a greater mortality rate than right-sided lesions.
One study suggested that polyhydramnios is also associated with a poorer outcome.
The potential for malignant transformation is recognized in all cases of CCAM 6. Whether or not complete resection of the affected area completely removes this risk is not known.
Other complications that have been described include the development of spontaneous pneumothorax, hemopneumothorax, and associated hemoptysis 4.
Figure 1. CCAM ultrasound
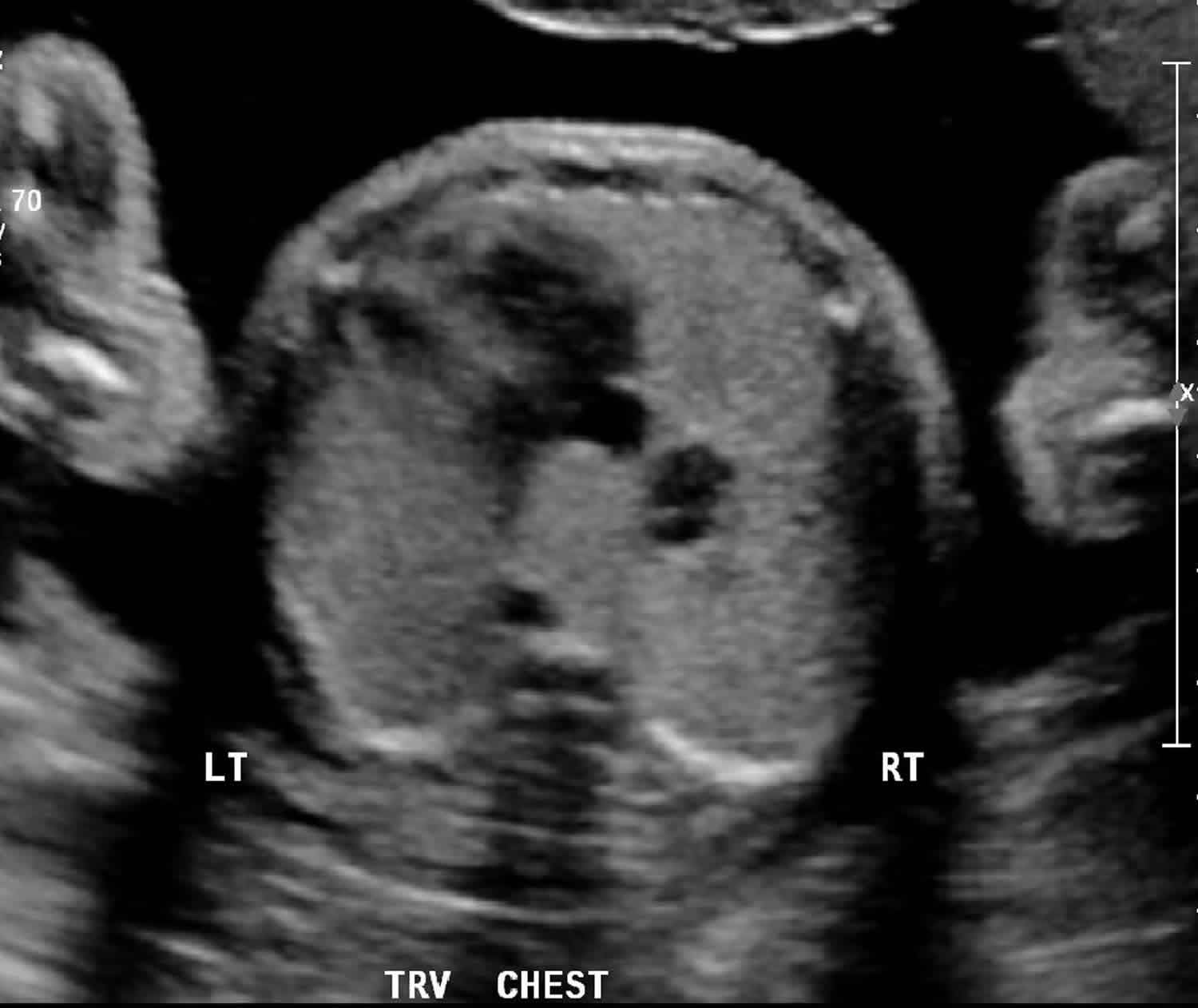
Footnote: The right lung is affected by the CCAM and has increased in echogenicity relative to the left. There has also been the development of a small cystic space in the lung. Mass effect from the malformation displaces the left atrium.
Can a CCAM be diagnosed before birth?
Yes, a CCAM can often be detected during a routine prenatal ultrasound, when doctors can see the location and size of the growth, which appears as a bright mass. The fetus’ heart may be shifted to the opposite side of her chest, and her normal lung tissue may be difficult to see on the side where the mass is located. Magnetic Resonance Imaging (MRI) may also be used to see the mass.
What happens during pregnancy?
In most cases, the fetus will do well and the mother will carry the baby to term without a problem. If your doctor suspects a CCAM, you will undergo a series of ultrasounds between your 20th to 30th week of pregnancy to see if it continues to grow. During that time, the fetus will grow rapidly, and the mass may appear smaller or unchanged. The CCAM may also shrink and even disappear during the third trimester of pregnancy.
However, in a small number of cases, the mass may grow to be life-threatening to the fetus. As it grows, there is the chance that it might compress blood vessels, making the heart have to pump harder to circulate blood, and possibly leading to heart failure (hydrops).
Can CCAM be treated prior to birth?
CCAM is often treated when the fetus develops hydrops or impending hydrops. Your doctor can monitor this closely by ultrasound. Signs of hydrops may include the following symptoms in the fetus:
- ascites (fluid in the abdomen)
- edema (extra fluid) of the skin and scalp
- pleural or pericardial effusion (fluid around the heart and lungs)
Doctors will also be paying close attention to the mother’s health. As the fetus develops hydrops, the mother may show symptoms of preeclampsia (a condition marked by high blood pressure and protein in the urine). If the mother is ill and the hydrops gets too severe, doctors may not be able to perform surgery.
If the fetus develops hydrops that isn’t too severe (or hydrops development seems imminent) and the mother’s health allows, prenatal intervention will be offered sometime prior to 28 weeks gestation. The procedure involved depends on the type of CCAM.
- If the mass is cystic in appearance, then it usually can be drained percutaneously (with a needle and a drain).
- If the fluid reaccumulates, a shunt (a method of redirecting fluid from one part of the body to another, where it can be absorbed) can be placed to continuously drain the cyst.
Sometimes when the mass is solid–and even sometimes when it is cystic–it will need to be removed during open fetal surgery. This procedure usually occurs prior to 28 weeks of pregnancy. During the procedure:
- The mother is placed under general anesthesia.
- The uterus is surgically opened.
- The fetus is partially taken out of the uterus and the mass is removed.
- The fetus is returned to the uterus, which is then closed.
- The mother and fetus are monitored closely after the procedure.
There is a risk for preterm labor when an incision is made in the uterus, but medications are available to help control the contractions.
What happens after the baby is born?
Doctors recommend that all babies with a CCAM, even those who underwent prenatal surgery, be delivered at a hospital with a Neonatal Intensive Care Unit (NICU). Often babies will have no symptoms after birth. When this happens, an x-ray will be performed and the baby will go home with mother.
When the baby is around 3 months old, she will have a special x-ray called a CT scan, which produces a 3-dimensional image of her lung and the growth.
Sometimes, babies have difficulties breathing after birth and require oxygen and assistance with breathing. Doctors may place a breathing tube and the baby will undergo an operation to remove the mass. After surgery, the baby will stay in the hospital for about a week.
Your doctor will schedule surgery to remove the CCAM during the baby’s is 3 to 6 months old. The hospital stay is usually about three to five days, and your pediatric surgeon will continue to see the baby after she is discharged from the hospital.
CCAM types
There are five types of CCAM lesions (types 0-4), some associated with cystic areas and adenomatous overgrowth of the terminal bronchioles 7.
Five CCAM subtypes are currently classified, mainly according to cyst size:
Type 0
- very rare, lethal postnatally
- acinar dysgenesis or dysplasia 8
- represents global arrest of lung development 9
Type 1
- most common: 70% of postnatal (after birth) cases 10
- large cysts that are few in number (1 to 4)
- one or more dominant cysts: 2-10 cm in size
- may be surrounded by smaller cysts
- Type 1 has a very favorable outcome.
Type 2
- 15-20% of cases 10
- cysts are <2 cm in diameter
- associated with other abnormalities
- renal agenesis or dysgenesis
- pulmonary sequestration
- congenital cardiac anomalies
- skeletal anomalies
- genitourinary anomalies
- hydrocephalus
- diaphragmatic hernia
- The prognosis for type 2 depends upon the severity of the associated anomalies.
Type 3
- ~10% of cases
- microcysts: <5 mm in diameter
- typically involves an entire lobe that cause shifting of the organs that are normally found in the chest. This means that the windpipe, heart and unaffected lung are shifted and compressed. For this reason, heart may not be able to function well. This can lead to the development of hydrops. Hydrops is an abnormal accumulation of fluid in at least two fetal cavities, such as in the abdomen, around the heart or lungs or under the skin.
- The prognosis for type 3 CCAM is poor, especially when hydrops occurs.
Type 4
- unlined cyst
- typically affects a single lobe
- indistinguishable from type 1 on imaging 8
Congenital cystic adenomatoid malformation causes
The cause of CCAM is unknown. CCAM not linked to a gene or to a chromosomal abnormality, and does not run in families (is not hereditary). Cystic adenomatoid malformation is caused by overgrowth of abnormal lung tissue that may form fluid-filled cysts. The cysts prevent the tissue from functioning as normal lung tissue. Resected CCAMs show signs of increased cell proliferation and decreased apoptosis 11. Studies have investigated the role of HOXB5 gene and protein expression, as well as other growth factors such as mesenchymal platelet–derived growth factor-BB 12. CCAMs typically increase in size until 26 – 28 weeks’ gestation, after which the size of many CCAMs plateau or even decrease.
Pathophysiology
The pathophysiologic effects of CCAM may be divided into prenatal and postnatal effects. Large lesions may be associated with the development of hydrops fetalis in as many as 40% cases and is a poor prognostic sign. Hydrops is thought to arise from compression of the inferior vena cava, which compromises venous return and leads to a decrease in cardiac output and the development of effusions. Fetal demise may result; premature delivery is attempted in order to salvage the fetus 13. The other main prenatal event is compromised pulmonary growth. Resultant pulmonary hypoplasia may lead to the postnatal development of respiratory distress.
Polyhydramnios has also been associated with CCAM. This develops as a result of elevated intrathoracic pressure that leads to esophageal compression and the inability to swallow 14.
CCAM may remain undiagnosed until it is discovered as an incidental finding later in life; however, its usual postnatal presentation is respiratory distress in the newborn period. This may be due to pulmonary hypoplasia, mediastinal shift, spontaneous pneumothorax, and pleural effusions secondary to hydrops. Recurrent chest infections may be a feature later in life 15. A risk of malignant transformation in later years is noted 6.
Prenatal regression and complete prenatal resolution have also been described 16.
CCAM symptoms
With the increasing use of prenatal ultrasonography as well as improvement in technology and skill, most cases of congenital cystic adenomatoid malformation (CCAM) are prenatally diagnosed. Prenatally diagnosed lesions can present with various symptoms.
Many babies with CCAM will appear “normal” at birth without any apparent complications. If CCAM is not diagnosed prenatally, these babies would typically go home and either never be diagnosed or would be diagnosed later after a chest X-ray or after recurrent respiratory infections.
At birth, some babies born with CCAM will not be able to breathe effectively due to the large mass in their chest. If babies are unable to breathe effectively on their own, it can have an effect on their heart rate. These babies need immediate intensive care to stabilize them until they can have surgery.
About 10 percent of the prenatally diagnosed masses will get smaller or even disappear on their own.
Respiratory distress
- This is the presenting symptom in most newborns with a diagnosis of CCAM. It may range in severity from grunting, tachypnea, and a mild oxygen requirement to fulminant respiratory failure requiring aggressive ventilator support or extracorporeal membrane oxygenation (ECMO).
- Multiple mechanisms account for the onset of respiratory difficulty. Pulmonary hypoplasia may arise as a consequence of a large CCAM, mediastinal shift may compromise cardiac and respiratory function, spontaneous pneumothoraces may occur, and air trapping within the cyst leads to compression of functional pulmonary tissue.
Recurrent infection
- Children in whom the CCAM has not been resected are at risk of recurrent pulmonary infections due to bronchial compression, air trapping, and inability to clear secretions.
Hemoptysis (coughing up blood)
- Hemoptysis has occasionally been described as a manifestation of CCAM in the older child.
Shortness of breath (dyspnea) and chest pain
- Dyspnea may be a feature of pneumothorax, which has been described as a presenting feature of CCAM.
Miscellaneous: Cough, fever, and failure to thrive have all been reported in association with the presentation of CCAM.
Generally, the physical signs observed in children with CCAM are nonspecific.
- Tachypnea: Tachypnea is the most common sign encountered in the newborn period, reflecting respiratory distress.
- Pneumothorax/air trapping: Signs consistent with a pneumothorax or air trapping may be elicited, including tracheal deviation, which indicates mediastinal shift, shifted heart sounds, and decreased air entry on the affected side.
- Cyanosis
- Accessory muscle use
- Grunting
- Failure to thrive
CCAM complications
- Fetal death caused by hydrops, fetal surgery, prematurity, or associated malformations
- Premature delivery due to polyhydramnios.
- Respiratory distress due to hydrops, pulmonary hypoplasia, pulmonary hypertension, pneumothorax, or prematurity
- Postnatal death due to respiratory distress, untreated hydrops, or pulmonary hypertension
- Recurrent pneumonia 17
- Pneumothorax
- Hemothorax
- Malignant change: Rhabdomyosarcoma, pulmonary blastomas, minute squamous cell carcinoma, and bronchioloalveolar carcinoma have all been described in association with CCAM 18.
CCAM diagnosis
Prenatal ultrasonography
With increasing use and technical ability of prenatal ultrasonography and sonographers, most cases of congenital lung anomalies are prenatally diagnosed 13. No specific diagnostic features of CCAM allow the ability to distinguish it unequivocally from other lung lesions such as congenital lobar emphysema or pulmonary sequestration.
Ultrasonography may demonstrate evidence of hydrops, such as fetal ascites or pleural effusions 19.
Type 1 lesions appear as multiple large cystic areas in the lung. In type 2 lesions, multiple small cysts are evident on ultrasonography. Because of the extremely small size of the cysts in type 3 lesions, the prenatal ultrasonographic appearance is often one of a homogenous mass.
Chest radiography
Chest radiography is essential in the workup of the child with suspected CCAM 20. The appearance of CPAMs will vary depending on the type.
Chest radiographs in type 1 and 2 CCAMs may demonstrate a multicystic (air-filled) lesion. Large lesions may cause a mass effect with resultant mediastinal shift, depression, and even inversion of the diaphragm. In the early neonatal period, the cysts may be completely or partially fluid-filled, in which case the lesion may appear solid or with air-fluid levels. Lesions may change in size on interval imaging (expand from collateral ventilation via pores of Kohn). Type 3 lesions appear solid.
CT scanning
- CT scanning of the thorax provides a safe and rapid means of defining the extent of CCAM in all age groups 21.
- The typical appearance is of multilocular cystic lesions with thin walls surrounded by normal lung parenchyma. The presence of superimposed infection with the lesion may complicate the appearance.
- Air fluid levels may be evident.
- CT scanning of the chest may outline additional coexisting lesions. Series have suggested a small percentage of cases thought to have resolved antenatally that demonstrated persistence of lesions on CT scan.
The definition of high-resolution chest tomography (HRCT) is sufficient to differentiate between microcystic and macrocystic lesions.
MRI
- MRI permits increased definition of a particular lesion, thereby enhancing the clinician’s ability to accurately diagnose and offer an informed prognosis.
- Additionally, maternal problems that prevent the optimal use of fetal ultrasonography, such as obesity, poor fetal lie, and oligohydramnios, pose no obstacle to MRI. MRI may be useful particularly in distinguishing CCAM from congenital diaphragmatic hernia.
- Lastly, no maternal or fetal exposure to ionizing radiation occurs in contrast to the use of CT scanning.
Other imaging studies
- Perform renal and cerebral ultrasonography in all newborns with CCAM in order to exclude coexisting renal and CNS anomalies.
- Perform echocardiography in all newborns with CCAM to rule out any coexisting cardiac lesions. Furthermore, in infants with respiratory distress, echocardiography may provide evidence of persistent pulmonary hypertension (eg, right-to-left shunting, increased pulmonary artery pressures).
CCAM treatment
No specific medical therapies are described for congenital cystic adenomatoid malformation (CCAM), aside from antibiotics in children with CCAM complicated by pneumonia and supportive care, ranging from oxygen supplementation to mechanical ventilation, in older children with respiratory distress 22. Solid microcystic lesions sometimes respond to steroids, which may be used to interrupt the growth of the lesion. Using steroids to slow the growth of the CCAM can prevent the need for fetal surgery in certain cases 23.
Some of the more severely affected infants may develop persistent pulmonary hypertension. Persistent pulmonary hypertension is a serious condition in which pressure in the lungs is high, forcing blood flow away from the lungs. This results in decreased levels of oxygen in the blood that goes to the body. High pressure in the lungs is normal for a fetus before birth because they are not using their lungs this way. However, after birth, this high pressure must decrease so that an adequate blood flow can pass through the lungs to rid the blood of carbon dioxide and pick up oxygen. Treatment for this condition may include inhaled nitric oxide or ECMO. ECMO stands for extracorporal membrane oxygenation. It is a machine that does work that the heart and lungs normally would do.
Management of CCAM during pregnancy
Most CCAMs are small and do not cause any stress on your baby’s heart before birth, or any problems with breathing after birth. These small CCAMs only require close monitoring before birth to ensure they remain small.
In rare instances, CCAMs grow to be large and may require treatment before birth. When the lung mass grows, it can take up valuable space in the chest. This can restrict normal lung growth and can lead to underdeveloped lungs which will not function adequately at birth. Large lesions can also shift the heart and impair blood flow. This can lead to fetal heart failure (fetal hydrops) and cause the buildup of fluid in the fetus and placenta.
If the fetus develops hydrops, you may “mirror” your baby’s illness. If you are carrying a baby diagnosed with CCAM, your pregnancy must be carefully monitored for high blood pressure and other signs and symptoms of the maternal mirror syndrome, or preeclampsia. Blood pressure and urine tests can monitor for signs of preeclampsia.
Delivery of babies with CCAM
Most commonly, you and your medical team will make a delivery plan for 32 weeks’ gestation. Your delivery will depend on the size of the CCAM and the course of prenatal treatment.
In the majority of cases, a mother may deliver her baby at her local hospital, without the need for high-risk neonatal care. These small lesions may no longer be visible on follow-up ultrasounds or imaging at birth, but the lesion is still present. The fetal surgeon who counseled you prenatally will determine when the lesion can be removed after birth. It is important for you to schedule a follow-up appointment once your baby is born.
Babies with larger or more complex lesions might have trouble breathing when they’re born, and should deliver in a center that offers expert care for both mother and baby in one location.
Surgical care
Surgical intervention is the mainstay of therapy for CCAM including fetal surgery and postnatal surgical approaches. These treatment options depend on the type of CCAM (microcystic or macrocystic) and the gestational age of your fetus. These options be discussed with you at the time of your evaluation with your doctor.
Fetal intervention
Fetal surgery should be considered in patients with large CCAMs and in cases complicated by hydrops, in which prognosis is poor 13.
Thoracocentesis allows drainage of a large cyst with immediate decompression of the CCAM; however, fluid rapidly reaccumulates, thus negating the benefit of the procedure 13.
Another option is to place a thoracoamniotic shunt that continually drains fluid from the CCAM to the amniotic space. This is most beneficial when the CCAM consists of a large fluid-filled cyst. Complications such as obstruction and shunt dislodgement may occur 13. The shunting procedure itself is performed under ultrasound guidance. A large trocar (hollow needle) is guided through the mother’s abdomen and uterus, and into the fetal chest. The shunt is passed through the trocar into the fluid-filled cyst. The goal is to divert fluid from the cyst to the amniotic sac. The shunt will remain until delivery. The goal of these procedures is to decrease the size of the mass to ward off heart failure (fetal hydrops).
Ex-utero intrapartum treatment (EXIT) procedure: If your baby has a large CCAM that continues to grow, but hydrops does not develop, a special delivery technique called an ex utero intrapartum treatment (EXIT) procedure may be required. This delivery technique is required because their lungs are compressed and they may not be able to breathe on their own when they’re born. In an EXIT procedure, a Special Delivery Unit team will partially deliver the baby so that they are still attached to the placenta and receiving oxygen through the umbilical cord. This procedure allows time for fetal surgeons to establish an airway and remove the CCAM 24. After the mass is removed and your baby can breathe on their own, they will be fully delivered.
C-section to resection: More commonly, babies with large lung lesions can be safely delivered by cesarean section and be carried immediately to the adjacent operating room where expert fetal surgeons will remove the mass.
Resection of the affected lobe (lobectomy) is an alternative procedure for cases with no dominant cyst available for draining. Fourteen cases treated with maternal-fetal surgery from the Children’s Hospital of Philadelphia suggest a 50% survival rate from the time of surgical intervention to infant discharge from the neonatal intensive care unit (NICU) 19. Complications included intraoperative bradycardia, the development of preterm labor and maternal mirror syndrome requiring early delivery, and postoperative intrauterine death. Survivors demonstrated residual lung growth and normal development.
Postnatal surgery
Resection of CCAM in all children is recommended to remove the risk of direct complications, such as recurrent infection and pneumothorax. Additionally, the malignant potential of CCAM in later life has long been recognized 18. Children with asymptomatic CCAM that was diagnosed antenatally can be followed without surgical intervention as some lesions may decrease in size or resolve without intervention. If surgery is recommended, most suggest it be completed before the child is aged 12 months to enhance compensatory lung growth. Studies have not confirmed this hypothetical difference in lung function by age at time of surgical resection 7.
Older patients
In one third of cases, the presence of pneumonia indicates the need for more extensive pulmonary resection 25.
When can my baby go home?
Your baby will go home when he or she can eat enough to maintain and gain weight and can breathe well without help. The length of stay for a baby with CCAM varies depending on the severity of his or her condition and his or her response to treatment.
If the cyst(s) is large, involves more than one lobe, or is bilateral (involves both lungs) and requires immediate intervention and surgery, the stay will be longer. The average stay for this course of care can range from 4 to 12 weeks.
This is compared to a baby who has milder symptoms or is able to wait 2 to 6 months for surgery. Their average stay can range from a few days in the newborn nursery to 1 or 2 weeks in the NICU. If there are associated anomalies (more common with type 2 CCAM), it can prolong or complicate the length of stay as well, no matter how large or small the CCAM is.
Follow-up care
Follow-up care for children with CCAM will depend on the treatment the child received.
Most children treated for small lesions after birth will only need monitoring for the first year after surgery to ensure normal lung growth and lung function. The majority will require no additional long-term follow-up care.
Children treated for more severe or complex lung lesions may require ongoing monitoring through childhood. For those who experience limited lung growth resulting in pulmonary hypoplasia, Pulmonary Hypoplasia Program offers comprehensive long-term care. The Pulmonary Hypoplasia Program team provides multidisciplinary care with a focus on improving your child’s pulmonary health, evaluating neurodevelopmental growth, meeting nutritional needs, monitoring for late onset hearing loss or any surgical issues, and more.
CCAM prognosis
Generally, following resection of the CCAM, anticipate full recovery. The vast majority of children with fetal lung lesions do extremely well and have normal lung function after their lesions are removed thanks to rapid compensatory lung growth that occurs during childhood. Having surgery early maximizes this compensatory growth.
Regular follow-up in patients who present after adolescence may be necessary in order to detect the presence of an associated malignancy 18.
In patients who underwent fetal surgery or postnatal resection of CCAM, no clinical evidence of respiratory problems is anticipated; however, a small study has shown that a slight reduction in lung volumes occurred in children who had CCAM postnatally resected 26. Few reports of long-term outcomes are available.
Prognosis
- The risk of mortality in fetuses with hydrops is high 13.
- Other indicators of poor prognosis include the type of lesion, with microcystic CCAM associated with much poorer outcomes 13.
- The overall size of the lesion has also been reported as being an important predictor of survival; however, this index may be compromised by the fact that CCAM may undergo involution and even disappear in utero 27.
- Polyhydramnios is also associated with a poorer outcome 13.
CCAM survival rate
Babies who have the CCAM removed either before or after birth usually do well afterwards. Babies’ lungs continue to grow until the child is 9 years old, leaving plenty of time for the development of normal healthy lungs.
In children who have had an uncomplicated resection of CCAM, no residual effects (based on currently available data) are anticipated.
Less data are available regarding the longer-term outcomes in children who underwent fetal surgery.
- Hamanaka R, Yagasaki H, Kohno M, Masuda R, Iwazaki M. Congenital cystic adenomatoid malformation in adults: Report of a case presenting with a recurrent pneumothorax and a literature review of 60 cases. Respir Med Case Rep. 2018;26:328–332. Published 2018 Feb 25. doi:10.1016/j.rmcr.2018.02.002 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6424061[↩]
- Stanton M, Njere I, Ade-Ajayi N, Patel S, Davenport M. Systematic review and meta-analysis of the postnatal management of congenital cystic lung lesions. J Pediatr Surg. 2009 May. 44(5):1027-33.[↩][↩]
- Wong A, Vieten D, Singh S, Harvey JG, Holland AJ. Long-term outcome of asymptomatic patients with congenital cystic adenomatoid malformation. Pediatr Surg Int. 2009 Jun. 25(6):479-85.[↩]
- Laberge JM, Flageole H, Pugash D, et al. Outcome of the prenatally diagnosed congenital cystic adenomatoid lung malformation: a Canadian experience. Fetal Diagn Ther. 2001 May-Jun. 16(3):178-86.[↩][↩][↩][↩]
- Usui N, Kamata S, Sawai T, et al. Outcome predictors for infants with cystic lung disease. J Pediatr Surg. 2004 Apr. 39(4):603-6.[↩]
- Sauvat F, Michel JL, Benachi A, Emond S, Revillon Y. Management of asymptomatic neonatal cystic adenomatoid malformations. J Pediatr Surg. 2003 Apr. 38(4):548-52.[↩][↩]
- Kotecha S, Barbato A, Bush A, Claus F, Davenport M, Delacourt C, et al. Antenatal and postnatal management of congenital cystic adenomatoid malformation. Paediatr Respir Rev. 2012 Sep. 13(3):162-70; quiz 170-1.[↩][↩]
- Biyyam DR, Chapman T, Ferguson MR et-al. Congenital lung abnormalities: embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics. 2010;30 (6): 1721-38. doi:10.1148/rg.306105508[↩][↩]
- Kao SW, Zuppan CW, Young LW. AIRP best cases in radiologic-pathologic correlation: type 2 congenital cystic adenomatoid malformation (type 2 congenital pulmonary airway malformation). Radiographics. 2011;31 (3): 743-8. doi:10.1148/rg.313105162[↩]
- Gross GW. Pediatric chest imaging. Curr Opin Radiol. 1992;4 (5): 36-43.[↩][↩]
- Wilson RD, Hedrick HL, Liechty KW, et al. Cystic adenomatoid malformation of the lung: review of genetics, prenatal diagnosis, and in utero treatment. Am J Med Genet A. 2006 Jan 15. 140(2):151-5.[↩]
- Wang X, Wolgemuth DJ, Baxi LV. Overexpression of HOXB5, cyclin D1 and PCNA in congenital cystic adenomatoid malformation. Fetal Diagn Ther. 2011. 29(4):315-20.[↩]
- Adzick NS, Harrison MR, Crombleholme TM, et al. Fetal lung lesions: management and outcome. Am J Obstet Gynecol. 1998 Oct. 179(4):884-9.[↩][↩][↩][↩][↩][↩][↩][↩]
- Adzick NS. Management of fetal lung lesions. Clin Perinatol. 2003 Sep. 30(3):481-92.[↩]
- Luján M, Bosque M, Mirapeix RM, Marco MT, Asensio O, Domingo C. Late-onset congenital cystic adenomatoid malformation of the lung. Embryology, clinical symptomatology, diagnostic procedures, therapeutic approach and clinical follow-up. Respiration. 2002. 69(2):148-54.[↩]
- Duncombe GJ, Dickinson JE, Kikiros CS. Prenatal diagnosis and management of congenital cystic adenomatoid malformation of the lung. Am J Obstet Gynecol. 2002 Oct. 187(4):950-4.[↩]
- Parikh D, Samuel M. Congenital cystic lung lesions: is surgical resection essential?. Pediatr Pulmonol. 2005 Dec. 40(6):533-7.[↩]
- Granata C, Gambini C, Balducci T, Toma P, Michelazzi A, Conte M. Bronchioloalveolar carcinoma arising in congenital cystic adenomatoid malformation in a child: a case report and review on malignancies originating in congenital cystic adenomatoid malformation. Pediatr Pulmonol. 1998 Jan. 25(1):62-6.[↩][↩][↩]
- Adzick NS, Harrison MR. Management of the fetus with a cystic adenomatoid malformation. World J Surg. 1993 May-Jun. 17(3):342-9.[↩][↩]
- Schwartz DS, Reyes-Mugica M, Keller MS. Imaging of surgical diseases of the newborn chest. Intrapleural mass lesions. Radiol Clin North Am. 1999 Nov. 37(6):1067-78, v.[↩]
- Kim WS, Lee KS, Kim IO, et al. Congenital cystic adenomatoid malformation of the lung: CT-pathologic correlation. AJR Am J Roentgenol. 1997 Jan. 168(1):47-53.[↩]
- Cystic Adenomatoid Malformation Treatment & Management. https://emedicine.medscape.com/article/1001488-treatment[↩]
- Effect of single and multiple courses of maternal betamethasone on prenatal congenital lung lesion growth and fetal survival. J Pediatr Surg. 2016 Jan;51(1):28-32. doi: 10.1016/j.jpedsurg.2015.10.018. Epub 2015 Oct 23. https://www.ncbi.nlm.nih.gov/pubmed/26526208[↩]
- Congenital Cystic Adenomatoid Malformation (CCAM). https://www.chop.edu/conditions-diseases/congenital-cystic-adenomatoid-malformation-ccam[↩]
- Makhija Z, Moir CR, Allen MS, Cassivi SD, Deschamps C, Nichols FC 3rd, et al. Surgical management of congenital cystic lung malformations in older patients. Ann Thorac Surg. 2011 May. 91(5):1568-73; discussion 1573.[↩]
- Frenckner B, Freyschuss U. Pulmonary function after lobectomy for congenital lobar emphysema and congenital cystic adenomatoid malformation. A follow-up study. Scand J Thorac Cardiovasc Surg. 1982. 16(3):293-8.[↩]
- Crombleholme TM, Coleman B, Hedrick H, et al. Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung. J Pediatr Surg. 2002 Mar. 37(3):331-8.[↩]

{kind=link}