
Dysmorphic features
Dysmorphic feature is a medical term referring to a difference of body structure that is suggestive of a congenital disorder, genetic syndrome, or birth defect. A dysmorphic feature can be a minor and isolated birth defect (e.g., clinodactyly, not accompanied by other features or problems. Alternatively it can be one of a combination of features signaling a serious multi-system syndrome (e.g., the epicanthal folds of Down’s syndrome). Dysmorphic features may include craniofacial dysmorphism, skeletal abnormalities, shortened proximal limbs, calcific stippling of epiphyses, and renal cysts in different disorders linked to peroxisomal dysfunction 1.
Dysmorphic features may result from a perturbation of human development 2. This perturbation can be a direct effect of a genetic mutation or can indirectly involve a genetic disturbance, such as in the case of gestational exposure to a teratogen. Figure 1 are examples of dysmorphic features that can be found on physical examination that may be helpful in the diagnosis of genetic syndromes.
A full dysmorphology examination is best conducted by a trained clinical geneticist; a primary care physician should conduct a thorough physical examination 3. An essential component of the physical examination for genetic syndromes is comparison with established normative values. In addition to weight, height, and head circumference, examples of other important measurements include interpupillary distance (the distance between the center of the pupils) if a midline defect is suspected (as in the case of holoprosencephaly, the most common malformation of the human forebrain), or the size of the limbs in the case of a skeletal dysplasia 4. These values must be interpreted in the context of the patient’s longitudinal growth, as well as the family background.
The absence or presence of certain features in family members can be a clue that a feature is either pathologic or merely a familial or ethnic variant. For example, inner epicanthal folds (small folds of skin over the medial eyes) can occur in persons with Down syndrome, and are also described in more than 50 other syndromes, including Noonan syndrome, Rubinstein-Taybi syndrome, and Smith-Lemli-Opitz syndrome. However, epicanthal folds are also a normal finding in many persons of Asian or Native American descent 5. Recently, an effort has been made to codify physical descriptors, which can help with communication and establishment of the differential diagnosis 3.
The presence of one obvious malformation should not limit the full evaluation, because additional, subtler findings will often be important in the differential diagnosis. For example, a newborn might be immediately noted to have a cleft palate, which has a broad differential diagnosis. A patient with a cleft palate; hypocalcemia; a ventricular septal defect; and subtle physical features, such as a broad nasal root, overfolded helices of the ears, and long, narrow fingers and toes, likely has 22q11.2 deletion syndrome (formerly called DiGeorge syndrome), which has a prevalence of approximately one in 4,000 live births 6.
Some genetic conditions are diagnosed on clinical grounds. Geneticists can assist in diagnosis, suggest additional testing and referrals if warranted, help direct medical care, and provide counseling for affected patients and their families. Physicians can locate a regional geneticist through the American College of Medical Genetics Web site at https://www.acmg.net/GIS.
For other patients, the diagnosis is made through genetic testing. There is a wide array of tests that may be used.8 The choice of test depends on the nature of the condition, the expense and availability of the test, and the specific clinical scenario (for example, the testing strategy may be different if a condition were suspected in an adult versus a fetus). Ultimately, whether or not a patient or family elects to undergo testing is a matter of personal choice, and patients should be counseled regarding what a test may or may not reveal.
Dysmorphic features key points
- A dysmorphology assessment requires a thorough and detailed physical examination.
- Ancillary investigations may be useful.
- Chromosome and genetic tests may be warranted in certain circumstances.
- Overcome any reluctance to discuss your assessment with the family, but make sure you use sensitive language.
Figure 1. Dysmorphic features

Footnote: Examples of dysmorphic features that can be found on physical examination that may be helpful in the diagnosis of genetic syndromes. (A) A black infant presents with pigmentary anomalies in the context of a genetic condition; notice the hypopigmented tips of the hair. (B) An iris coloboma can be associated with many genetic syndromes, or can occur as an isolated feature. (C) A single maxillary central incisor can occur in conditions that include midline deficits, and can be a clue to the presence of accompanying brain malformations. (D) A small ear with an overfolded helix can be a familial variant or can occur as part of a genetic syndrome. (E) Fifth-finger clinodactyly (incurving of the fifth finger) occurs in many genetic conditions, and is classically seen in persons with Down syndrome. (F) An unusual shape of the digits, such as tapered digits, can occur in many conditions; this patient has Coffin-Lowry syndrome. (G) Various types of syndactyly (fusion) of the fingers or toes may be seen; second- and third-toe syndactyly, which occurs in Smith-Lemli-Opitz syndrome, is shown here. (H) Examination of the skin is an important part of the physical examination; this child with neurofibromatosis 1 has multiple café au lait macules. (I) This lesion, visible with a Wood lamp, has irregular borders and is typical of sporadic café au lait macules.
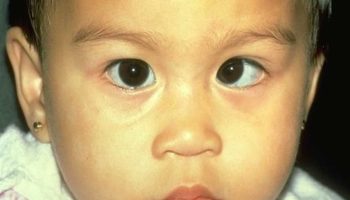
[Source 7 ]Figure 2. Facial dysmorphic features
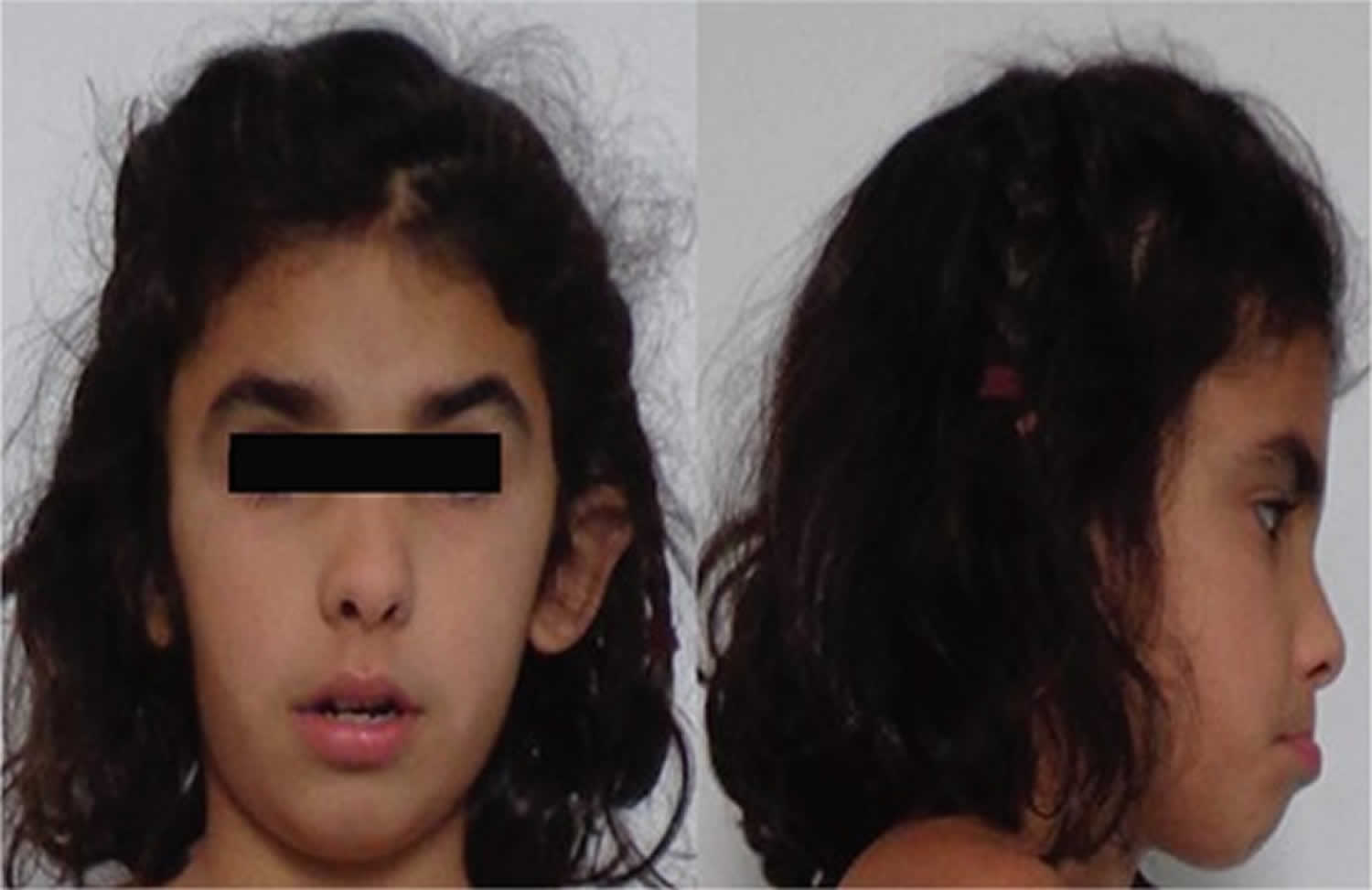
Footnote: 7-year-old girl referred to our diabetes unit for hyperglycemia associated with facial dysmorphic features (triangular face, short front, depressed nasal bridge, low hair implantation, bushy eyebrows, synophridia, and microretrognathia), intellectual disability, and cerebral cavernomas. Given the clinical picture and the multiplex ligation-dependent probe amplification findings, array based comparative genomic hybridization was performed showing a monoallelic deletion of 7.23 Mb in the short arm of chromosome 7 (7p13-p12.1). The deleted intervals contain 39 genes listed in the Online Mendelian Inheritance in Man list, including GCK associated with MODY 2, CCM2 associated with type 2 cerebral cavernous malformations, IGFBP-3 associated with decrease in postnatal growth, and OGD associated with alpha-ketoglutarate dehydrogenase deficiency, with cognitive impairment and movement abnormalities. This previously unreported deletion was considered to explain the clinical picture of the patient. Also, the findings suggest that 7p13-p12.1 contains genes involved in intellectual disability and craniofacial development.
[Source 8 ]Dysmorphic features clinical assessment
A dysmorphology assessment of a newborn focuses on aspects of history, physical examination and investigations that may lead to a syndrome diagnosis.
A dysmorphology assessment should be carried out on a child with any of the following:
- a congenital abnormality
- growth abnormalities
- dysmorphic features.
Checklists below will assist with:
- taking a history and examining an infant with a dysmorphology focus
- descriptions of investigations that the pediatrician should consider as part of a dysmorphology work-up.
For many doctors, discussing issues relating to syndrome diagnosis and dysmorphism with parents can be difficult, and some suggestions are outlined below.
History checklist
Use this checklist to take a detailed history of the mother and infant:
- obstetric history
- recurrent miscarriages
- uterine abnormalities
- pregnancy history
- note exposure to any teratogens
- history of maternal alcohol or recreational drug exposure
- amniotic fluid volume
- results of ultrasound and amniocentesis/chorionic villus sampling
- fetal growth and movements
- maternal illness, medical diagnoses and medications taken
- birth history
- Apgar scores, resuscitation required
- family history of abnormalities, stillbirths, childhood deaths
- consanguinity.
Examination checklist
- The following focuses on the examination for dysmorphic features in a baby.
- A thorough examination of all systems is vital when considering a syndrome diagnosis.
- Initial observation will enable assessment of general muscle tone, movements, postural abnormalities and abnormal body proportions.
Growth
Assess whether the baby’s growth parameters are in proportion as well as the percentiles:
- birthweight
- length
- head circumference.
Ectodermal features
Examine the skin and hair:
- skin
- texture
- color
- birthmarks
- redundancy
- defects
- hair
- scalp hair
- body hair
- color
- distribution
- position of anterior and posterior scalp hairline.
Skull
Examine the skull:
- shape
- symmetry
- sutures (over-riding/normal/widely open)
- fontanelle size and number.
Face overall impression
In examining the face, it can be useful to first gain an overall impression of the facial appearance. Sometime, an overall gestalt can be diagnostic (for example, Down syndrome).
If no diagnosis is made it is important to divide the face into sections to examine it thoroughly.
You may divide the face into the forehead, midface and oral region. It can sometimes help to cover parts of the face with your hand, in order to isolate the section of the face you are assessing.
In assessing the face, it is important to view the face from the front and from the lateral view. The depth or height of structures such as the nasal bridge, the position of the mandible relative to the maxilla and the development of the midface are best assessed by the lateral view.
Examination needs to be considered in the context of family features so it is useful to see parents and any siblings.
Face shape
Examine the overall face shape, symmetry and facial muscle movement:
Forehead region
When examining the forehead assess:
- forehead shape – (broad/bitemporal narrowing/tall)
- eyes
- palpebral fissure length (short/long)
- palpebral fissure slant (up/down)
- epicanthic folds – a fold of skin which arcs from below the eye into the upper lid
- eye spacing (use a rough guide of 1:1:1 for the ratio of left palpebral fissure length: inner canthal distance: right palpebral fissure length)
- palpebral fissure shape
- red reflex
- iris colour
- pupil shape
- retina
- globe position (assessed from lateral view: protuberant vs deep set globes).
Midfacial region
When examining the midfacial region assess:
- nose – divide the nose into three sections from the lateral view from superior to inferior into the nasal root, bridge and tip:
- root
- bridge (depressed/prominent/broad)
- tip
- columella (the vertical ridge separating the nostrils)
- nostrils – patency, position (anteverted nostrils often reflect a short nose)
- ears – ear rotation is normally 15 degrees posterior to the vertical plane of the head
- ear shape and structure
- ear position.
Oral region
When examining the oral region of the face note:
- mouth size and shape
- lip shape, thickness
- gum thickness
- philtrum definition and length
- jaw position (prognathia/micrognathia)
- palate shape
- oral cavity – natal teeth/frenulum/tongue size and morphology.
Hands and feet
Examine hands and feet carefully:
- overall shape and size of hand and foot
- digit number
- digit shape (for example, clinodactyly) and length
- webbing between digits
- palmar, plantar and digit creases
- nail morphology.
Joints and skeleton
Examine joints and skeleton for:
- contractures
- limb shortening
- joint range of movement
- soft tissue webbing across joints (pterygium)
- sternum length and shape (pectus carinatum / pectus excavatum)
- shape of thoracic cage
- spine length, straight/curved
- neck length, webbing.
Genitalia and anus
Check the following:
- phallus size, morphology
- development of scrotum and palpation of testes
- development of labia
- position of anus relative to genitalia
- patency of anus
- note any genital ambiguity.
Examine family members
Examination of other family members (siblings and parents) may be crucial to determine whether any dysmorphic features noted are familial or syndromic.
Investigations – when to do what?
In cases where the history and clinical features suggest a specific diagnosis, it may be possible to order a confirmatory test.
Tests and examinations to use in the syndrome work-up include:
- renal ultrasound
- echocardiogram
- cranial ultrasound
- MRI
- midline abnormalities tend to cluster together, so, for example, an echo may be indicated when there is a cleft palate and dysmorphic features
- eye examinations are useful for clues to make a syndrome diagnosis
- skeletal radiographs are indicated when there is disproportionate short stature or other abnormalities in the skeletal system
- x-rays may be useful to diagnose a skeletal dysplasia, a disorder caused by a primary abnormality of bone growth/development, or to assist in diagnosing a dysmorphic syndrome which can have skeletal abnormalities associated with it.
Genetic skeletal survey
A genetic skeletal survey includes:
- Anterior-posterior (AP) and lateral X rays of the skull
- Anterior-posterior (AP) and lateral pelvis and spine (cervical to sacrum)
- Anterior-posterior (AP) of one arm
- Anterior-posterior (AP) both hands
- Anterior-posterior (AP) of one leg and AP of both feet.
In a neonate, it may be sufficient to obtain a ‘baby-gram’ (x-ray of the baby) and a separate x-ray of the hands and feet.
Lab tests
- Routine hematology and biochemistry
- Blood chromosomes are indicated when:
- there are multiple congenital abnormalities +/- dysmorphic features
- there is one congenital abnormality in the presence of dysmorphic features and/or growth restriction.
Chromosome abnormalities are more likely when there are abnormalities of growth, most commonly growth restriction and microcephaly, in association with dysmorphic features and congenital abnormalities.
Chromosome analysis issues to note:
- A normal chromosome analysis does not exclude a single gene mutation or a micro deletion syndrome.
- A normal antenatal chromosome analysis does not completely exclude a chromosome abnormality as the resolution of chromosome banding may be greater on a postnatal sample than samples from chorionic villus sampling (CVS) or amniocentesis.
- If a chromosome abnormality is strongly suspected it is indicated to repeat chromosomes in the postnatal period.
- A chromosome test takes a minimum of five days and the time taken to obtain a result depends on the growth of cells in culture.
- If an infant has been transfused, there is a small risk that there may be circulating lymphocytes from the blood donor which may lead to an ambiguous result.
- Most laboratories recommend delaying a karyotype until one week following a transfusion.
Flourescence in situ hybridation (FISH)
- For trisomies 13/18/21, FISH is used to expedite diagnosis when trisomy of a specific chromosome is suspected.
- A result is usually available within 48 hours.
- FISH for submicroscopic deletion syndromes are tests using a probe that detects small chromosome deletions not visible on routine chromosome analysis.
- 22 q FISH should be considered in babies with heart defects, particularly those with cleft palate and dysmorphic features.
- The commonest cardiac defects seen are conotruncal (abnormalities of cardiac outflow tracts) heart defects and ventricular septal defect.
- 7 q FISH (Williams’ syndrome) should be considered in babies with supravalvular aortic stenosis and/or hypercalcaemia.
Fragile X testing
Fragile X testing is rarely indicated in the neonatal period in the absence of a family history.
Single gene tests
Single gene tests may be indicated, depending on the syndrome being considered. Such tests usually require liaison with the clinical geneticist.
Microarray
Chromosome Microarray (molecular karyotype) is a detailed genetic test that looks for extra or missing segments of DNA in the chromosomes. The result are available in 10-14 days.
Saliva can be used as an alternative to blood for microarray testing – simple, quick and painless.
Biochemical tests
Biochemical tests such as 7-dehdrocholesterol assays if considering Smith-Lemli-Opitz as a diagnosis.
Cluster of Findings
Many genetic conditions are suggested through a combination of clinical features, including the physical appearance of the patient, laboratory abnormalities, and aspects of family history 9. For example, an adolescent presenting for a sports physical examination who is noted to have arachnodactyly (long, thin fingers) and pectus excavatum, and whose father died of an aortic dissection would be suspected of having Marfan syndrome 10. Short stature and thumb anomalies in a child who also has aplastic anemia may suggest Fanconi anemia, and confirmation would be important to institute surveillance for long-term complications 11.
There are several possible explanations for the presence of a cluster of findings in a patient with a genetic syndrome. One common reason is pleiotropy, in which a mutation in a single gene results in multiple effects on separate organ systems. For example, in Holt-Oram syndrome, sometimes called heart-hand syndrome, mutations in the gene TBX5 cause congenital heart and limb malformations.18 Another possible explanation for the presence of a cluster of findings is that the patient has a contiguous gene syndrome, in which he or she has deletions (missing genetic material) or duplications (extra genetic material) involving a certain portion of a chromosome. Because all genes in the deleted or duplicated intervals are affected, the involvement of many genes can result in a complicated clinical picture. The 22q11.2 deletion syndrome is a well-known example of a contiguous gene syndrome 6.
Table 1 lists resources that can be helpful in establishing a differential diagnosis based on the presence of several distinct features.
Table 1. Web-Based Resources for Clinical Genetics
| Resource | Web address | Description |
|---|---|---|
American Academy of Family Physicians | Provides links to published resources pertaining to genetics, including key articles and resources for patients, in the “AFP By Topic” module on genetics: https://www.aafp.org/afp/genetics | |
American Academy of Pediatrics | Provides guidelines and algorithms on the management of relatively common genetic conditions in the “Committee on Genetics” section: http://www2.aap.org/visit/cmte18.htm | |
American College of Medical Genetics | Provides many resources, including algorithms for treating affected patients and managing abnormal newborn screening test results (http://www.acmg.net/AM/Template.cfm?Section=ACT_Sheets_and_Confirmatory_Algorithms&Template=/CM/HTMLDisplay.cfm&ContentID=5661), and a database of available regional genetic services (http://www.acmg.net/GIS/) | |
ClinicalTrials.gov | Registry of federally and privately supported clinical trials, including those dedicated to determining the genetic causes of disease, conducted in the United States and internationally | |
GeneTests | Expert-written reviews, which include contact information for the authors of each article, on many genetic conditions, as well as links to clinical and research laboratories that provide testing | |
Genetics Home Reference | Provides many resources, including short descriptions of conditions and explanations of genetic terms; may be a useful source for families with less familiarity with medical terminology | |
London Medical Databases | http://www.lmdatabases.com/about_lmd.html (subscription required) | Series of searchable databases of genetic conditions; can be especially useful for developing a differential diagnosis |
National Human Genome Research Institute’s Elements of Morphology: Human Malformation Terminology | A source for newly standardized terms used in human dysmorphology, with links to source articles | |
National Organization for Rare Disorders | Serves patients with rare diseases; includes links to support groups, ways to apply for help (with treatment costs), booklets on a few disorders, and links to physicians with expertise in specific disorders | |
Online Mendelian Inheritance in Man | Database of genes associated with human disease, as well as conditions with proven and hypothetical genetic underpinnings; searchable by multiple phenotypic and genetic terms | |
Possum Web | http://www.possum.net.au/ (subscription required) | Dysmorphology database consisting of multiple malformations, and metabolic, teratogenic, chromosomal, and skeletal syndromes, which can be useful for the establishment of a differential diagnosis |
Table 2. Overgrowth in the neonatal period and associated conditions
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Beckwith-Wiedemann syndrome | Macroglossia Abdominal wall defects Hemihyperplasia Neonatal hypoglycemia Visceromegaly Posterior helical ear pits Anterior linear ear lobe creases | Blood glucose level monitoring Abdominal ultrasound Alpha-fetoprotein level | Methylation analysis of 11p15 |
| Chromosomal abnormalities | Congenital heart defects Ophthalmologic abnormalities Genitourinary abnormalities | Echocardiogram Ophthalmologic evaluation Renal ultrasound | Chromosomal microarray |
| Infant of a diabetic mother | Holoprosencephaly Spina bifida Congenital heart defects Neonatal small left colon Vertebral defects Tibial hemimelia with preaxial polydactyly Caudal regression syndrome | Cranial ultrasound or head MRI Echocardiogram Renal ultrasound Sacral ultrasound AP and lateral radiographs of the entire spine | None |
Table 3. Fetal growth restriction and associated genetic conditions
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Chromosomal abnormalities | (See Table 2) | (See Table 2) | (See Table 2) |
| Trisomy 13 | Holoprosencephaly Microphthalmia/ colobomas Congenital heart defects Cutis aplasia | Head ultrasound Ophthalmologic evaluation Echocardiogram Renal ultrasound | Karyotype |
| Trisomy 18 | Prominent occiput Micrognathia Congenital heart defects Horseshoe kidney Overlapping fingers | Echocardiogram Renal ultrasound | Karyotype |
Table 4. Aplasia cutis congenita (congenital absence of the skin) and associated genetic conditions
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Adams-Oliver | Limb defects Cutis marmorata telangiectasia congenita CNS abnormalities Cardiovascular abnormalities | Brain imaging Limb radiographs Echocardiogram | Sequencing of ARHGAP31, DOCK6, RBPJ, EOGT |
| Scalp or midline back ACC (without multiple anomalies) | May have underlying bony or neural tube defects | Infectious work up Skull radiograph Head MRI Spinal ultrasound or MRI | None |
| Trisomy 13 | (See Table 3) | (See Table 3) | (See Table 3) |
Table 5. Holoprosencephaly (a structural brain abnormality resulting from the incomplete cleavage of the forebrain into the right and left hemispheres) and associated conditions
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Chromosomal abnormalities | (See Table 2) | (See Table 2) | (See Table 2) |
| Infant of diabetic mother | (See Table 2) | (See Table 2) | (See Table 2) |
| Single gene disorder | Microcephaly Hypotelorism Nasal hypoplasia Midline cleft lip with or without palate Single central incisor | Head MRI imaging Dental evaluation in those where teeth have erupted | Sequencing of SHH, ZIC2, SIX3, TGIF1, GLI2, PTCH |
| Trisomy 13 | (See Table 3) | (See Table 3) | (See Table 3) |
| Trisomy 18 | (See Table 3) | (See Table 3) | (See Table 3) |
Table 6. Asymmetric crying facies (presents with drooping of the corner of the mouth on the unaffected side when crying or grimacing) and associated conditions
| Syndromes/conditions to consider | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Isolated congenital absence or hypoplasia of depressor anguli oris muscle | Congenital heart defects | Echocardiogram | None |
| 22q11 deletion | Laterally built up nose Aplasia/hypoplasia of thymus Hypocalcemia Congenital heart defects Long fingers and toes Renal anomalies | Ionized calcium and intact parathyroid hormone levels Thyroid function tests Immunology evaluation Echocardiogram Hearing screen Renal ultrasound Ophthalmologic evaluation | Chromosomal microarray or FISH for 22q11 deletion |
Table 7. Conditions associated with preauricular ear tags
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Craniofacial Microsomia | External ear anomalies Hearing loss Cleft palate Maxillary and/or mandibular hypoplasia Renal anomalies | Audiology evaluation Renal ultrasound | Chromosomal microarray |
| Isolated | May have a positive family history | Audiology evaluation | None |
Table 8. Conditions associated with preauricular ear pits
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Branchio-otorenal syndrome | External ear anomalies Brachial cleft fistulae Renal anomalies | Audiology evaluation Renal ultrasound | Sequencing of EYA1, SIX5, SIX1 |
| Craniofacial Microsomia | (See Table 7) | (See Table 7) | (See Table 7) |
Table 9. Cleft palate with or without cleft lip associated genetic conditions
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Holoprosencephaly (if cleft is midline) | (See Table 5) | (See Table 5) | (See Table 5) |
| Isolated cleft lip/palate | None | Audiology evaluation Feeding assessment | None |
| Trisomy 13 | (See Table 3) | (See Table 3) | (See Table 3) |
| Van der Woude | Lower lip pits | Feeding assessment | Sequencing of IRF6 |
Table 10. Cleft palate WITHOUT cleft lip and associated conditions
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| 22q11 deletion | (See Table 6) | (See Table 6) | (See Table 6) |
| CHARGE | Coloboma Ear anomalies Cardiac defects Choanal atresia Genitourinary abnormalities Omphalocele | Audiology evaluation ENT evaluation Echocardiogram Ophthalmologic evaluation Renal ultrasound | Sequencing of CHD7 |
| Isolated cleft palate | None | None | None |
| Smith-Lemli-Optiz | Microcephaly Characteristic facial features Cataracts Hypospadias Postaxial polydactyly 2-3 toe syndactyly | 7-dehydrocholesterol and total cholesterol levels Echocardiogram Ophthalmologic evaluation | Sequencing DHCR7 |
| Stickler | Myopia Cataract Retinal detachment Hearing loss Spondyloephiphyseal dysplasia | Audiology evaluation Ophthalmologic evaluation | Sequencing of COL2A1, COL9A1, COL9A2, COL11A1, and COL11A2 |
| Treacher-Collins | Lower eyelid abnormalities Microtia and other external ear abnormalities Zygomatic bone hypoplasia | Airway and feeding evaluations Audiology evaluation | Sequencing of TCOF1, POLR1C, and POLR1D |
Table 11. Cardiac defects and associated genetic syndromes
| Cardiac Defect | Genetic syndrome | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|---|
| Atrial septal defect | Holt-Oram | Upper limb malformation Cardiac conduction disease | Upper limb radiographs Echocardiogram | Sequencing of TBX5 |
| Atrioventricular canal | Down (trisomy 21) | Up-slanting palpebral fissures 5th finger clinodactyly Single transverse palmar creases Increased gap between 1st and 2nd toes | Audiology evaluation Complete blood count Ophthalmologic evaluation Thyroid function tests | Karyotype |
| Coarctation of the aorta | Kabuki | Long palpebral fissures Large ears Spinal column abnormalities Postnatal growth deficiency | Ophthalmologic evaluation Renal ultrasound Spine radiographs | Sequencing of KMT2D and KDM6A |
| Turner | Webbed posterior neck Broad chest with wide-spaced nipples Lymphedema of hands and feet | Audiology evaluation Renal ultrasound Thyroid function tests | Karyotype | |
| Hypoplastic left heart syndrome | Turner | (see above) | (see above) | (see above) |
| Interrupted aortic arch | 22q11 deletion | (See Table 6) | (See Table 6) | (See Table 6) |
| Peripheral pulmonary artery stenosis | Alagille | Bile duct paucity Butterfly vertebrae Posterior embryotoxon | Abdominal ultrasound Chest radiographs Liver function tests Ophthalmologic evaluation | Sequencing of JAG1 |
| Pulmonary valve stenosis | Noonan | Tall forehead Hypertelorism Down-slanting palpebral fissures Low-set, posteriorly rotated ears Excess nuchal skin Low posterior hairline | Ophthalmologic evaluation Renal ultrasound | Molecular testing: at least 12 genes, including PTPN11 (multigene panel testing available) |
| Supravavular aortic stenosis | Williams | Hypercalcemia Hypotonia Peripheral pulmonic stenosis Failure to thrive Renal artery stenosis | Bladder and kidney ultrasound Calcium level Ophthalmologic evaluation | Microarray or deletion testing for 7q11.23 |
| Tetralogy of Fallot | 22q11 deletion | (See Table 6) | (See Table 6) | (See Table 6) |
| Ventricular septal defect | Down (trisomy 21) | (see above) | (see above) | (see above) |
| 22q11 deletion | (See Table 6) | (See Table 6) | (See Table 6) |
Table 12. Tracheoesophageal fistula and associated conditions
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Infant of a diabetic mother | (See Table 2) | (See Table 2) | (See Table 2) |
| Down (trisomy 21) | (See Table 11) | (See Table 11) | (See Table 11) |
| CHARGE | (See Table 10) | (See Table 10) | (See Table 10) |
| Chromosomal abnormalities | (See Table 2) | (See Table 2) | (See Table 2) |
| Fanconi anemia | Microcephaly Short stature Pigmentary abnormalities Thumb abnormalities (absent/hypoplastic, bifid, duplicated, etc) Other upper extremity abnormalities Lower extremity abnormalities Genitourinary abnormalities Pancytopenia | Hematologic studies including complete blood count and bone marrow aspirate Renal ultrasound | Chromosomal breakage studies Molecular testing; at least 16 genes, including FANCA and BRCA2 |
| Trisomy 18 | (See Table 3) | (See Table 3) | (See Table 3) |
| VACTERL association | Vertebral defects Anal atresia/ imperforate anus Cardiac defects Tracheoesophageal fistula Limb anomalies Renal anomalies | Abdominal x-rays AP and lateral radiographs of the entire spine Echocardiogram Radiographs of affected limbs Renal ultrasound | None |
Table 13. Ventral wall defects (omphalocele and gastroschisis are the most common congenital ventral wall defects) and associated conditions
| Type of defect | Genetic syndrome | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|---|
| Omphalocele | Beckwith-Wiedemann | (See Table 2) | (See Table 2) | (See Table 2) |
| CHARGE | (See Table 10) | (See Table 10) | (See Table 10) | |
| Trisomy 13 | (See Table 3) | (See Table 3) | (See Table 3) | |
| Trisomy 18 | (See Table 3) | (See Table 3) | (See Table 3) | |
| VACTERL | (See Table 12) | (See Table 12) | (See Table 12) | |
| Gastroschisis | None | Cardiac anomalies Intestinal atresia Genitourinary anomalies Musculoskeletal anomalies | Abdominal radiographs Echocardiogram Renal ultrasound Skeletal radiographs | None |
Table 14. Preaxial polydactyly and associated conditions
| Differential diagnosis | Associated features | Potential evaluations | Potential genetic studies |
|---|---|---|---|
| Fanconi anemia | Microcephaly Short stature Pigmentary abnormalities Thumb abnormalities (absent/hypoplastic, bifid, duplicated, etc) Other upper extremity abnormalities Lower extremity abnormalities Genitourinary abnormalities Pancytopenia | Hematologic studies including complete blood count and bone marrow aspirate Renal ultrasound | Chromosomal breakage studies Molecular testing; at least 16 genes, including FANCA and BRCA2 |
| VACTERL association | (See Table 12) | (See Table 12) | (See Table 12) |
Neurocognitive impairment
In many genetic conditions, neurocognitive impairment may be the first and most obvious sign of disease 12. Genetic conditions can produce neurocognitive impairment in a number of different ways, including via structural brain malformations, aberrant signaling involving genes that play important neurologic roles, and inborn errors of metabolism, the latter of which can cause neurologic disease because of an inadequate amount of a needed substrate or accumulation of a toxic metabolite. Phenylketonuria, galactosemia, methylmalonic acidemia, and maple syrup urine disease are examples of conditions caused by inborn errors of metabolism that are included in newborn screening 13. In the assessment of developmental delay and/or neurocognitive impairment, neuroimaging is warranted when a structural, degenerative, or metabolic process is suspected 14.
Neurocognitive impairment related to genetic conditions may be recognized in childhood; however, some conditions, such as Huntington disease or some types of Charcot-Marie-Tooth disease, as well as familial forms of complex disorders, such as Parkinson disease and Alzheimer disease, may manifest much later in life 15. Key historical items should include the degree and type of dysfunction; the timing, both in terms of when the dysfunction was first noticed as well as how the impairment has changed with time (including whether or not there has been regression, or loss of developmental milestones); and if there are any recognized triggers.
Inborn errors of metabolism, such as phenylketonuria, deserve special attention, largely because medical interventions in these disorders can be especially important for preventing neurocognitive impairment 14. For example, it may be necessary to remove a type of food source that cannot be properly metabolized (phenylalanine in the case of phenylketonuria). These interventions may influence the degree of cognitive impairment, and can be a matter of life or death.
- Poll-The B. T., Aubourg P., Wanders R. J. A. Peroxisomal disorders. In: Saudubray J. M., van den Berghe G., Walter J. H., editors. Inborn Metabolic Diseases. 5th. Berlin, Germany: Springer; 2011.[↩]
- Hunter AG. Medical genetics: 2. The diagnostic approach to the child with dysmorphic signs. CMAJ. 2002;167(4):367–372.[↩]
- Allanson JE, Biesecker LG, Carey JC, Hennekam RC. Elements of morphology: introduction. Am J Med Genet A. 2009;149A(1):2–5.[↩][↩]
- Solomon BD, Mercier S, Vélez JI, et al. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C(1):133–141.[↩]
- Cassidy SB, Allanson JE. Management of Genetic Syndromes. 3rd ed. Hoboken, NJ: Wiley-Blackwell; 2010.[↩]
- Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370(9596):1443–1452.[↩][↩]
- When to Suspect a Genetic Syndrome. Am Fam Physician. 2012 Nov 1;86(9):826-833. https://www.aafp.org/afp/2012/1101/p826.html[↩]
- López GP, Quispe BV, Cabrejas Núñez MJ, Castaño L, Barrio R. Dysmorphic Features, Frontal Cerebral Cavernoma, and Hyperglycemia in a Girl with a De Novo Deletion of 7.23 Mb in Region 7p13-p12.1. J Clin Res Pediatr Endocrinol. 2017;9(4):355–359. doi:10.4274/jcrpe.4324 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5785643[↩]
- Jones KL, Smith DW. Smith’s Recognizable Patterns of Human Malformation. 6th ed. Philadelphia, Pa.: Elsevier-Saunders; 2006.[↩]
- Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965–1976.[↩]
- Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 200729–39.[↩]
- Moeschler JB. Genetic evaluation of intellectual disabilities. Semin Pediatr Neurol. 2008;15(1):2–9.[↩]
- Zschocke J, Hoffmann GF, Milupa AG. Vademecum Metabolicum: Manual of Metabolic Paediatrics. 2nd ed. Friedrichsdorf, Germany: Schattauer; 2004.[↩]
- Kahler SG, Fahey MC. Metabolic disorders and mental retardation. Am J Med Genet C Semin Med Genet. 2003;117C(1):31–41.[↩][↩]
- Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26(suppl 1):S1–S58.[↩]

{kind=link}