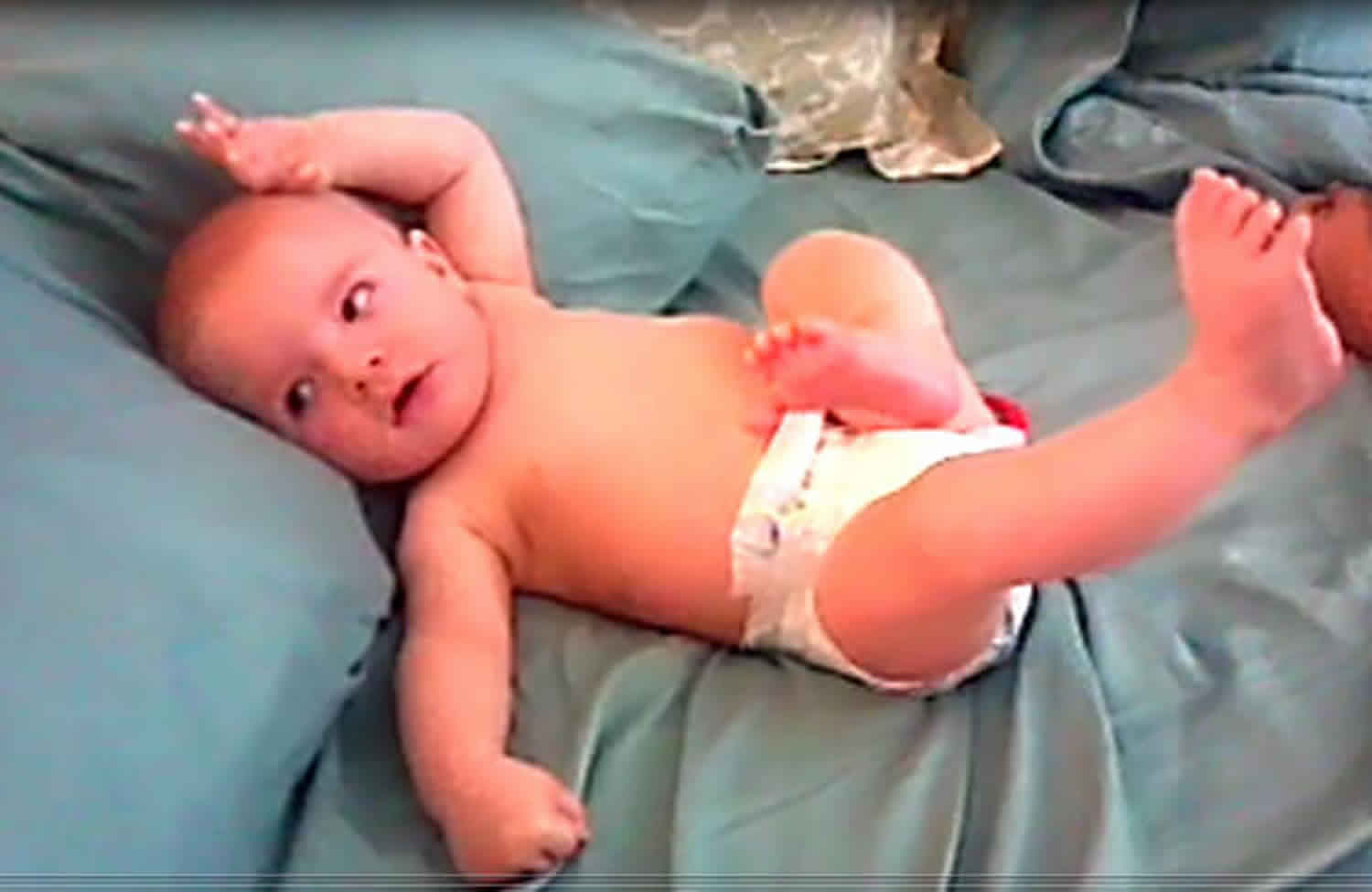
Epileptic spasms
An epileptic spasm is a sudden flexion, extension or mixed flexion-extension of proximal and truncal muscles, lasting 1-2 seconds i.e. longer than a myoclonic jerk (which lasts milliseconds) but not as long as a tonic seizure (which lasts > 2 seconds) 1. Epileptic spasms typically occur in a series, usually on wakening. Subtle forms may occur with only chin movement, grimacing, or head nodding. Epileptic spasms may be bilaterally symmetric, asymmetric, or unilateral, depending on whether they are generalised onset or focal onset. Epileptic spasms usually occur in a series (several in a cluster), if singular, consider other seizure types.
Epileptic spasm is an age related disorder. Epileptic spasm is the most common epileptic syndrome in infancy 2. The incidence of infantile spasms has been estimated to range 2–5/10,000 newborne. Studies from high income countries showed wide range incidence rate (0.05–0.6/1000 liveborne) higher reported incidence were reported from the higher geographic latitudes; Sweden, Finland and Denmark and lowest incidence in United States of America, Britain and Korea. It is not clear if this difference were due to environmental factors or specific genetic predisposition. The age specific prevalence is around 1–2/10,000 children by age of 10 years. Like incidence the highest prevalence values also corresponds to high geographical latitude 3. There are scant report from sub-Saharan Africa on the incidence or prevalence of epileptic spasms. In the review of the epidemiology of epilepsy in resource limited countries Senanayaka and Roman did not include epileptic spasm among the seizure types reviewed 4, while in a survey of childhood epilepsy in rural Uganda, though none of the 440 children reviewed then had epileptic spasms, 7 of them had previous history suggestive of epileptic spasms 5.
The age of onset is reported to vary from the first week of life up to 3 years. The peak is between 4 and 7 months, age of onset is within one year in 94% of cases. Almost all cases occur within 3 years of age. However, rare cases of epileptic spasm with onset at up to 14 years of age are reported, hence the new preferred term of epileptic spasm which was first suggested in the 1991 workshop of the International League Against Epilepsy commission on paediatric epilepsy 6.
Whilst studies suggest a slight male predominance in the prevalence of epileptic spasms in the average ratio of 6:4, this finding is not consistent. The reason for this differences is not clear, Brna et al. 7 suggested that the observed male predominance in some studies simply reflects the predominance in males in the referring population. An alternate explanation is the increased complication rate in predisposing conditions such as neonatal hypoglycaemia and hypoxic-ischemic encephalopathy reported to occur in male infants 8.
Epileptic spasms are most commonly accompanied by a high voltage triphasic sharp or slow wave followed by low amplitude fast activity and voltage attenuation. The EEG pattern may be generalized (for generalized epileptic spasms) or seen focally (for focal epileptic spasms). This EEG pattern may be seen in sleep with or without clinical seizures seen 9.
Epileptic spasms is classically accompanied by an interictal EEG pattern called hypsarrhythmia, characterized by very high voltage, irregular, asynchronous slow waves with overriding multifocal independent epileptiform discharges 10. However, multiple variants of hypsarrhythmia are commonly seen 11 and not all cases of epileptic spasms exhibit hypsarrhythmia 12. As a result, the identification of this EEG pattern suffers from poor inter-rater reliability 13, yet it is a standard clinical criteria used for both diagnosis and assessment of treatment response. While the presence or absence of hypsarrhythmia prior to treatment is unrelated to the likelihood of favorable short-term response, children who exhibit hypsarrhythmia are more likely to receive first-line treatment, which is strongly associated with favorable response to treatment 14. Therefore, there is a need for objective interictal EEG markers of epileptic spasms that are independent from visually-dominating patterns such as hypsarrhythmia 15.
Recent clinical studies demonstrate that functional network characteristics associated with epileptic spasms are good candidates for this marker 15. Multiple neurophysiologic approaches, including EEG source analysis 16, fMRI 17, PET 18, and SPECT 19, all find that hypsarrhythmia is likely generated subcortically, with predominant cortical expression in the parietal and occipital cortices. This suggests that a common network may underlie EEG patterns associated with epileptic spasms, despite a seemingly chaotic appearance on standard clinical review. This is supported by the fact that hypsarrhythmia is associated with increased EEG coherence in long-distance connections 20, and nonlinear time series analysis demonstrates that hypsarrhythmia contains only weakly nonlinear structures and is not strictly chaotic 21. However, studies of functional connectivity in epileptic spasms have focused only on patients exhibiting hypsarrhythmia, and the changes in functional networks following treatment have never been systematically evaluated 20.
IMPORTANT: Epileptic spasms may occur as a generalized onset, focal onset or unknown onset seizure. They may also occur as a later feature in a seizure, rather than at onset. Shorter time to diagnosis and control of epileptic spasms is associated with better developmental outcomes in young children. Focal onset epileptic spasms have particular importance, as identification of a structural cause can allow consideration of curative epilepsy surgery. Clues to a focal origin of epileptic spasms include asymmetry of the motor features (especially in initial spasms in the cluster), lateral head/eye version, focal emphasis to the interictal or ictal EEG and presence of focal structural brain abnormality.
Epileptic spasms related syndromes:
- West syndrome
- Early myoclonic encephalopathy
- Ohtahara syndrome
- Lennox-Gastaut syndrome
Epileptic spasms related causes:
- Structural brain abnormalities
- Gene abnormalities eg CDKL5, ARX
Epileptic spasms often leads to devastating neurocognitive consequences, and over 50% of patients with epileptic spasms will develop other forms of highly refractory epilepsy 22. The impact of these outcomes, both on the patients’ families and the healthcare system, is tremendous 23. Although a majority of children suffer poor outcomes – especially those with severe underlying causes, early age of onset, delayed treatment, or developmental delay prior to the onset of epileptic spasms – superior outcomes accompany prompt diagnosis and successful treatment 24.
Epileptic spasms causes
A study of 269 infants with epileptic spasms in a national childhood encephalopathy study, found that 34% had antecedent factors which may have caused the spasms, the commonest of these were perinatal hypoxia in 38 cases and tuberous sclerosis complex in 16 cases 25. This case control analysis showed no significant association between epileptic spasms and pertussis immunisation in the 28 days before onset. There was some clustering of cases immunised with either diphtheria, tetanus and pertussis (DTP) or DT vaccines in the 7 days before onset. This study was important to emphasize and support that vaccinations did not cause epileptic spasms but could trigger their onset in infants in whom the disorder was predestined to develop.
A further study of 207 infants with epileptic spasms found that, 127 (61%) had a proven aetiology, 68 (33%) had no identified aetiology, and 12 (6%) were not fully investigated 26. Causes were prenatal in 63, perinatal in 38, postnatal in 8, and 18 had other causes. The most common causes were: hypoxic-ischemic encephalopathy [n = 21] (10%), chromosomal [n = 16] (8%), malformations [n = 16] (8%), stroke [n = 16] (8%), tuberous sclerosis complex [n = 15] (7%), and periventricular leukomalacia or haemorrhage [n = 11] (5%). The remaining 32 causes were all individually uncommon.
The National Infantile Spasms Consortium in North America prospectively evaluated the cause of new-onset epileptic spasms and evaluated the yield of genetic and metabolic investigations in those without obvious cause after initial clinical evaluation and magnetic resonance imaging (MRI) 27. Twenty-one United States paediatric epilepsy centres prospectively enrolled infants with newly diagnosed West syndrome in a central database. A total of 251 infants were enrolled (53% male). A cause was identified in 161 (64.4%) of 250 cases (genetic, 14.4%; genetic-structural, 10.0%; structural-congenital, 10.8%; structural-acquired, 22.4%; metabolic, 4.8%; and infectious, 2.0%). An obvious cause was found after initial clinical assessment (history and physical examination) and/or MRI in 138 of 161, whereas further genetic and metabolic studies were revealing in another 23 cases. Of 112 subjects without an obvious cause after initial evaluation and MRI, 81 (72.3%) had undergone genetic testing, which showed a causal abnormality in 23.5% and a variant of unknown significance in 14.8%. Although metabolic studies were done in the majority, these revealed an aetiology in only five cases (4.5%). The group concluded that the clinical evaluation and MRI provided a specific diagnosis in 55% of children presenting with West syndrome. They recommended a cost-effective workup for those without obvious cause, after initial clinical evaluation and MRI,that should include an array comparative genomic hybridization (aCGH) followed by an epilepsy gene panel if the microarray is not definitive, as well as serum lactate, serum amino acids, and urine organic acids.
Genetics causes are increasingly recognized as a cause of epileptic spasms. Genetic causes can either be disorders of genomic imbalance (e.g. Down’s syndrome, Palister–Killian syndrome, Williams syndrome or Miller–Dieker syndrome) or single gene disorders such as mutations in CDKL5, STXBP1, or ARX. Recent discoveries of responsible gene mutations, such as in GRIN2B that codes for the NR2B sub-unit of the N-methyl-d-aspartate (NMDA) receptor and results in a gain of function, raise the possibility of novel treatments that may be directed at the molecular pathology e.g. NMDA receptor antagonists 28. A recent study of 73 infants with epileptic spasms and no clear cause underwent array-comparative genomic hybridization and molecular analysis of 5 genes (CDKL5, STXBP1, KCNQ2, GRIN2A and MAGI2) 29. A disease-causing mutation or CNV (Copy Number Variation) was identified in 15% of the patients. Which included 6 point mutations found in CDKL5 (n = 3) and STXBP1 (n = 3), 3 microdeletions (10 Mb in 2q24.3, 3.2 Mb in 5q14.3 including the region upstream to MEF2C, and 256 kb in 9q34 disrupting EHMT1), and 2 microduplications (671 kb in 2q24.3 encompassing SCN2A, and 11.93 Mb in Xq28). In addition, 3 Copy Number Variations as potential risk factors, including one 16p12.1 deletion, one intronic deletion of the NEDD4 gene, and one intronic deletion of CALN1 gene.
Metabolic aetiologies are rare but also recognized. Pyridoxine dependency, biotinidase deficiency, PEHO syndrome, mitochondrial disorders, molybdenum co-factor deficiency and non-ketotic hyperglycinaemia have all been described 30.
Epileptic spasms pathogenesis
The underlying pathogenesis of epileptic spasms is not fully understood. Epileptic spasms is proposed to be a derangement of a network, or a system epilepsy. The mechanism for the associated encephalopathy is still not fully elucidated. It is hypothesised that the encephalopathy is a reflection of the background slowing and disruption in the normal brain rhythms due to a disturbance in brain networks. The infant is especially vulnerable to the development of epileptic spasms based on their stage of brain maturation and the time window that this places them in. Hence a wide range of aetiologies have the capacity of leading to the same outcome, namely epileptic spasms and often West syndrome, they have the equivalent mechanism of flipping a switch (which may have been predestined in a vulnerable child or directly operational in instigating the ripple effect of damage) 31. A common mechanism involved in the diverse cases of epileptic spasms is proposed to be due to brainstem pathology 32. An infant with hydrancephaly was able to generate epileptic spasms which was clinically identical to that seen in infants with intact nervous systems and supported that the brainstem is able to generate spasms 33. Further supporting data was evident from other studies assessing MRI and evoked potential results, and when reviewing the progress of neonates who suffered hypoxic-ischemic injuries to their subcortical and brainstem regions and subsequently developed epileptic spasms 34. Further concepts arose that spasms could be triggered by an interaction between the cortical grey and subcortical structures. Once activated the subcortical, brainstem or both could become generators of epileptic spasms 35. These findings support the idea that the pathogenesis is more complex and more likely related to widely disrupted networks at a particular stage of development and that this process is implicit in the associated encephalopathy. The encephalopathy precedes the development of the spasms 36. The EEG background pattern for children with epileptic spasms and the other epileptic encephalopathies is typically extremely disrupted, independent of the ictal events, electrodecrements and periods of discontinuity which occur 37. Extending on these findings, the disruption in the resting state networks of the brain by chaotic brain activity could be responsible for the global cognitive dysfunction seen in children with epileptic encephalopathies, especially those with epileptic spasms 38.
Epileptic spasms symptoms
Epileptic spasms are brief and abrupt contractions followed by less intense and sustained tonic phase lasting up to 1–2 seconds which involves the muscles of the neck, trunk, upper and lower limbs. They are more prolonged than a myoclonic jerk but less sustained than a tonic seizure. The spasms may be flexor, extensor or mixed. The flexure spasms is the most common, there is however wide individual variability in both the intensity and type of jerks 3. Epileptic spasms could be symmetrical or asymmetrical, focal, multifocal or generalized. Children with underlying cortical damage may have pre-existing focal neurological signs e.g. hemiparesis that inevitably mean the spasms will not be symmetrical. Infact on account of the uncertainty in the true characteristics of epileptic spasms, in the new International League Against Epilepsy classification of epileptic seizures epileptic spasms is not classified either as focal or generalized 39. The clinical significance of subtle spasm with features such as yawning, gasping, isolated eye movement and transient focal motor activities which has been reported is unknown but they occur in the context of classical EEG pattern of infantile spasms–hypsarrythmia 40.
Clinical phenomena that may be associated with the motor spasm before, during or after the attack include cyanosis, pallor, eye deviation and or change in respiratory pattern. Cry or scream may precede or follow the ictal phase. Often infants will be disturbed or upset by epileptic spasms.
Epileptic spasms usually occur in clusters; this was observed by West in his original description. Approximately 80% of epileptic spasms occur in clusters and 88% of patients report clustering phenomenon. Studies have shown that there is little diurnal variation in frequency of spasm/cluster over a 24 hour period. However, epileptic spasms do not occur in sleep but occur most frequently on awakening or just before sleep.
Epileptic spasms diagnosis
The classic hypsarrhythmia seen in patients with infantile spasms is an EEG pattern of a poorly organized, high amplitude (500–1000 μV), slow background, with accompanying multifocal epileptiform discharges, seen interictally, with generalized electrodecrement seen ictally during the spasms 41. It is however not present in all cases of infantile spasms and variation or modification of hypsarrhythmia is reported.
There are children with infantile spasms whose inter-ictal EEG does not show hypsarrythmia or any of its variants. Caraballo et al. 42 followed up 16 such cases and observed focal spikes in seven cases, bilateral spikes and spike and waves in five patients, multifocal spikes in two and normal inter-ictal EEG in two patients.
Benign non-epileptic infantile spasms has been reported by some workers and these children have an excellent prognosis with a normal EEG. According to current knowledge a normal EEG excludes the diagnosis of infantile spasms 43.
Epileptic spasms treatment
Epileptic spasms treatment involves treating the underlying cause. There are significant challenges associated with standardized clinical decision making for the treatment of epileptic spasms. Epileptic spasms is associated with a wide range of causes, including focal and diffuse pathologies 44 and it often co-occurs with a pre-existing epilepsy.
With regards to interventions, the first report of corticoadrenal hormones used therapeutically in epilepsy was published by McQuarrie et al. 45. McQuarrie 45 observed seizures induced in epileptic patients by increasing water intake and giving ADH. Deoxycortisone was proposed to cause opposite effects and therefore could have antiepileptic properties. They administered the intervention to one patient, with complete resolution of seizures. Further studies specific to the role of corticosteroids in the treatment of epileptic spasms, added to the wealth of data relating to this condition and the combined findings led to recommendations from the American Academy of Neurology, as well as the Cochrane database, for hormonal therapies to be the optimal intervention 46.
Data from The National Infantile Spasms Consortium of North America supported the need to follow accepted standardized protocols namely adrenocorticotrophic hormone (ACTH), oral corticosteroids or vigabatrin (VBG) 47. The paper stated that more favourable responses occurred in the ACTH treated group but this was a prospective case series and not a scientific clinical trial and therefore the results need to be viewed with some caution because of the possibility of bias.
The role of vigabatrin was reported in a study in which 192 out of 250 infants with classic epileptic spasms were retrospectively reviewed 48. Median follow-up time was 7.6 months (range 0.5–28.6 months). Initial suppression of epileptic spasms occurred in 131 (68%) infants. In the group with tuberous sclerosis complex this was 27/28 (96%). In infants under 3 months of age at the time of the spasms onset resolution occurred in 18/20 (90%). Response time was 4 days and mean dose was 99 mg/kg/day. Comparison of vigabatrin versus hydrocortisone in a prospective randomised multicentre study compared responses in 22 patients with tuberous sclerosis complex 49. The study supported the superior efficacy and tolerability of vigabatrin in infants with tuberous sclerosis complex and epileptic spasms when compared with hydrocortisone. However, it should be noted that hydrocortisone is rarely used elsewhere in the treatment of infantile spasms. Another study of 42 patients found no significant difference between vigabatrin and ACTH. In the vigabatrin group 11/23 became spasm free with more efficacy seen for the patients with tuberous sclerosis complex and or malformations, with 13% reporting side effects 50. 14/19 administered ACTH responded with earlier resolution on EEG and side effects reported by 37%.
Concern relating to vigabatrin and visual field defects has affected confidence in prescribing the product 51. A study of 91 children aged between 5.6 and 17.9 years, found visual field constriction in 17/91 (which was not statistically significant). But significant correlation was found with development of constriction in children who were treated for longer periods. The constricted group were treated for 46.1 months compared to the normal visual field group who were treated for 33.5 months. Also the cumulative dose was a statisically significant positive risk indicator.
The multicentre, randomised controlled trial, the United Kingdom Infantile Spasms Study compared prednisolone or tetracosactide versus vigabatrin in the treatment of infantile spasms 52. In this study children with tuberous sclerosis complex were excluded. The primary clinical outcome was the proportion with no spasms at days 13 and 14. This was found in 73% of the hormonal group and 54% of the vigabatrin group which was statistically significant. Adverse events were reported in 55% of the hormonal group and 54% of the vigabatrin group. Follow up data at 14 months based on the absence of spasms at final clinical assessment found that this was the case for 41/55 of the hormonal group (75%) compared to 39/51 of the vigabatrin group (76%) 53. The mean VABS score for the hormone group was 78.6 and the vigabatrin group was 77.5; also not significant. However, there was a significant aetiology–treatment interaction with respect to developmental outcome with hormonal therapies being associated with a significantly better developmental outcome than vigabatrin in those children with no proven aetiology for their spasms.
In resource limited settings access to ACTH or vigabatrin may not be viable. Indeed even in many parts of the US the cost of ACTH often precludes its use as therapy. Data is evolving supportive of high dose prednisolone (8 mg/kg/day, or 40–60 mg per day, for 2 weeks of therapy followed by a 2 week taper) with equivalent responses to those seen with ACTH 54.
The ketogenic diet has been proposed as an alternate intervention for infants with epileptic spasms but studies have provided inconsistent results 55. Most studies introduce the intervention after failure of standard therapy. The ideal study is yet to be performed to clarify if the ketogenic diet is a viable option in the management of epileptic spasms.
Precision medicine is a concept which, whilst relatively novel, may have a role for isolated patients with infantile spasms. An example could be in the setting of patients with infantile spasms related to KCNT1 mutations 56. Effective response to quinidine is reported in one case study 57Brain Dev. 2016;)). Some caution is needed however as there is variable response for patients with KCNT1 mutations and early onset epileptic encephalopathies 58.
Focusing on the developmental outcome, no difference in developmental outcome was found between the two treatment groups (hormonal treatment versus vigabatrin) for all infants 59. In the subgroup of infants with no identified aetiology, hormonal treatments had a significantly higher composite VABS scores than those allocated vigabatrin when measured both at 14 months and at 4 years.
The study was extended into The International Collaborative Infantile Spasms Study, which measured cessation of spasms (days 14–42), cessation of spasms and resolution of hypsarrhythmia (electro-clinical outcome), development at 18 months and 3.5 years. The primary clinical outcome of cessation of seizures between days 14 and 42 was seen in 108 (56.6%) out of 191 allocated hormonal treatment compared to 133 (71.9%) out of 185 allocated combination therapy (vigabatrin and hormonal therapy). Difference in response was 15.3%. The median response time in responders for those on combination therapy was 2 days compared to a median response time of 4 days for those responding to hormonal therapy alone. The implication being that optimal management would be through combination of hormonal as well as vigabatrin treatment.
Another key factor associated with developmental outcome is the “lead time to treatment” i.e. the time from seizure onset to initiation of treatment 60. In this study that looked at participants in United Kingdom Infantile Spasms Study (UKISS) 53 lead-time to treatment was categorised as 7 days or less, 8–14 days, 15 days–1 month, 1–2 months and greater than two months. The study showed that the earlier the intervention the better, each increase in category of lead time duration was associated with a 3.9 decrease in VABS score.
Additional influences on outcome relate to the impact of the underlying aetiology. In general symptomatic cases do worse than infants with cryptogenic causes 61. Further, certain genetic mutations can have specific outcomes, such as ARX mutations which impact on autistic outcome 62. The prompt response to treatment and a short duration of hypsarrythmia is also an indicator 63. This has also been reported in patients with infantile spasms and tuberous sclerosis complex 64 as well as those with infantile spasms and Trisomy 21 65.
Epilepsy surgery
There is a subset of children with epileptic spasms who may be suitable for epilepsy surgery. Focal lesions in infantile spasms are supported by findings in EEG recordings in 110/149 in one study 66 and in 28/67 in another 67. Also on neuroimaging (CT/MRI) in 17/37 infants 68, and following positron emission tomography (PET) in 5/13 infants 69 and 30/97 infants 70. Epilepsy surgery was undertaken in 23 children between 1–10 months of age (mean 9.5 months), based on supporting data from PET and EEG which was lateralised/localised in all 69. Fifteen children underwent cortical resection and 8 hemispherectomy. At follow-up between 4–67 months, 15 were seizure free, 4 had between 75–90% seizure freedom and 4 continued to have seizures.
With regards to developmental outcome, a study of 24 children with infantile spasms (15 girls) who underwent epilepsy surgery found a significant increase in developmental level at 2 years postsurgery compared with presurgical levels 71. Younger age at operation correlates with a better outcome 72. The longer the period of seizure duration before surgical intervention the lower Vineland DQ 73. This is further supported by the study by Loddenkemper et al. which found that better developmental outcome was found in the early surgical intervention group 74.
- Epileptic spasms. https://www.epilepsydiagnosis.org/seizure/epileptic-spasms-overview.html[↩]
- Epileptic spasms — 175 years on: Trying to teach an old dog new tricks. Wilmshurst, Jo M. et al. Seizure – European Journal of Epilepsy, Volume 44, 81 – 86 https://doi.org/10.1016/j.seizure.2016.11.021[↩]
- Pavone, P., Striano, P., Falsaperla, R., Pavone, L., and Ruggieri, M. Infantile spasms syndrome, West syndrome and related phenotypes: what we know in 2013. Brain Dev. 2014; 36: 739–751[↩][↩]
- Senanayake, N. and Román, G.C. Epidemiology of epilepsy in developing countries. Bull World Health Organ. 1993; 71: 247–258[↩]
- Duggan, M.B. Epilepsy in rural Ugandan children: seizure pattern, age of onset and associated findings. Afr Health Sci. 2010; 10: 218–225[↩]
- International CoPEot & Epilepsy. LA. Workshop on infantile spasms. Epilepsia. 1992; 33: 195[↩]
- Brna, P.M., Gordon, K.E., Dooley, J.M., and Wood, E.P. The epidemiology of infantile spasms. Can J Neurol Sci. 2001; 28: 309–312[↩]
- Tundidor, D., García-Patterson, A., María, M.A., Ubeda, J., Ginovart, G., Adelantado, J.M. et al. Perinatal maternal and neonatal outcomes in women with gestational diabetes mellitus according to fetal sex. Gend Med. 2012; 9: 411–417[↩]
- Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017 Apr;58(4):522-530. doi: 10.1111/epi.13670. Epub 2017 Mar 8. https://doi.org/10.1111/epi.13670[↩]
- Gibbs F Atlas of Electroencephalography. Cambridge, MA: Addison-Wesley; 1952.[↩]
- Kramer U, Sue WC, Mikati MA. Hypsarrhythmia: frequency of variant patterns and correlation with etiology and outcome. Neurology. 1997;48:197–203.[↩]
- Caraballo RH, Fortini S, Reyes G, Carpio Ruiz A, Sanchez Fuentes SV, Ramos B. Epileptic spasms in clusters and associated syndromes other than West syndrome: A study of 48 patients. Epilepsy Res. 2016;123:29–35.[↩]
- Hussain SA, Kwong G, Millichap JJ, Mytinger JR, Ryan N, Matsumoto JH, et al. Hypsarrhythmia assessment exhibits poor interrater reliability: a threat to clinical trial validity. Epilepsia. 2015;56:77–81.[↩]
- Demarest ST, Shellhaas RA, Gaillard WD, Keator C, Nickels KC, Hussain SA, et al. The impact of hypsarrhythmia on infantile spasms treatment response: Observational cohort study from the National Infantile Spasms Consortium. Epilepsia. 2017;58:2098–103.[↩]
- Shrey DW, Kim McManus O, Rajaraman R, Ombao H, Hussain SA, Lopour BA. Strength and stability of EEG functional connectivity predict treatment response in infants with epileptic spasms. Clin Neurophysiol. 2018;129(10):2137–2148. doi:10.1016/j.clinph.2018.07.017 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6193760[↩][↩]
- Japaridze N, Muthuraman M, Moeller F, Boor R, Anwar AR, Deuschl G, et al. Neuronal networks in west syndrome as revealed by source analysis and renormalized partial directed coherence. Brain Topogr. 2013;26:157–70.[↩]
- Siniatchkin M, van Baalen A, Jacobs J, Moeller F, Moehring J, Boor R, et al. Different neuronal networks are associated with spikes and slow activity in hypsarrhythmia. Epilepsia. 2007;48:2312–21.[↩]
- Chugani HT, Shewmon DA, Sankar R, Chen BC, Phelps ME. Infantile spasms: II. Lenticular nuclei and brain stem activation on positron emission tomography. Ann Neurol. 1992;31:212–9.[↩]
- Chiron C, Dulac O, Bulteau C, Nuttin C, Depas G, Raynaud C, et al. Study of regional cerebral blood flow in West syndrome. Epilepsia. 1993;34:707–15.[↩]
- Burroughs SA, Morse RP, Mott SH, Holmes GL. Brain connectivity in West syndrome. Seizure. 2014;23:576–9.[↩][↩]
- Van Putten MJAM Stam CJ. Is the EEG really “chaotic” in hypsarrhythmia. IEEE Eng Med Biol Mag. 2001;20:72–9.[↩]
- Pavone P, Striano P, Falsaperla R, Pavone L, Ruggieri M. Infantile spasms syndrome, West syndrome and related phenotypes: what we know in 2013. Brain Dev. 2014;36:739–51.[↩]
- Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, et al. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51:2175–89.[↩]
- Gaily E, Lommi M, Lapatto R, Lehesjoki AE. Incidence and outcome of epilepsy syndromes with onset in the first year of life: A retrospective population-based study. Epilepsia. 2016;57:1594–601.[↩]
- Bellman, M.H., Ross, E.M., and Miller, D.L. Infantile spasms and pertussis immunisation. Lancet. 1983; 1: 1031–1034[↩]
- Osborne, J.P., Lux, A.L., Edwards, S.W., Hancock, E., Johnson, A.L., Kennedy, C.R.C. et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia. 2010; 51: 2168–2174[↩]
- Wirrell, E.C., Shellhaas, R.A., Joshi, C., Keator, C., Kumar, S., Mitchell, W.G. et al. How should children with West syndrome be efficiently and accurately investigated? Results from the National Infantile Spasms Consortium. Epilepsia. 2015; 56: 617–625[↩]
- Lemke, J.R., Hendrickx, R., Geider, K., Laube, B., Schwake, M., Harvey, R.J. et al. GRIN2 B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol. 2014; 75: 147–154[↩]
- Boutry-Kryza, N., Labalme, A., Ville, D., de Bellescize, J., Touraine, R., Prieur, F. et al. Molecular characterization of a cohort of 73 patients with infantile spasms syndrome. Eur J Med Genet. 2015; 58: 51–58[↩]
- Alrifai, M.T., AlShaya, M.A., Abulaban, A., and Alfadhel, M. Hereditary neurometabolic causes of infantile spasms in 80 children presenting to a tertiary care center. Pediatr Neurol. 2014; 51: 390–397[↩]
- Pellock, J.M., Hrachovy, R., Shinnar, S., Baram, T.Z., Bettis, D., Dlugos, D.J. et al. Infantile spasms: a U. S. consensus report. Epilepsia. 2010; 51: 2175–2189[↩]
- Hrachovy, R.A. and Frost, J.D. Infantile spasms. Pediatr Clin North Am. 1989; 36: 311–329[↩]
- Neville, B.G. The origin of infantile spasms: evidence from a case of hydranencephaly. Dev Med Child Neurol. 1972; 14: 644–647[↩]
- Gano, D., Sargent, M.A., Miller, S.P., Connolly, M.B., Wong, P., Glass, H.C. et al. MRI findings in infants with infantile spasms after neonatal hypoxic-ischemic encephalopathy. Pediatr Neurol. 2013; 49: 401–405[↩]
- Guggenheim, M.A., Frost, J.D., and Hrachovy, R.A. Time interval from a brain insult to the onset of infantile spasms. Pediatr Neurol. 2008; 38: 34–37[↩]
- Philippi, H., Wohlrab, G., Bettendorf, U., Borusiak, P., Kluger, G., Strobl, K. et al. Electroencephalographic evolution of hypsarrhythmia: toward an early treatment option. Epilepsia. 2008; 49: 1859–1864[↩]
- Nordli, D.R. Pediatric epilepsy syndromes. in: J.S. Ebersole, A.M. Hussain, D.R. Nordli (Eds.) Current practice of clinical electroencephalography. 4th edition. Wolters Kluwer Health, Philadelphia; 2014[↩]
- Nordli, D.J. et al. Infantile spasms semiology and pathophysiology: have we made progress?. in: S.L. Moshe, J.H. Cross, J. De Bellescize (Eds.) Seizures and syndromes of onset in the two first years of life. 1st edition. John Libbey Eurotext, Montrouge, France; 2013[↩]
- Berg, A.T., Berkovic, S.F., Brodie, M.J., Buchhalter, J., Cross, J.H., van Emde Boas, W. et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010; 51: 676–685[↩]
- Lux, A.L. and Osborne, J.P. A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: consensus statement of the West Delphi group. Epilepsia. 2004; 45: 1416–1428[↩]
- Hrachovy, R.A. and Frost, J.D. Jr. Infantile epileptic encephalopathy with hypsarrhythmia (infantile spasms/West syndrome). J Clin Neurophysiol. 2003; 20: 408–425[↩]
- Caraballo, R.H., Ruggieri, V., Gonzalez, G., Cersósimo, R., Gamboni, B., A, Rey, A. et al. Infantile spasms without hypsarrhythmia: a study of 16 cases. Seizure. 2011; 20: 197–202[↩]
- Dravet, C., Giraud, N., Bureau, M., Roger, J., Gobbi, G., and Dalla Bernardina, B. Benign myoclonus of early infancy or benign non-epileptic infantile spasms. Neuropediatrics. 1986; 17: 33–38[↩]
- Osborne JP, Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia. 2010;51:2168–74.[↩]
- McQuarrie, I., Anderson, J.A., and Ziegler, M.R. Observation of the antagonistic effects of posterior pituitary and cortico-adrenal hormones in the epileptic subjects. J Clin Endocrin Metab. 1942; 2: 1[↩][↩]
- Go, C.Y., Mackay, M.T., Weiss, S.K., Stephens, D., Adams-Webber, T., Ashwal, S. et al. Evidence-based guideline update: medical treatment of infantile spasms. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2012; 78: 1974–1980[↩]
- Knupp, K.G., Coryell, J., Nickels, K.C., Ryan, N., Leister, E., Loddenkemper, T. et al. Response to treatment in a prospective national infantile spasms cohort. Ann Neurol. 2016; 79: 475–484[↩]
- Aicardi, J., Mumford, J.P., Dumas, C., and Wood, S.I. Vigabatrin: as initial therapy for infantile spasms: a European retrospective survey. Sabril IS Investigator and Peer Review Groups. Epilepsia. 1996; 37: 638–642[↩]
- Chiron, C., Dumas, C., Jambaqué, I., Mumford, J., and Dulac, O. Randomized trial comparing vigabatrin and hydrocortisone in infantile spasms due to tuberous sclerosis. Epilepsy Res. 1997; 26: 389–395[↩]
- Vigevano, F. and Cilio, M.R. Vigabatrin versus ACTH as first-line treatment for infantile spasms: a randomized, prospective study. Epilepsia. 1997; 38: 1270–1274[↩]
- Vanhatalo, S., Nousiainen, I., Eriksson, K., Rantala, H., Vainionpää, L., Mustonen, K. et al. Visual field constriction in 91 Finnish children treated with vigabatrin. Epilepsia. 2002; 43: 748–756[↩]
- Lux, A.L., Edwards, S.W., Hancock, E., Johnson, A.L., Kennedy, C.R., Newton, R.W. et al. The United Kingdom Infantile Spasms Study comparing vigabatrin with prednisolone or tetracosactide at 14 days: a multicentre, randomised controlled trial. Lancet. 2004; 364: 1773–1778[↩]
- Lux, A.L., Edwards, S.W., Hancock, E., Johnson, A.L., Kennedy, C.R., Newton, R.W. et al. The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: a multicentre randomised trial. Lancet Neurol. 2005; 4: 712–717[↩][↩]
- Hussain, S.A., Shinnar, S., Kwong, G., Lerner, J.T., Matsumoto, J.H., Wu, J.Y. et al. Treatment of infantile spasms with very high dose prednisolone before high dose adrenocorticotropic hormone. Epilepsia. 2014; 55: 103–107[↩]
- Hussain, S.A., Shin, J.H., Shih, E.J., Murata, K.K., Sewak, S., Kezele, M.E. et al. Limited efficacy of the ketogenic diet in the treatment of highly refractory epileptic spasms. Seizure. 2016; 35: 59–64[↩]
- Ohba, C., Kato, M., Takahashi, N., Osaka, H., Shiihara, T., Tohyama, J. et al. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia. 2015; 56: e121–e128[↩]
- Fukuoka, M., Kuki, I., Kawawaki, H., Okazaki, S., Kim, K., Hattori, Y. et al. Quinidine therapy for West syndrome with KCNTI mutation: a case report. (pii: S0387-7604(16)30106-1 (Epub ahead of print[↩]
- Chong, P.F., Nakamura, R., Saitsu, H., Matsumoto, N., and Kira, R. Ineffective quinidine therapy in early onset epileptic encephalopathy with KCNT1 mutation. Ann Neurol. 2016; 79: 502–503[↩]
- Darke, K., Edwards, S.W., Hancock, E., Johnson, A.L., Kennedy, C.R., Lux, A.L. et al. Developmental and epilepsy outcomes at age 4 years in the UKISS trial comparing hormonal treatments to vigabatrin for infantile spasms: a multi-centre randomised trial. Arch Dis Child. 2010; 95: 382–386[↩]
- O’Callaghan, F.J., Lux, A.L., Darke, K., Edwards, S.W., Hancock, E., Johnson, A.L. et al. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: evidence from the United Kingdom Infantile Spasms Study. Epilepsia. 2011; 52: 1359–1364[↩]
- Riikonen, R. Epidemiological data of West syndrome in Finland. Brain Dev. 2001; 23: 539–541[↩]
- Turner, G., Partington, M., Kerr, B., Mangelsdorf, M., and Gecz, J. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am J Med Genet. 2002; 112: 405–411[↩]
- Rener-Primec, Z., Lozar-Krivec, J., Krivec, U., and Neubauer, D. Head growth in infants with infantile spasms may be temporarily reduced. Pediatr Neurol. 2006; 35: 197–203[↩]
- Jambaqué, I., Chiron, C., Dumas, C., Mumford, J., and Dulac, O. Mental and behavioural outcome of infantile epilepsy treated by vigabatrin in tuberous sclerosis patients. Epilepsy Res. 2000; 38: 151–160[↩]
- Eisermann, M.M., DeLaRaillere, A., Dellatolas, G., Tozzi, E., Nabbout, R., Dulac, O., and et al. Infantile spasms in Down syndrome?effects of delayed anticonvulsive treatment. Epilepsy Res. 2003; 55: 21–27[↩]
- Riikonen, R. A long-term follow-up study of 214 children with the syndrome of infantile spasms. Neuropediatrics. 1982; 13: 14–23[↩]
- Kramer, U., Sue, W.C., and Mikati, M.A. Focal features in West syndrome indicating candidacy for surgery. Pediatr Neurol. 1997; 16: 213–217[↩]
- Shields, W.D., Shewmon, D.A., Chugani, H.T., and Peacock, W.J. Treatment of infantile spasms: medical or surgical?. Epilepsia. 1992; 33: S26–S31[↩]
- Chugani, H.T., Shewmon, D.A., Shields, W.D., Sankar, R., Comair, Y., Vinters, H.V. et al. Surgery for intractable infantile spasms: neuroimaging perspectives. Epilepsia. 1993; 34: 764–771[↩][↩]
- Chugani, H.T. and Conti, J.R. Etiologic classification of infantile spasms in 140 cases: role of positron emission tomography. J Child Neurol. 1996; 11: 44–48[↩]
- Asarnow, R.F., LoPresti, C., Guthrie, D., Elliott, T., Cynn, V., Shields, W.D. et al. Developmental outcomes in children receiving resection surgery for medically intractable infantile spasms. Dev Med Child Neurol. 1997; 39: 430–440[↩]
- Koo, B., Hwang, P.A., and Logan, W.J. Infantile spasms: outcome and prognostic factors of cryptogenic and symptomatic groups. Neurology. 1993; 43: 2322–2327[↩]
- Favatà, I., Leuzzi, V., and Curatolo, P. Mental outcome in West syndrome: prognostic value of some clinical factors. J Ment Defic Res. 1987; 31: 9–15[↩]
- Loddenkemper, T., Holland, K.D., Stanford, L.D., Kotagal, P., Bingaman, W., E, and Wyllie, E. Developmental outcome after epilepsy surgery in infancy. Pediatrics. 2007; 119: 930–935[↩]
{kind=link}