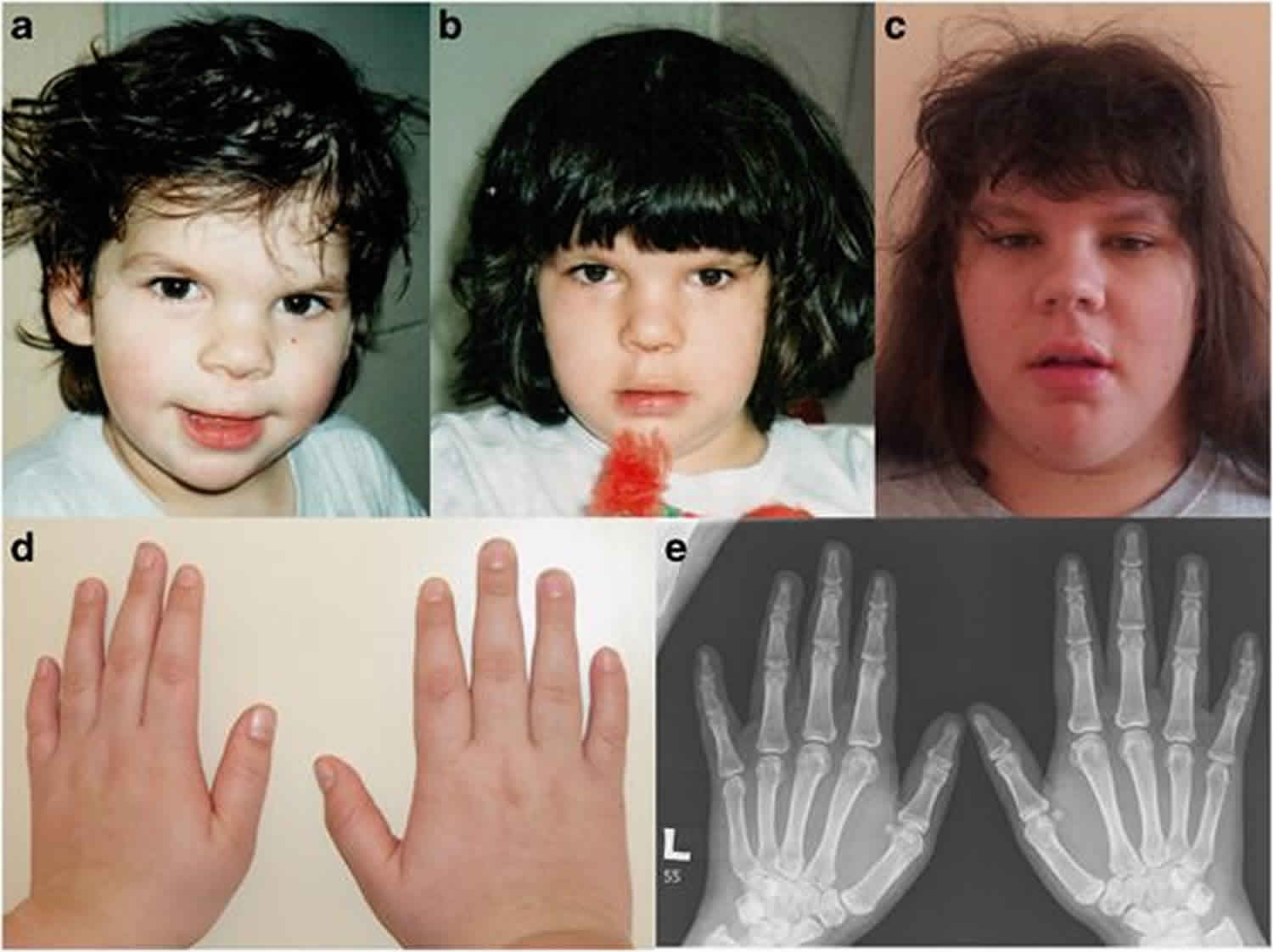
Coffin Siris syndrome
Coffin-Siris syndrome also called dwarfism-onychodysplasia, fifth digit syndrome or short stature-onychodysplasia, is a rare genetic condition that affects several body systems. Although there are many variable signs and symptoms, hallmarks of Coffin-Siris syndrome include variable degrees of learning disability, developmental delays, underdeveloped fifth (pinky) fingers or toes, and characteristic facial features.
Most affected individuals have mild to severe intellectual disability or delayed development of speech and motor skills such as sitting and walking. Another feature of Coffin-Siris syndrome is underdevelopment (hypoplasia) of the tips of the fingers or toes, or hypoplasia or absence of the nails. These abnormalities are most common on the fifth fingers or toes. In addition, most affected individuals have facial features described as coarse. These typically include a wide nose with a flat nasal bridge, a wide mouth with thick lips, and thick eyebrows and eyelashes. Affected individuals can have excess hair on other parts of the face and body (hirsutism), but scalp hair is often sparse. There is a range of facial features seen in people with Coffin-Siris syndrome, and not all affected individuals have the typical features. In addition, people with this condition may have an abnormally small head (microcephaly).
Additionally, some infants and children with Coffin-Siris syndrome have frequent respiratory infections, difficulty feeding, and an inability to gain weight at the expected rate (failure to thrive). Other signs and symptoms that may occur in people with this condition include short stature, low muscle tone (hypotonia), and abnormally loose (lax) joints. Abnormalities of the eyes, brain, heart, and kidneys may also be present.
Coffin-Siris syndrome is a rare condition that is diagnosed in females more frequently than in males. Approximately 140 cases have been reported in the medical literature 1.
Coffin-Siris syndrome can be caused by a change (mutation) in any of several genes including the ARID1A, ARID1B, SMARCA4, SMARCB1, DPF2 or SMARCE1 genes 2. Coffin-Siris syndrome follows an autosomal dominant pattern of inheritance, however it usually occurs for the first time in a family due to a new (de novo) mutations 1. Other researchers indicate that Coffin-Siris syndrome may be inherited as an autosomal recessive trait 3. In recessive disorders, the condition does not appear unless a person inherits the same defective gene for the same trait from each parent. If an individual receives one normal gene and one abnormal gene, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier is 50% with each pregnancy. The chance for a child to receive a normal gene from each parent for that particular trait is 25%. The risk is the same for males and females.
Diagnosis of Coffin-Siris syndrome is largely based upon the presence or absence of common signs and symptoms in the individual. While formal diagnostic criteria have not been established, most individuals with a clinical diagnosis of Coffin-Siris syndrome have certain features in common 4.
Occupational, physical, and/or speech therapy can help affected individuals reach their full potential 4.
Developmental pediatricians may be helpful in recommending and coordinating therapeutic and educational interventions. Additional specialty care may be needed depending on the symptoms in the individual, such as by gastrointestinal, eye, kidney, heart, and hearing specialists 4.
Figure 1. Coffin-Siris syndrome
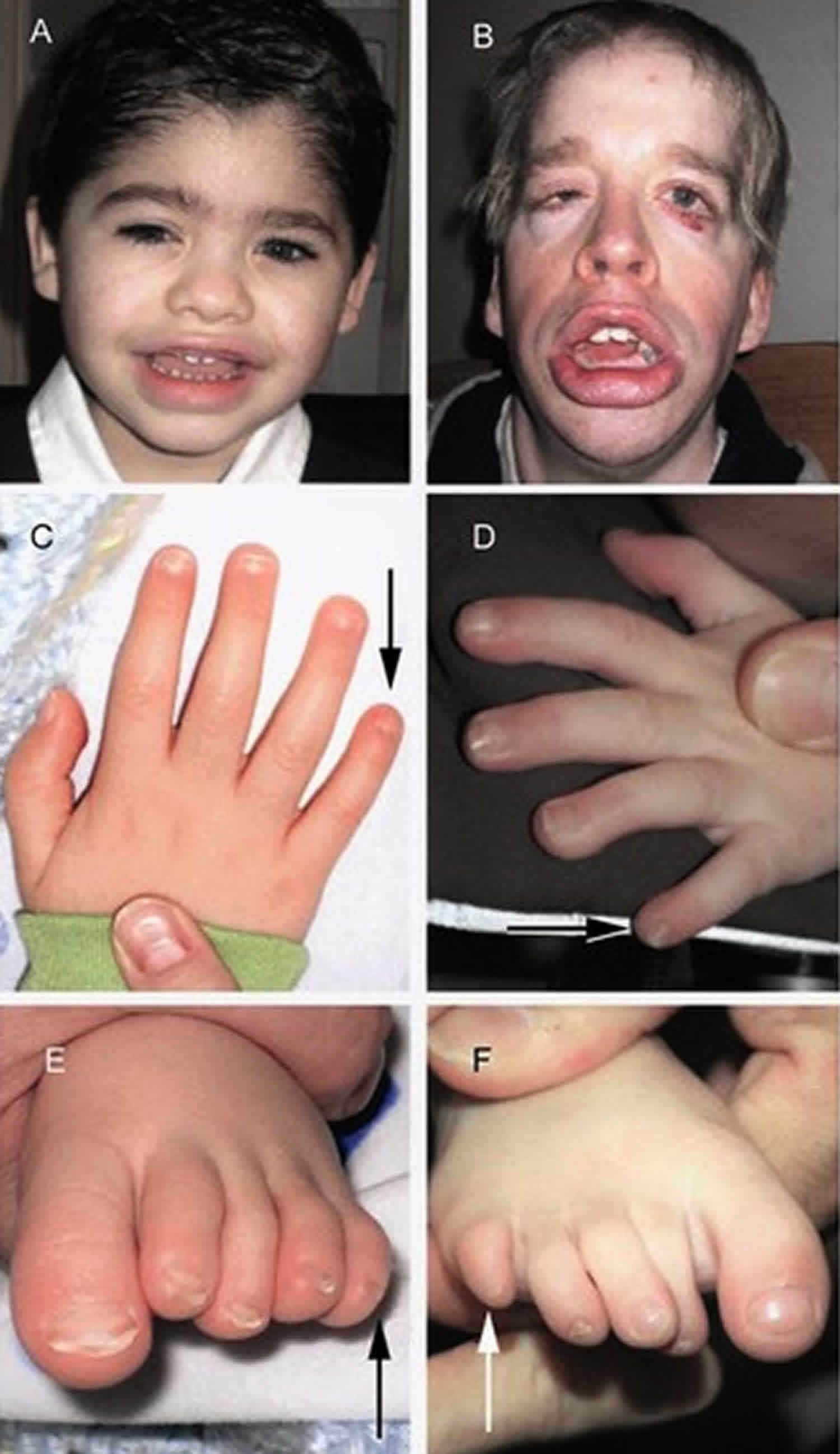
Footnote: Individuals with Coffin-Siris syndrome with some features highlighted. (A) a 5-year old affected boy; (B) a 29-year old affected man; (C) fifth finger nail hypoplasia; (D) hypoplasia of the tip of the fifth finger; (E) fifth toenail hypoplasia; (F) hypoplasia of the tip of the fifth toe.
[Source 4 ]Coffin Siris syndrome cause
Coffin-Siris syndrome is caused by mutations in the ARID1A, ARID1B, ARID2, SMARCA4, SMARCB1, SMARCE1 and SOX11 gene 3. Each of these genes provides instructions for making one piece (subunit) of several different SWI/SNF protein complexes. SWI/SNF complexes regulate gene activity (expression) by a process known as chromatin remodeling. Chromatin is the network of DNA and protein that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly regions of DNA are packaged. Chromatin remodeling is one way gene expression is regulated during development; when DNA is tightly packed, gene expression is often lower than when DNA is loosely packed.
In some cases, no genetic mutation can be identified and the cause of Coffin-Siris syndrome in the family remains unknown.
Through their ability to regulate gene activity, SWI/SNF complexes are involved in many processes, including repairing damaged DNA; copying (replicating) DNA; and controlling the growth, division, and maturation (differentiation) of cells.
Although it is unclear what effect mutations in these genes have on SWI/SNF complexes, researchers suggest that the mutations result in abnormal chromatin remodeling. Disturbance of this process alters the activity of many genes and disrupts several cellular processes, which could explain the diverse signs and symptoms of Coffin-Siris syndrome.
Not all affected individuals have mutations in the ARID1A, ARID1B, SMARCA4, SMARCB1, SMARCE1, or SOX11 genes 3. It is likely that there are additional genes that may cause Coffin-Siris syndrome. Some researchers also suggest that isolated (sporadic) and familial cases of Coffin-Siris syndrome may be due to unknown chromosomal abnormalities. Further research is required to fully determine the disorder’s underlying cause and potential mode of transmission.
Mutations in the genes that cause Coffin-Siris syndrome have also been linked to other disorders (allelic disorders). Mutations in the ARID1B gene have been reported in several individuals with isolated intellectual disability and absence of other physical features of Coffin-Siris syndrome. Mutations in the SMARCA4 and SMARCB1 genes have been reported to carry a potential increased risk toward the growth of rhabdoid tumors (tumors of muscle tissue) and atypical teratoid and rhabdoid tumors (tumors typically located in the brain and other areas of the central nervous system). Overall, the risk of tumor growth is very low; further research is required to better assess the cancer risk in individuals with these mutations.
Coffin-Siris syndrome inheritance pattern
Coffin-Siris syndrome appears to follow an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder. However, Coffin-Siris syndrome is not usually inherited from an affected parent, but occurs from new (de novo) mutations in the gene that likely occur during early embryonic development.
Other researchers indicate that Coffin-Siris syndrome may be inherited as an autosomal recessive trait 3. In recessive disorders, the condition does not appear unless a person inherits the same defective gene for the same trait from each parent. If an individual receives one normal gene and one abnormal gene, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier is 50% with each pregnancy. The chance for a child to receive a normal gene from each parent for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are related by blood (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder. Parents of some individuals with Coffin-Siris syndrome have been closely related by blood.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Coffin Siris syndrome symptoms
Coffin-Siris syndrome is characterized by distinctive abnormalities of the head and facial (craniofacial) region with affected individuals often described as having coarse facial features that become more prominent with age. Affected individuals may have an unusually small or large head (micro- or macrocephaly); a wide mouth with full, prominent lips; a broad nasal tip; a low nasal bridge; and an abnormally long vertical groove between the nose and the upper lip (philtrum). Additional features may include thick eyebrows, long eyelashes, and generalized excessive hair growth (hypertrichosis) with the exception of the scalp hair, which tends to be relatively sparse (scalp hypotrichosis). Reports suggest that sparse scalp hair improves with age.
Individuals with Coffin-Siris syndrome also have characteristic skeletal abnormalities. For example, certain fingers and toes (digits), particularly the fifth fingers (“pinkies”) and toes, may be unusually short due to absence or underdevelopment (hypoplasia) of the end bones (terminal phalanges) within these digits. The fingernails and toenails may also be underdeveloped or absent. Additional abnormalities may include dislocation of the inner forearm bone (radius) at the elbow, deformity of the hip (coxa valga), or unusually small or absent knee caps (patellae). However, there are individuals with Coffin-Siris syndrome who do not have the classic fifth digit findings.
Early in life, infants with Coffin-Siris syndrome typically experience feeding difficulties, vomiting, slow growth and weight gain (failure to thrive) which may have begun while the infant was still in the womb (intrauterine growth retardation), and frequent respiratory infections. In addition, affected infants and children may have hypotonia, abnormally loose joints, delayed bone age (2 to 3 years behind the chronological age), and mild to severe intellectual disability. Affected infants and children may also have mild to severe speech delays, where expressive language is affected more severely than receptive language, as well as moderate to severe delays in motor skills such as sitting and walking. Reports suggest that on average, affected children learn to sit up at 12 months (typically occurs at 6 to 8 months), walk at 30 months (typically occurs at 9 to 18 months), and speak at 24 months (typically begins around 12 months).
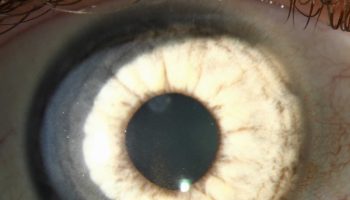
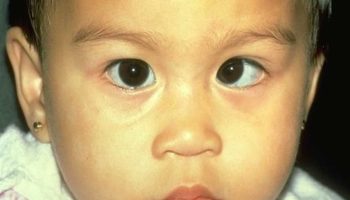
Affected individuals may also have eye (ophthamologic) abnormalities. This can include drooping of the upper eyelid (ptosis), clouding of the lens of the eye (cataracts), and misalignment of the eyes (strabismus, commonly known as “lazy eye”).
Coffin-Siris syndrome has been reported to manifest kidney (renal) or genitourinary abnormalities in some affected individuals. There have been reports of affected individuals with fused kidneys at the lower end (horseshoe kidney) and the urethra – the tube through which urine drains from the bladder to exit the body – opening on the underside of the penis instead of at the tip (hypospadias).
Individuals with Coffin-Siris syndrome may also have gastric abnormalities which may include one portion of the bowel sliding into the next like a telescope (intussusception) or an opening in the diaphragm allowing abdominal organs to push up into the chest cavity (diaphragmatic hernia).
Less commonly, affected individuals may have additional physical abnormalities, such as choanal atresia, a malformation in which a bony or thin layer of tissue blocks the passageway between the nose and throat, leading to breathing difficulties. Some individuals with Coffin-Siris syndrome may also have heart abnormalities at birth. In addition, a brain abnormality known as Dandy-Walker malformation has been reported in some cases. This condition is characterized by cystic malformation and expansion of one of the cavities in the brain (fourth ventricle). Dandy-Walker malformation is usually associated with an abnormal accumulation of cerebrospinal fluid (CSF) in the skull (hydrocephalus), resulting in increased fluid pressure, a rapid increase in head size, abnormal prominence of the back region of the head (occiput), and/or other associated findings. Some individuals with Coffin-Siris syndrome may also have partial or complete absence of the band of nerve fibers that joins the two hemispheres of the brain (agenesis of the corpus callosum) and fewer folds in their brain (gyral simplification). Some affected individuals may also experience hearing loss, seizures and tics. There have been reports of liver cancer (hepatoblastoma) in affected individuals, but the link between Coffin-Siris syndrome and tumor risk needs to be further investigated.
Genotype-phenotype correlations
Genotype-phenotype correlations have been seen in clinically diagnosed individuals with pathogenic variants in ARID1A, ARID1B, SMARCA4, SMARCB1, SMARCE1, and SOX11 5.
- ARID1A. Individuals with a pathogenic variant in ARID1A displayed a wide spectrum of severity; some exhibited only mild intellectual disability while others had severe intellectual disability. Some individuals also had serious medical complications (e.g., aspiration pneumonia, seizures) leading to death.
- ARID1B. Individuals with pathogenic ARID1B variants are typically at the milder end of the spectrum of Coffin-Siris syndrome and often have normal growth. Moderately severe feeding problems are noted in two thirds, seizures in one third, and hypoplasia of the corpus callosum in one third. Facial gestalt is consistent with Coffin-Siris syndrome, albeit at times milder, with hypertelorism and anteverted nares more commonly noted. Distal digital hypoplasia is usually limited to the fifth digit.
- SMARCA4. Individuals with a pathogenic variant in SMARCA4 appear to have growth impairment that is mild prenatally and mild to moderate postnatally; sucking/feeding difficulty is almost always observed. While individuals can sometimes have severe developmental delays, significant behavioral challenges tend to be more characteristic. Facial features have demonstrated less coarseness, while hypoplastic fifth fingers or toes and hypoplastic fifth fingernails or toenails are a near-constant finding (and hypoplasia of other fingernails or toenails an occasional finding). Prominence of interphalangeal joints and distal phalanges is also noted in some.
- SMARCB1. Individuals with a pathogenic variant in SMARCB1 typically have a more severely affected phenotype and all have growth impairment, usually mild prenatally and moderate to severe postnatally, with sucking/feeding difficulty. Structural CNS abnormalities with hypotonia and seizures are typical findings accompanied by severe developmental delay/intellectual disability; individuals are typically nonverbal. Typical skeletal findings include hypoplastic fifth fingers or toes, hypoplastic other fingernails or toenails, prominent distal phalanges, and scoliosis. Some individuals may walk independently. Gastrointestinal complications and hernia as well as cardiovascular and genitourinary complications are common.
- SMARCE1. Individuals with pathogenic SMARCE1 variants tend to have severe intellectual disability, typical facial gestalt, and hypoplastic or absent fifth finger- and toenails associated with hypoplasia of other nails. The hands are characterized by long and slender fingers. Individuals are typically small for gestational age and have postnatal short stature and severe microcephaly, complex congenital heart defects, feeding difficulties, and seizures.
- SOX11. Individuals display mild to severe intellectual and developmental delay, along with fifth-digit nail and distal phalangeal hypoplasia. Neurodevelopmental abnormalities tend to be more prevalent than organ-system or physical complications.
Coffin Siris syndrome diagnosis
Coffin-Siris syndrome should be suspected in individuals with the following findings 6:
- Fifth-digit nail / distal phalanx hypoplasia/aplasia. Typically, individuals with a clinical diagnosis of Coffin-Siris syndrome have either aplasia or hypoplasia of the distal phalanx or absence of the nail, typically involving the fifth finger, but other digits may also be affected (see Figure 1C, D, E, F). Toes can also be affected, where the finding tends to involve multiple digits.
- Developmental or cognitive delay of variable degree
- Facial features. Individuals with typical features demonstrate a wide mouth with thick, everted upper and lower lips, broad nasal bridge with broad nasal tip, thick eyebrows, and long eyelashes 7. Together, these features can give a suggestion of coarseness in individuals with Coffin-Siris syndrome (see Figure 1A, B).
- Hypotonia that is central in origin
- Hirsutism/hypertrichosis. Hair growth in atypical areas (e.g., the back) or excessive hair growth on the arms or face
- Sparse scalp hair, especially in infancy, particularly in the temporal regions
Coffin-Siris syndrome should be suspected in newborns with underdeveloped nails and short fifth fingers and distinctive facial features. The facial features may become more apparent as the child grows. A diagnosis is based upon a thorough clinical evaluation and characteristic physical findings. However, physical features of Coffin-Siris syndrome may be more variable as more individuals are diagnosed. Specialized testing may be conducted to detect certain findings that may be associated with the disorder. Diagnostic criteria were proposed in 2012 noting that most affected individuals have short fifth fingers with absent or underdeveloped nails, developmental and/or cognitive delays, and facial features such as a wide mouth and broad nose. Given the recent discovery of the genetic mutations causing Coffin-Siris syndrome, diagnostic criteria will likely evolve to include clinical evaluations and molecular testing.
It is possible that a diagnosis of Coffin-Siris syndrome may be suggested before birth (prenatally) based upon specialized tests such as ultrasound. During fetal ultrasonography, reflected sound waves are used to generate an image of the developing fetus. Ultrasound studies may reveal characteristic findings, such as cardiac or kidney malformations and intrauterine growth retardation, which may be associated with the disorder.
If a disease causing mutation has been identified in an affected family member, molecular testing can be done on the fetus. This involves the removal of fetal cells through chorionic villus sampling (performed at 10 to 12 weeks gestation with cells removed from the placenta) or amniocentesis (performed at 15 to 18 weeks gestation with cells removed from the amniotic fluid). DNA extracted from the fetal cells is then examined to see if the mutation is present in the fetus. Molecular genetic testing is available as a diagnostic service at specialized laboratories.
Clinical testing and workup
If indicated, further examinations and specialized imaging techniques are recommended to establish the extent of the disorder. For example, an MRI (magnetic resonance imaging) may be used to detect structural abnormalities, such as in the brain. During an MRI, radio waves and a magnetic field are used to generate an image. X-rays of the hands can be performed to confirm the underdevelopment or absence of the end bones in the fifth fingers. Echocardiograms, which are a type of ultrasound, can be used to generate images of the heart to detect any cardiac abnormalities that may be present. Other examinations can include developmental examinations, dietary evaluations, and eye and hearing examinations.
Once diagnosed, individuals with Coffin-Siris syndrome should have yearly follow-up exams. This includes evaluation by a pediatrician to assess developmental progress and to determine the need for any educational or therapeutic interventions and follow-ups with other specialists to track any feeding, gastrointestinal, vision, or hearing abnormalities.
Establishing the diagnosis
The diagnosis of Coffin-Siris syndrome is established in a proband with identification of a heterozygous pathogenic variant in one of the genes listed in Table 1.
Table 1. Molecular genetic testing used in Coffin-Siris syndrome
| Gene | Proportion of Coffin-Siris syndrome Attributed to Pathogenic Variants in This Gene 1 | Proportion of Pathogenic Variants Detected by Method 2 | |
|---|---|---|---|
| Sequence analysis 3 | Gene-targeted deletion/duplication analysis 4 | ||
| ARID1A | 8/172 (5%) | 8/8 | 0/8 |
| ARID1B | 64/172 (37%) | 59/64 | 3/60 5 |
| SMARCA2 6 | 5/172 (2%) | 4/5 | 1/5 |
| SMARCA4 | 12/172 (7%) | 12/12 | 0/12 |
| SMARCB1 | 12/171 (7%) | 12/12 | 0/13 |
| SMARCE1 | 3/171 (2%) | 3/3 | 0/3 |
| SOX11 | 2/92 (2%) 8 | 5/12 5 | Unknown 11; 7 individuals w/deletions & a CSS phenotype have been reported 5 |
| PHF6 8 | 2/37 (5%) 9 | 2/2 | 0/2 |
| Unknown 9 | 69/172 (40%) | NA | |
Footnotes:
1. Numbers represent a compilation of unique cases in reports of cohorts of clinically ascertained patients with Coffin-Siris syndrome 10 except as indicated with a footnote.
2. Number of individuals with an identified pathogenic variant / number of individuals tested
3. Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
4. Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications. Mosaic pathogenic variants have been noted for ARID1A 9.
5. Microdeletions of chromosome 6q25.3 that include ARID1B have been reported in: (a) children with Coffin-Siris syndrome ascertained prior to the understanding of the molecular basis of Coffin-Siris syndrome 11; (b) children ascertained with a microdeletion containing ARID1B and secondarily noted to have features similar to Coffin-Siris syndrome 12; and (c) individuals with mildly or variably syndromic intellectual disability 13 for whom available clinical information is insufficient to determine the similarity to Coffin-Siris syndrome. Of note, these individuals may have complex clinical findings due to the involvement of additional genes surrounding the ARID1B locus.
6. Reevaluation of an individual initially thought to have Coffin-Siris syndrome concluded that findings were more consistent with Nicolaides-Baraitser syndrome 14; however, since a number of individuals with SMARCA2 pathogenic variants were initially ascertained as Coffin-Siris syndrome, the authors have included these numbers for comparison purposes. See Differential Diagnosis.
7. No data on detection rate of gene-targeted deletion/duplication analysis are available.
8. Individuals initially ascertained as Coffin-Siris syndrome when younger have been found to have pathogenic variants in PHF6. Most of these have acquired facial features more consistent with Borjeson-Forssman-Lehmann syndrome as they age [Wieczorek et al 2013]. See Differential Diagnosis.
9. In these studies, approximately 40% (69/172) of individuals with Coffin-Siris syndrome did not have a pathogenic variant in one of the known genes 10.
Coffin Siris syndrome treatment
To establish the extent of disease and needs of an individual diagnosed with Coffin-Siris syndrome, the following evaluations are recommended:
- Consultation with a clinical geneticist and/or genetic counselor
- Neurologic and/or developmental examination to record developmental milestones and identify neurologic symptoms or deficits
- Evaluation for occupational, speech, or physical therapy as needed
- Gastrointestinal evaluation for feeding difficulties or poor growth
- Dietary evaluation by a nutritionist as needed
- Ophthalmologic examination, including a dilated fundus examination and visual acuity
- Audiology evaluation with auditory brain stem response testing and otoacoustic emission testing to assess for hearing loss
- Echocardiogram to evaluate for structural cardiac defects
- Renal ultrasonography to evaluate for structural kidney or genitourinary anomalies
The treatment of Coffin-Siris syndrome is directed toward the specific features of each individual. Such treatment may require the coordinated efforts of a team of medical professionals who may need to systematically and comprehensively plan an affected child’s treatment. These professionals may include pediatricians; physicians who specialize in disorders of the skeleton, joints, muscles, and related tissues (orthopedists); physicians who diagnose and treat heart abnormalities (cardiologists); physicians who specialize in digestive abnormalities; physical therapists; geneticists and/or other health care professionals.
In some affected individuals, treatment may include surgical repair of certain craniofacial, skeletal, cardiac, or other abnormalities potentially associated with the disorder. The surgical procedures performed will depend upon the severity of the anatomical abnormalities, their associated symptoms, and other factors.
In addition, in those with choanal atresia, surgery or other appropriate methods may be required to decrease the airway obstruction or correct the malformation. If affected individuals have Dandy-Walker malformation, treatment may include surgical implantation of a specialized device (shunt) to drain excess cerebrospinal fluid (CSF) away from the brain and into another part of the body where the CSF can be absorbed. During infancy, treatment may also require measures to help prevent or aggressively treat respiratory infections.
Early intervention may be important in ensuring that affected children reach their potential. Special services that may be benefit developmental outcomes include special education, physical, speech, or occupational therapy, or other social, and/or vocational services. Additional treatments to assist affected children can include eyeglasses, hearing aids, and nutritional supplements. If needed, the placement of a gastrostomy tube (a tube inserted through the abdomen to deliver nutrition directly to the stomach) can help with feeding difficulties.
Genetic counseling will also be of benefit for individuals with Coffin-Siris syndrome and their families. Other treatment is symptomatic and supportive.
Surveillance
Surveillance includes the following:
- Yearly evaluation by a developmental pediatrician to assess developmental progress and therapeutic and educational interventions
- Annual follow up with a gastroenterologist and feeding specialists as needed to monitor feeding and weight gain
- Regular follow up of ophthalmologic and/or audiologic abnormalities
Because of the rarity of tumors in Coffin-Siris syndrome, the utility of tumor surveillance has not been determined.
Coffin Siris syndrome prognosis
In the absence of long-term studies, information on life span in individuals with Coffin-Siris syndrome is not available. Children have been reported to die of aspiration pneumonia and/or seizures, although this is not common 15. Efforts are in progress by the Coffin-Siris Syndrome International Consortium 16 to better understand prognosis in individuals with Coffin-Siris syndrome.
- Coffin-Siris syndrome. https://ghr.nlm.nih.gov/condition/coffin-siris-syndrome[↩][↩]
- Knapp K, Poke G, Jenkins D, Truter N, Bicknell L. Expanding the phenotype spectrum associated with DPF2: A new case report. American Journal of Medical Genetics. June 17, 2019; 1-5. https://www.ncbi.nlm.nih.gov/pubmed/31207137[↩]
- Coffin Siris Syndrome. https://rarediseases.org/rare-diseases/coffin-siris-syndrome[↩][↩][↩][↩]
- Schrier Vergano S, Santen G, Wieczorek D, et al. Coffin-Siris Syndrome. 2013 Apr 4 [Updated 2018 Feb 8]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK131811[↩][↩][↩][↩]
- Hempel A, Pagnamenta AT, Blyth M, Mansour S, Mcconnell V, Kou I, Ikegawa S, Tsurusaki Y, Matsumoto N, Lo-Castro A, Plessis G, Albrecht B, Battaglia A, Taylor JC, Howard MF, Keays D, Sohal AS, Kuhl SJ, Kini U, Mcneill A, et al. Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin-Siris syndrome. J Med Genet. 2016;53:152–62.[↩][↩][↩]
- Santen GW, Clayton-Smith J, Consortium ABC. The ARID1B phenotype: what we have learned so far. Am J Med Genet C Semin Med Genet. 2014;166C:276–89.[↩]
- Schrier SA, Bodurtha JN, Burton B, Chudley AE, Chiong MA, D’avanzo M G, Lynch SA, Musio A, Nyazov DM, Sanchez-Lara PA, Shalev SA, Deardorff MA. The Coffin-Siris syndrome: a proposed diagnostic approach and assessment of 15 overlapping cases. Am J Med Genet A. 2012;158A:1865–76[↩]
- Tsurusaki Y, Koshimizu E, Ohashi H, Phadke S, Kou I, Shiina M, Suzuki T, Okamoto N, Imamura S, Yamashita M, Watanabe S, Yoshiura K, Kodera H, Miyatake S, Nakashima M, Saitsu H, Ogata K, Ikegawa S, Miyake N, Matsumoto N. De novo SOX11 mutations cause Coffin-Siris syndrome. Nat Commun. 2014a;5:4011.[↩]
- Wieczorek D, Bogershausen N, Beleggia F, Steiner-Haldenstatt S, Pohl E, Li Y, Milz E, Martin M, Thiele H, Altmuller J, Alanay Y, Kayserili H, Klein-Hitpass L, Bohringer S, Wollstein A, Albrecht B, Boduroglu K, Caliebe A, Chrzanowska K, Cogulu O, Cristofoli F, Czeschik JC, Devriendt K, Dotti MT, Elcioglu N, Gener B, Goecke TO, Krajewska-Walasek M, Guillen-Navarro E, Hayek J, Houge G, Kilic E, Simsek-Kiper PO, Lopez-Gonzalez V, Kuechler A, Lyonnet S, Mari F, Marozza A, Mathieu Dramard M, Mikat B, Morin G, Morice-Picard F, Ozkinay F, Rauch A, Renieri A, Tinschert S, Utine GE, Vilain C, Vivarelli R, Zweier C, Nurnberg P, Rahmann S, Vermeesch J, Ludecke HJ, Zeschnigk M, Wollnik B. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet. 2013;22:5121–35.[↩][↩]
- Tsurusaki Y, Okamoto N, Ohashi H, Mizuno S, Matsumoto N, Makita Y, Fukuda M, Isidor B, Perrier J, Aggarwal S, Dalal AB, Al-Kindy A, Liebelt J, Mowat D, Nakashima M, Saitsu H, Miyake N, Matsumoto N. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin Genet. 2014b;85:548–54.[↩][↩]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44:376–8.[↩]
- Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, Den Hollander NS, Ruivenkamp CA, Van Ommen GJ, Breuning MH, Den Dunnen JT, Van Haeringen A, Kriek M. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012;44:379–80.[↩]
- Michelson M, Ben-Sasson A, Vinkler C, Leshinsky-Silver E, Netzer I, Frumkin A, Kivity S, Lerman-Sagie T, Lev D. Delineation of the interstitial 6q25 microdeletion syndrome: refinement of the critical causative region. Am J Med Genet A. 2012;158A:1395–9.[↩]
- Van Houdt JK, Nowakowska BA, Sousa SB, Van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, Van Den Boogaard MJ, Bottani A, Castori M, Cormier-Daire V, Deardorff MA, Filges I, Fryer A, Fryns JP, Gana S, Garavelli L, Gillessen-Kaesbach G, Hall BD, Horn D, Huylebroeck D, Klapecki J, Krajewska-Walasek M, Kuechler A, Lines MA, Maas S, Macdermot KD, Mckee S, Magee A, De Man SA, Moreau Y, Morice-Picard F, Obersztyn E, Pilch J, Rosser E, Shannon N, Stolte-Dijkstra I, Van Dijck P, Vilain C, Vogels A, Wakeling E, Wieczorek D, Wilson L, Zuffardi O, Van Kampen AH, Devriendt K, Hennekam R, Vermeesch JR. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet. 2012;44:445–9.[↩]
- Schrier SA, Bodurtha JN, Burton B, Chudley AE, Chiong MA, D’avanzo M G, Lynch SA, Musio A, Nyazov DM, Sanchez-Lara PA, Shalev SA, Deardorff MA. The Coffin-Siris syndrome: a proposed diagnostic approach and assessment of 15 overlapping cases. Am J Med Genet A. 2012;158A:1865–76.[↩]
- Kosho T, Miyake N, Carey JC. Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am J Med Genet C Semin Med Genet. 2014a;166C:241–51.[↩]


{kind=link}