Abetalipoproteinemia
Abetalipoproteinemia also known as Bassen-Kornzweig syndrome or Bassen-Kornzweig disease, is a very rare genetic disorder that affects fat and vitamin absorption by the intestines and liver, leading to very low low-density lipoprotein (LDL)-cholesterol and malnutrition. Abetalipoproteinemia typically presents in infancy with failure to gain weight and grow at the expected rate (failure to thrive), diarrhea, fatty foul-smelling stools (steatorrhea), vomiting, malabsorption of fat and poor growth 1. Hematologic manifestations may include acanthocytosis (irregularly spiculated red blood cells) resulting in low levels of circulating red blood cells (anemia) with abnormally star-shaped red blood cells (acanthocytosis) and hemolysis with resultant hyperbilirubinemia. Malabsorption of fat-soluble vitamins (A, D, E, and K) can result in an increased international normalized ratio (INR) resulting in difficulty forming blood clots, which can cause abnormal bleeding. In some cases, a condition called fatty liver develops, which can cause liver damage.
Neuromuscular findings in untreated individuals which typically manifest in the first or second decades of life include disturbances in nerve function that may lead to poor muscle coordination and difficulty with balance and movement (ataxia). They can also experience progressive loss of deep tendon reflexes, vibratory sense and proprioception, impaired speech (dysarthria), tremors or other involuntary movements (motor tics), a loss of sensation in the extremities (peripheral neuropathy), or muscle weakness 2. The muscle problems can disrupt skeletal development, leading to an abnormally curved lower back (lordosis), a rounded upper back that also curves to the side (kyphoscoliosis), high-arched feet (pes cavus), or an inward- and upward-turning foot (clubfoot).
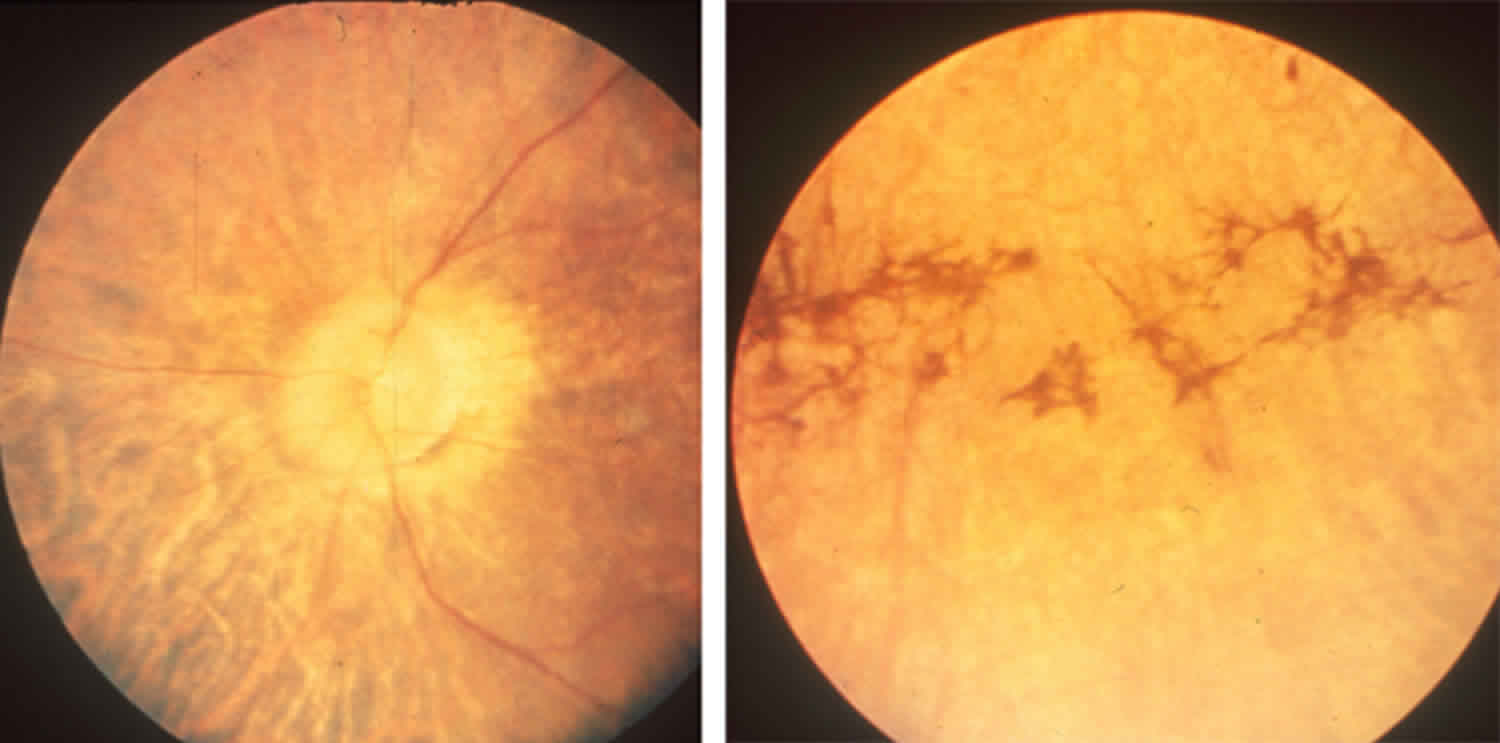
Untreated individuals may also develop an eye disorder called retinitis pigmentosa, in which breakdown of the light-sensitive layer (retina) at the back of the eye can cause progressive loss of night vision and/or color vision in adulthood. In individuals with abetalipoproteinemia, the retinitis pigmentosa can result in complete vision loss. People with abetalipoproteinemia may also have other eye problems, including involuntary eye movements (nystagmus), eyes that do not look in the same direction (strabismus), and weakness of the external muscles of the eye (ophthalmoplegia).
Abetalipoproteinemia is diagnosed based on clinical exam, laboratory tests showing abnormally low cholesterol, and confirmed by genetic testing 3. Abetalipoproteinemia is caused by mutations in the microsomal triglyceride transfer protein (MTTP) gene and is inherited in an autosomal recessive pattern 1.
Abetalipoproteinemia has been treated with a low fat diet and vitamin supplements 3. Most people with abetalipoproteinemia who are treated do not develop complications 4. The long-term outcome can be difficult to predict.
Abetalipoproteinemia is a ultra-rare disorder. The exact prevalence and incidence of abetalipoproteinemia is unknown, but it is estimated to affect less than 1 in 1,000,000 people in the general population. More than 100 cases have been described worldwide 5. Abetalipoproteinemia affects both males and females. There are no known racial or ethnic preferences for the disorder. Abetalipoproteinemia is more prevalent in populations with a high incidence of consanguineous marriages. Symptoms usually become apparent during infancy.
Abetalipoproteinemia is sometimes classified as a neuroacanthocytosis syndrome, which refers to a group of disorders characterized by spiky or burr-shaped red blood cells (acanthocytosis) and neurological disorders, especially movement disorders.
How can I reach out to other families who have a child with abetalipoproteinemia?
The following organizations are focused on helping individuals and families touched by a rare disease. Support and advocacy groups can help you connect with other patients and families, and they can provide valuable services. Many develop patient-centered information and are the driving force behind research for better treatments and possible cures. They can direct you to research, resources, and services. Many organizations also have experts who serve as medical advisors or provide lists of doctors/clinics. Visit the group’s website or contact them to learn about the services they offer.
- Genetic Metabolic Dietitians International (https://gmdi.org)
- Foundation Fighting Blindness: Retinitis Pigmentosa (https://www.fightingblindness.org/diseases/retinitis-pigmentosa)
- Global Genes (https://globalgenes.org)
- RareConnect is an online resource where rare disease patients and families can develop communities and conversations across the world (https://www.rareconnect.org/en)
- GenomeConnect is a patient portal, or registry, that is working to build the knowledge base about genetics and health that will allow researchers and doctors to study the impact of genetic variation on health conditions. This group recently announced that interested participants may now connect with one another to find others with a similar diagnosis (https://www.genomeconnect.org)
Abetalipoproteinemia causes
Abetalipoproteinemia is caused by mutations in the microsomal triglyceride transfer protein (MTTP) gene, which provides instructions for making a protein called microsomal triglyceride transfer protein (MTTP or MTP). This protein is essential for creating and secretion of molecules called beta-lipoproteins (apoB-containing lipoproteins) in the liver and intestines. Beta-lipoproteins transport fats, cholesterol, and fat-soluble vitamins from the intestine to the bloodstream so these nutrients can be taken up by tissues throughout the body. Sufficient levels of fats, cholesterol, and vitamins are necessary for normal growth, development, and maintenance of the body’s cells and tissues. Mutations of the MTTP gene lead to low levels of functional MTTP or MTP, which in turn, hinders the liver and intestines from making and secreting apoB-containing lipoproteins. This, in turn, results in the inability to properly absorb and transport fats and fat soluble vitamins throughout the body. Therefore, a deficiency in MTP results in the absence of lipoproteins such as very low density lipoproteins (VLDLs), low density lipoproteins (LDLs), and chylomicrons in the blood. Lipoproteins are macromolecular complexes consisting of lipids and proteins. These lipid and protein complexes act as transporters that carry fats and fat soluble vitamins (e.g. vitamin E) throughout the body. The symptoms of abetalipoproteinemia are caused by the lack of these apoB-containing lipoproteins in the plasma.
Most MTTP gene mutations lead to the production of microsomal triglyceride transfer protein with reduced or absent function and unable to help in the formation of beta-lipoproteins. A lack of beta-lipoproteins causes severely reduced absorption (malabsorption) of dietary fats and fat-soluble vitamins from the digestive tract into the bloodstream. These nutritional deficiencies lead to health problems in people with abetalipoproteinemia.
Recent research has determined that MTP is also involved in the maturation of a family of proteins known as CD1, which are involved in lipid antigen-presentation to immune cells. More research is necessary to determine the complete functions of the MTP protein and the exact underlying mechanisms that cause disease in abetalipoproteinemia.
Additionally, several studies have shown that MTP is expressed in the heart and is involved in exporting lipids out of the heart. Low levels of MTP may lead to fat accumulation in the heart and affect heart function.
Abetalipoproteinemia inheritance pattern
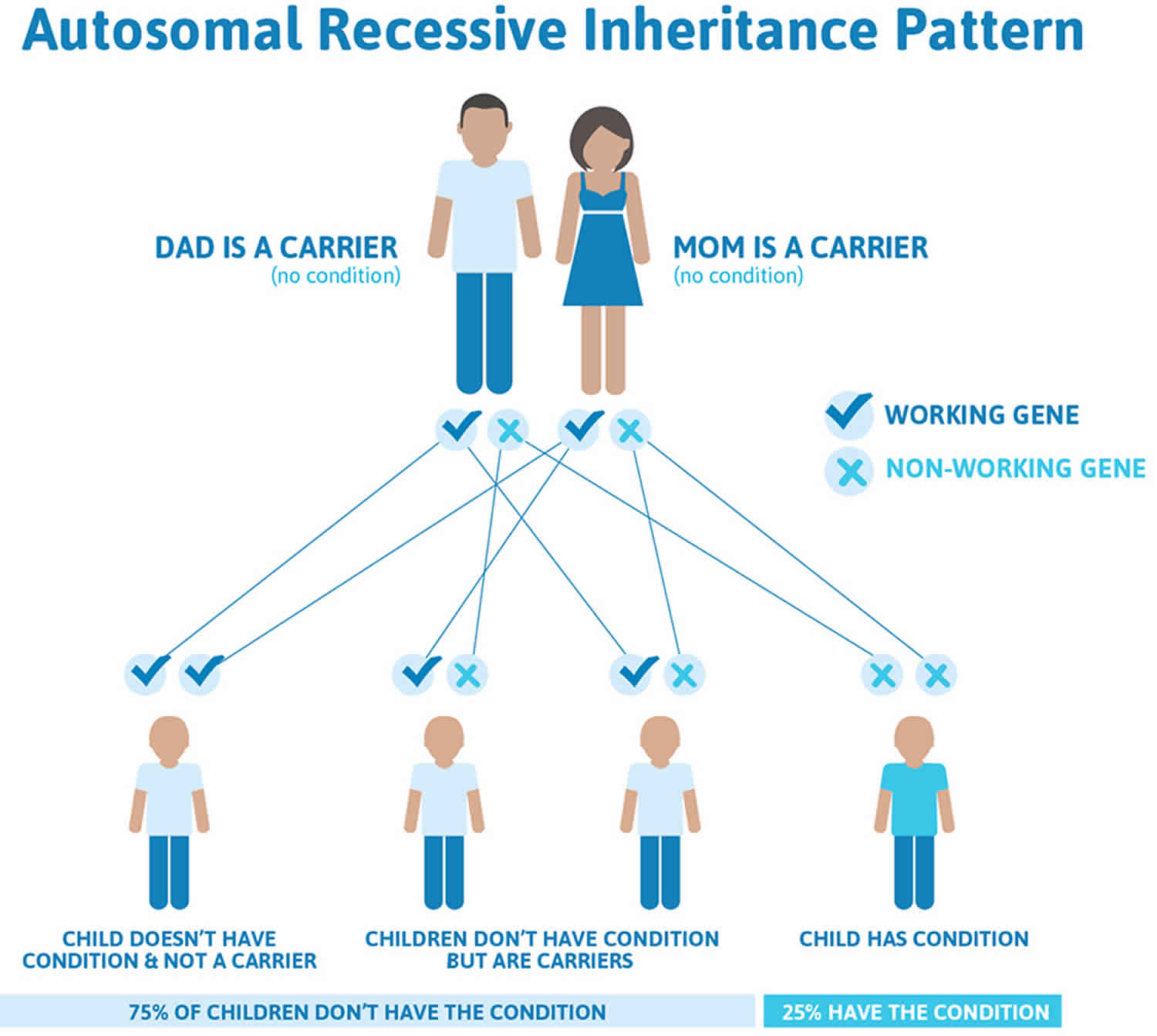
Abetalipoproteinemia is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Abetalipoproteinemia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Abetalipoproteinemia symptoms
Individuals with abetalipoproteinemia may experience a wide variety of symptoms affecting various parts of the body including the gastrointestinal tract, neurological system, eyes, and blood.
The signs and symptoms of abetalipoproteinemia usually appear in the first few months of life 1. They can include:
- Inability to absorb fats and some vitamins. Lipid malabsorption in infancy results in a deficiency of the fat-soluble vitamins A, E, and, less commonly, K. Although lipid is able to enter the intestinal mucosal epithelium, its exit is blocked. Fat-soluble vitamins cannot be normally incorporated into chylomicra for transportation to the liver from the intestine.
- Poor growth in infancy
- Digestive symptoms such as diarrhea and steatorrhea (foul-smelling stools). Steatorrhea with abdominal distension, failure to thrive, malnutrition, and spinal curvature beginning in the first few months of life.
- Abnormal, star-shaped red blood cells (acanthocytosis)
Gastroenterology symptoms:
- Steatorrhea and diarrhea
- Abdominal distention
Neurologic symptoms:
- Slow mental growth
- Deep tendon reflexes are absent
- Ataxia
- Slurred speech
- Peripheral neuropathy.
- Intention tremors.
Ophthalmologic symptoms:
- Retinitis pigmentosa by adolescence (due to deficiency of vitamin A)
- Decreased night and color vision.
- Blindness may occur
Affected infants often present with symptoms relating to gastrointestinal disease, which occur secondary to poor fat absorption. Such symptoms include pale, bulky foul-smelling stools (steatorrhea), diarrhea, vomiting, and swelling (distension) of the abdomen. Affected infants often fail to gain weight and grow at the expected rate (failure to thrive). These symptoms result from poor absorption of fat from the diet. In addition to poor fat absorption, fat-soluble vitamins such as vitamins A, E, and K are also poorly absorbed potentially resulting in fat-soluble vitamin deficiency. Further, patients do not have any apoB-containing lipoproteins in their plasma, and consequently they have very low levels of triglycerides, cholesterol and phospholipids. Thus, lipids and fat soluble vitamins are inadequately transported throughout the blood stream. Some patients may also have reduced non-apoB-containing lipoproteins (high density lipoproteins) or apoA1 levels in their plasma.
Between the ages of 2 and 20 years, a variety of neurological complications occur that resemble spinocerebellar degeneration, a general term for a group of disorders characterized by progressive impairment of the ability to coordinate voluntary movements due to degeneration of certain structures in the brain (cerebellar ataxia). Ataxia results in a lack of coordination and, eventually, difficulty in controlling the range of voluntary movement (dysmetria). Additional neurological symptoms include loss of deep tendon reflexes such as at the kneecap, difficulty speaking (dysarthria), tremors, motor tics, and muscle weakness. Intelligence is usually normal, but developmental delays or intellectual disability has been reported.
In some people, the damage or malfunction of the peripheral nervous system (peripheral neuropathy) may occur. The peripheral nervous system contains all of the nerves outside of the central nervous system. The associated symptoms can vary greatly from one person to another, but can include weakness of the muscles of the arms and legs or abnormal sensations such as tingling (paresthesias), burning or numbness.
Some individuals with abetalipoproteinemia may develop skeletal abnormalities including backward curvature (lordosis) or backward and sideways curvature of the spine (kyphoscoliosis), a highly arched foot (pes cavus) or clubfoot. These skeletal abnormalities may result from muscle imbalances during crucial stages of bone development. Eventually, affected individuals may be unable to stand or to walk unaided due to progressive neurological and skeletal abnormalities.
Some affected individuals may develop a rare eye condition called retinitis pigmentosa in which progressive degeneration of the nerve-rich membrane lining the eyes (retina) results in tunnel vision, loss of color vision, night blindness, and loss of peripheral vision. Affected individuals may eventually develop loss of visual acuity. Retinitis pigmentosa occurs most often around the age of 10 years and may be due to vitamin A and/or E deficiency. If left untreated, visual acuity may deteriorate to virtual blindness by the fourth decade of life.
Less often, additional symptoms that affect the eyes have been reported including rapid, involuntary eye movements (nystagmus), droopy upper eyelid (ptosis), crossed eyes (strabismus), unequal size of the pupils (anisocoria), and weakness or paralysis of muscles that control eye movements (ophthalmoplegia).
Individuals with abetalipoproteinemia may also have blood abnormalities including a condition called acanthocytosis in which deformed (i.e., burr-shaped) red blood cells (acanthocytes) are present in the body. Acanthocytosis may result in low levels of circulating red blood cells (anemia). Anemia may result in tiredness, increased need for sleep, weakness, lightheadedness, dizziness, irritability, palpitations, headaches, and pale skin color. Additional blood abnormalities may be due to vitamin K deficiency. Blood clotting factor levels may be reduced resulting in bleeding tendencies such as severe gastrointestinal bleeding.
Patients may have fatty liver, which can cause liver damage. In rare cases, fibrosis or scarring of the liver (cirrhosis) has also been reported.
Abetalipoproteinemia diagnosis
A diagnosis of abetalipoproteinemia is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests including tests to measure lipid and apoB-containing lipoproteins in the plasma, determine the form and structure (morphology) of red blood cells and an eye (ophthalmological) exam.
- Complete blood count (CBC): Complete blood count shows anemia, thrombocytopenia, or pancytopenia 6
- Blood smear: Blood smear shows acanthocytes (Burr cells).
- Fasting lipid profile: Fasting lipid profile shows low VLDLs, LDLs, and total cholesterol.
- Stool study: Stool studies show fat malabsorption and absence of evidence for other possible causes.
- Imaging studies:
- Hepatic scan or ultrasonography to assess changes of fatty liver
- Magnetic resonance imaging (MRI) of the spinocerebellar region may show degeneration
- Eye and retinal examination and imaging to check for retinal damage
Blood tests will detect low levels of both lipids, such as cholesterol and triglycerides, and lipid-soluble vitamins such as A, E, and K. ApoB-containing lipoproteins, such as chylomicrons or very low density lipoproteins, are not detectable in the plasma.
The identification of malformed red blood cells (acanthocytosis) may also be detected by blood tests.
A complete neurological assessment, an eye examination, an endoscopy, and a liver (hepatic) ultrasound may be performed to evaluate the presence of potentially associated symptoms.
Molecular genetic testing to detect mutations in the MTTP gene is available to confirm the diagnosis.
Abetalipoproteinemia treatment
The treatment of abetalipoproteinemia is directed toward the specific symptoms that are apparent in each individual. Individuals with abetalipoproteinemia have been treated with a low fat diet with strict restriction of long-chain fatty acids 7 and large doses of fat-soluble vitamins (very large doses of oral vitamin E and vitamin A supplementation is instituted if an elevated prothrombin time suggests vitamin K depletion). Treatment may require the coordinated efforts of a team of specialists. Neurologists, liver specialists (hepatologists), eye specialists (ophthalmologists), specialists in the study of fats (lipidologists), gastroenterologists, nutritionists, and other healthcare professionals may need to systematically, comprehensively, and collaboratively plan an affected child’s treatment. Patients should be closely monitored every 6-12 months. Neurological and eye exams should be performed routinely to measure any ophthalmological or neurological deteriorations. Further, amino transaminases and albumin in the blood should be measured every year to determine if there is liver damage. Hepatic ultrasound can be performed to detect the presence of fatty liver. Echocardiography should be performed every three years to ensure the heart is working properly.
Most affected individuals respond to dietary therapy consisting of a diet low in fat especially long-chain saturated fatty acids. The reduction of the intake of dietary fats generally relieves gastrointestinal symptoms. Patients should receive frequent dietary counseling. Diets in infants may be supplemented with medium chain fatty acids, which can be transported in the blood without apoB-containing lipoproteins, in order to promote normal growth and development.
The oral administration of high doses of fat-soluble vitamins (e.g., A, E, K) helps to prevent or improve many of the symptoms associated with abetalipoproteinemia. For example, treatment with vitamin E (i.e. tocopherol therapy) and vitamin A supplementation may prevent the neurological and retinal complications associated with abetalipoproteinemia. Vitamin D supplementation may help alleviate some of the symptoms associated with bone growth. Blood levels of fat soluble vitamins should be measured at each follow up because the blood levels do not always correlate with the amount of vitamins ingested. Doses should be adjusted based on the results of blood panels, neurological exams, and ophthalmological exams.
The prognosis of patients is highly variable. Early detection, treatment, and fat soluble vitamin supplementation can help curtail some of the neurological and ophthalmological deficiencies. Patients should be carefully monitored if receiving fat soluble drug treatments (i.e. for diseases unrelated to abetalipoproteinemia) as their pharmacokinetics, absorption, and transport may also be affected. Additional treatment is symptomatic and supportive.
Genetic counseling is recommended for families of children with abetalipoproteinemia.
Abetalipoproteinemia prognosis
Abetalipoproteinemia prognostic factors 4:
- Age at diagnosis
- The onset of treatment with low-fat diet and vitamin replacement therapy
- Type of MTTP mutation and APOE genotype with the long-term outcome of patients with abetalipoproteinemia
Because abetalipoproteinemia is extremely rare, the course of the disease is difficult to predict. Life expectancy is reduced, and death often occurs from cardiac arrhythmias 8.
Abetalipoproteinemia is associated with very high morbidity and thus it is vital to have an integrated pathway of management with close interaction with a number of health professionals. Besides a gastroenterologist, an interprofessional approach is essential if one is to improve outcomes:
- Ophthalmologist to regularly assess for retinal degeneration and ophthalmoplegia
- Internist to assess the cholesterol profile and the abetalipoproteinemia.
- Neurologist: to assess and monitor the patient for spinocerebellar degeneration
- Hematologist to assess the anemia and acanthocytosis
- Pharmacist to determine the need for pyridoxine and total parenteral nutrition or supplementation with vitamins
- Dietitian to assess the patient for failure to thrive, assess malabsorption of fat-soluble vitamins and the need to change diet
- Genetic counseling for patients and their first-degree relatives
- Social worker and nurse to assess the patients for nutritional needs, failure to thrive and referral to a specialist.
Because of the rarity of the disorder, there is a lack of randomized clinical trials on the long-term benefits of vitamins. However, small case series and anecdotal reports indicate that when patients are diagnosed and referred early to a clinic which specializes in the management of this lipid disorder, the outcomes are improved 9. There is ample evidence indicating that prompt supplementation with fat-soluble vitamins (vitmain A, D, E and K) can improve the prognosis and outcome 10. However, the long-term outlook for many of these patients is guarded as many go on to develop vision loss and permanent blindness.
- Burnett JR, Hooper AJ, Hegele RA. Abetalipoproteinemia. 2018 Oct 25. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532447[↩][↩][↩]
- Paquette M, Dufour R, Hegele RA, Baass A. A tale of 2 cousins: An atypical and typical case of abetalipoproteinemia. J Clin Lipidol. Jul-Aug 2016; 10(4):1030-1034. https://pubmed.ncbi.nlm.nih.gov/27578136[↩]
- Lee J, Hegele RA. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J Inherit Metab Dis. May 2014; 37(3):333-9. https://pubmed.ncbi.nlm.nih.gov/24288038[↩][↩]
- Junaid Z, Patel K. Abetalipoproteinemia. [Updated 2020 Aug 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513355[↩][↩]
- Abetalipoproteinemia. https://ghr.nlm.nih.gov/condition/abetalipoproteinemia[↩]
- Boltshauser E, Weber KP. Laboratory investigations. Handb Clin Neurol. 2018;154:287-298.[↩]
- Cuerq C, Henin E, Restier L, Blond E, Drai J, Marçais C, Di Filippo M, Laveille C, Michalski MC, Poinsot P, Caussy C, Sassolas A, Moulin P, Reboul E, Charriere S, Levy E, Lachaux A, Peretti N. Efficacy of two vitamin E formulations in patients with abetalipoproteinemia and chylomicron retention disease. J. Lipid Res. 2018 Sep;59(9):1640-1648.[↩]
- Abetalipoproteinemia. https://www.aao.org/disease-review/abetalipoproteinemia[↩]
- Gaudet LM, MacKenzie J, Smith GN. Fat-soluble vitamin deficiency in pregnancy: a case report and review of abetalipoproteinemia. J Obstet Gynaecol Can. 2006 Aug;28(8):716-719.[↩]
- Chardon L, Sassolas A, Dingeon B, Michel-Calemard L, Bovier-Lapierre M, Moulin P, Lachaux A. Identification of two novel mutations and long-term follow-up in abetalipoproteinemia: a report of four cases. Eur. J. Pediatr. 2009 Aug;168(8):983-9.[↩]

{kind=link}