POLG
POLG also called POLG-related disorders is a group of mitochondrial genetic diseases caused by mutations in the polymerase gamma (pol γ) or PolG gene that have overlapping signs and symptoms affecting muscle-, nerve-, and brain-related functions 1, 2, 3, 4, 5. POLG typically affects multiple organs, primarily the brain, nerves, muscles, and liver, and can affect vision due to involvement of brain structures 4, 6, 7, 8, 9, 10, 11. POLG is also one of the most common inherited mitochondrial disease 12. Up to 2% of those of Northern European decent may carry disease-causing POLG gene mutations 13 and the frequency of POLG disease is estimated to be 1 in 10,000 2. POLG gene mutations inheritance may be autosomal recessive meaning two mutations are required for disease expression or autosomal dominant meaning only one mutation is required for disease expression.
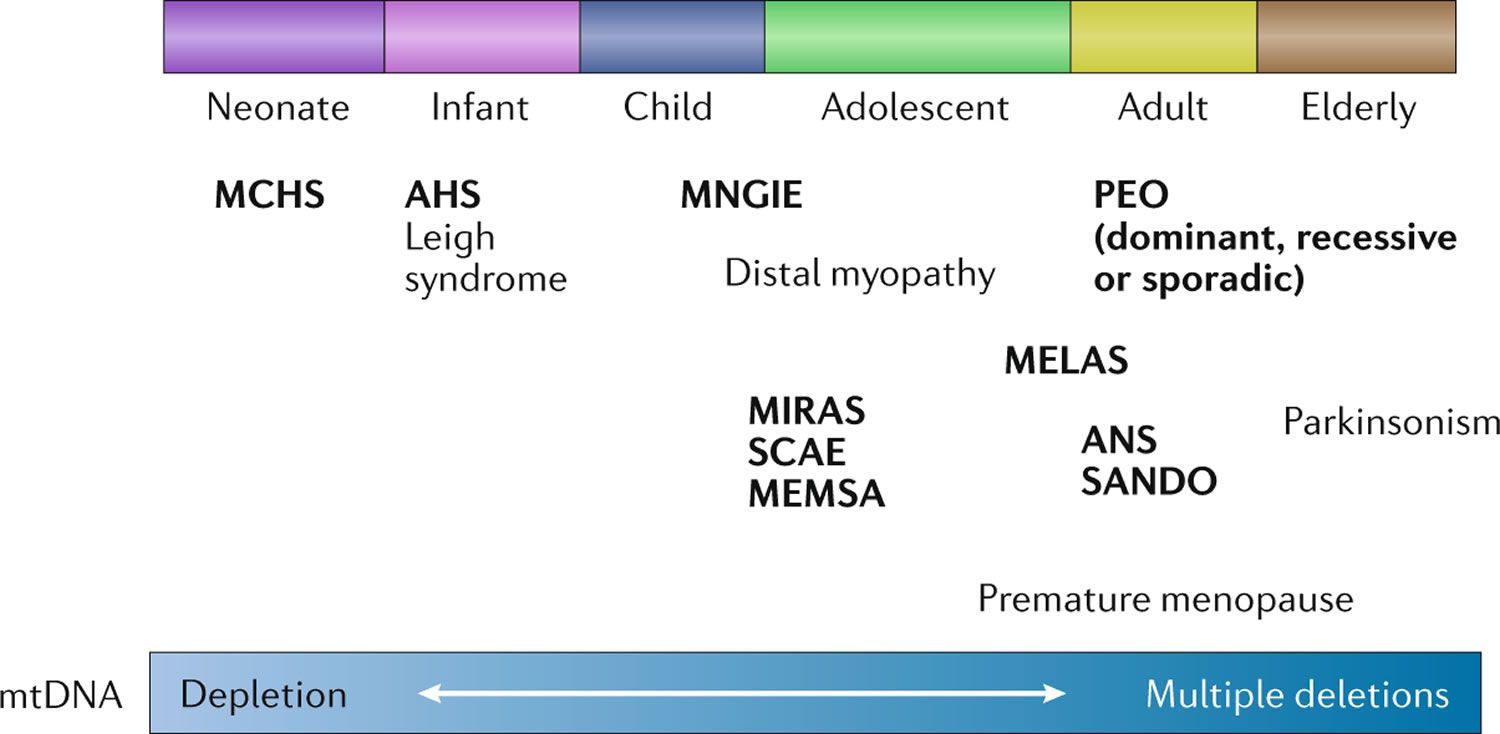
The signs and symptoms of POLG disease can appear anywhere between infancy and late adulthood and can vary widely among affected individuals 4:
- In individuals with early-onset disease prior to age 12 years, liver involvement, feeding difficulties, seizures (epilepsy), low muscle tone (hypotonia), and muscle weakness are the most common clinical features. This group has the worst prognosis.
- In the juvenile or adult-onset form with onset between the age 12 to 40 years, disease is typically characterized by peripheral nerves damaged (peripheral neuropathy), impaired balance and coordination (ataxia), seizures, stroke-like episodes, and, in individuals with longer survival, progressive external ophthalmoplegia (PEO). Progressive external ophthalmoplegia (PEO) is a condition characterized by weakness of the eye muscles that control eye movement, leading to progressive inability to move the eyes and eyelids, often with drooping eyelids (ptosis). This group generally has a better prognosis than the early-onset group.
- Late-onset disease with onset after age 40 years is characterized by drooping eyelids (ptosis) and progressive external ophthalmoplegia (PEO), with additional features such as peripheral nerves damaged (peripheral neuropathy), impaired balance and coordination (ataxia), and muscle weakness. This group overall has the best prognosis.
In general, earlier onset of POLG disease corresponds with more severe symptoms and poorer outcomes 4, 6.
POLG disease is caused by inherited mutations in the POLG gene 4, 6, 2. The POLG gene provides instructions for making the active piece, called the alpha subunit, of a protein called polymerase gamma (pol γ). To be most effective, the alpha subunit attaches to two copies of another protein called the beta subunit to form polymerase gamma (pol γ). Polymerase gamma (pol γ) is a DNA polymerase, which is a type of enzyme that “reads” sequences of your DNA and uses them as templates to produce new DNA. DNA polymerases are important for copying (replicating) cells’ genetic material. DNA polymerases also play critical roles in DNA repair.
Polymerase gamma (pol γ) functions in your mitochondria and is responsible for mitochondrial DNA (mtDNA) maintenance 12, 14. Mitochondria are structures within your cells, often called the “powerhouses” of the cell, in which a process called oxidative phosphorylation converts the energy from food you eat into a form called ATP (adenosine triphosphate) that your cells can use 15. Mitochondria use oxygen to break down glucose and other nutrients, releasing energy that is then stored in ATP. Your mitochondria also contain their own DNA known as mitochondrial DNA (mtDNA), which is essential for the normal function of these structures and is different from the DNA in your other cells nucleus. Polymerase gamma (pol γ) is the only DNA polymerase that is active in mitochondria and that can replicate mitochondrial DNA (mtDNA). This mitochondrial DNA (mtDNA) encodes for some proteins and RNAs necessary for mitochondrial function. Mutations in POLG gene compromise the functioning of the polymerase gamma (pol γ) enzyme responsible for mitochondrial DNA (mtDNA) maintenance. Poor DNA maintenance reduces the amount of mitochondrial DNA (mtDNA) present in the cell and/or introduces mutations in the mitochondrial DNA (mtDNA) 16.
Beyond energy production, your mitochondria also play other crucial roles, including regulating cell death (apoptosis) and contributing to the production of various molecules like proteins, DNA, RNA, lipids, hormones and neurotransmitters and calcium storage reservoirs, which are important for various cellular functions like muscle contraction and neurotransmitter release. Little is known about what exactly causes the clinical appearance of POLG disease and what external factors contribute to the development of symptoms and their severity.
POLG disease is sometimes divided into subtypes that includes 4, 6, 1, 5:
- Alpers-Huttenlocher syndrome also known as Alpers syndrome
- Alpers-Huttenlocher syndrome is characterized by seizures, loss of mental (dementia) and movement abilities (psychomotor regression), spasticity, blindness, and liver disease 17. The liver disease in Alpers-Huttenlocher syndrome can be brought on or made worse by valproic acid and sodium divalproate (divalproex), a common treatment for seizures 18.
- Alpers-Huttenlocher syndrome typically becomes apparent in children between ages 2 and 4. People with Alpers-Huttenlocher syndrome usually have 3 characteristic features: recurrent seizures that do not improve with treatment (intractable epilepsy), loss of mental and movement abilities (psychomotor regression), and liver disease 17.
- People with Alpers-Huttenlocher syndrome usually have additional signs and symptoms. Most have problems with coordination and balance (ataxia) and disturbances in nerve function (neuropathy). Neuropathy (nerve damage) can lead to abnormal or absent reflexes (areflexia). In addition, affected individuals may develop weak muscle tone (hypotonia) that worsens until they lose the ability to control their muscles and movement. Some people with Alpers-Huttenlocher syndrome lose the ability to walk, sit, or feed themselves. Other movement-related symptoms in affected individuals can include involuntary muscle twitches (myoclonus), uncontrollable movements of the limbs (choreoathetosis), or a pattern of movement abnormalities known as parkinsonism.
- People with Alpers-Huttenlocher syndrome may also have other brain-related signs and symptoms. Migraine headaches, often with visual sensations or auras, are common. Additionally, people with Alpers-Huttenlocher syndrome may have decreased brain function that is demonstrated as sleepiness, inability to concentrate, irritability, or loss of language skills or memory. Some people with the condition may lose their eyesight or hearing. People with Alpers-Huttenlocher syndrome can survive from a few months to more than 10 years after the condition first appears.
- The most common POLG gene mutation in Alpers-Huttenlocher syndrome replaces the amino acid alanine with the amino acid threonine at position 467 written as Ala467Thr or A467T 5. This POLG gene Ala467Thr mutation blocks the ability of the alpha subunit to attach to the beta subunits and reduces polymerase gamma (pol γ) ability to synthesize DNA. The Ala467Thr variant is also common in other POLG-related disorders. The different conditions may be determined, in part, by the variant in the other copy of POLG, but there are still some variant combinations that can cause more than one of the disorders. It is unclear how the same variant can lead to different conditions. Although the mechanism is unknown, many people with Alpers-Huttenlocher syndrome have fewer copies of mtDNA (mtDNA depletion). This abnormality is seen only in the tissues affected by the disease. MtDNA depletion leads to impaired oxidative phosphorylation and a decrease in cellular energy. These impairments affect tissues whose cells do not divide continually, such as brain, muscle, and liver. These tissues are most affected because they are more dependent on oxidative phosphorylation for energy, and impaired cells in these tissues are not generally replaced by new cells. The lack of energy supplies in these tissues could account for the signs and symptoms of Alpers-Huttenlocher syndrome.
- Alpers-Huttenlocher syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
- Alpers-Huttenlocher syndrome treatment is limited to symptom management and supportive care and family education should be addressed as soon as the family is able to absorb the diagnosis.
- Childhood Myocerebrohepatopathy Syndrome (MCHS)
- Childhood Myocerebrohepatopathy Syndrome (MCHS) is a severe usually fatal POLG-related disorder that affects the muscles (myo-), brain (cerebro-), and liver (hepato-) 19. Childhood Myocerebrohepatopathy Syndrome (MCHS) key 3 symptoms are myopathy (muscle diseases that affect skeletal muscles that control voluntary movements in the body) or low muscle tone (hypotonia), developmental delay or severe brain dysfunction (encephalopathy), and liver dysfunction. Childhood Myocerebrohepatopathy Syndrome (MCHS) typically presents in the first few months to 3 years of life, characterized by muscle weakness, developmental delay, and liver dysfunction. Other signs and symptoms of Childhood Myocerebrohepatopathy Syndrome (MCHS) include toxic buildup of lactic acid in the body (lactic acidosis), unable to gain weight and grow at the expected rate (failure to thrive), a form of kidney disease called renal tubular acidosis, inflammation of the pancreas (pancreatitis), recurrent episodes of nausea and vomiting (cyclic vomiting) and hearing loss 19. Seizures occur in about 75% of affected individuals. Liver failure typically occurs before the age of 1 year, and the condition is usually fatal with a median age of death in one study of 15.8 months (range: 1 to 184.6 months) 4. Major causes of death include liver failure, sepsis, and status epilepticus (abnormally prolonged seizure or a series of seizures without a period of recovery between them) 20, 21, 12.
- While Childhood Myocerebrohepatopathy Syndrome (MCHS) can present similarly to Alpers Syndrome or Alpers-Huttenlocher syndrome, which also involves liver failure and developmental delay, Childhood Myocerebrohepatopathy Syndrome (MCHS) generally lacks the specific liver histopathological features seen in Alpers-Huttenlocher syndrome. Additionally, Childhood Myocerebrohepatopathy Syndrome (MCHS) seizures are less common and severe, and the liver failure typically occurs before the age of 1 year 22.
- Many variants in the POLG gene can cause Childhood Myocerebrohepatopathy Syndrome (MCHS). Most of these POLG gene variants change single amino acids in the alpha subunit of polymerase gamma (pol γ). These variants reduce the activity of polymerase gamma (pol γ), decreasing mtDNA replication. As in other POLG-related disorders, people with Childhood Myocerebrohepatopathy Syndrome (MCHS) typically have mtDNA depletion in muscle, brain, or liver tissue. MtDNA depletion impairs oxidative phosphorylation in these tissues and decreases the energy available to the cells, which may cause the signs and symptoms of Childhood Myocerebrohepatopathy Syndrome (MCHS).
- Childhood Myocerebrohepatopathy Syndrome (MCHS) is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
- Childhood myocerebrohepatopathy syndrome (MCHS) treatment focuses on managing symptoms like seizures, lactic acidosis, and liver failure and providing supportive care.
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) also known as spinocerebellar ataxia with epilepsy (SCAE)
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) is a genetic disorder characterized by ataxia (lack of coordination), myoclonus (muscle jerks), and myopathy (muscle weakness), as well as seizures 23.
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) signs and symptoms typically appear during young adulthood. The first symptom of MEMSA is usually cerebellar ataxia, which refers to problems with coordination and balance due to defects in the part of the brain that is involved in coordinating movement (cerebellum). Recurrent seizures (epilepsy) usually develop later, often in combination with uncontrollable muscle jerks (myoclonus). The seizures usually begin in the right arm and spread to become generalized throughout the body. Additionally, affected individuals may have severe brain dysfunction (encephalopathy) or muscle weakness (myopathy). The myopathy can affect muscles close to the center of the body (proximal), such as the muscles of the hips, thighs, upper arms, or neck, or muscles farther away from the center of the body (distal), such as the muscles of the hands or feet. The myopathy may be especially noticeable during exercise (exercise intolerance).
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) is primarily caused by mutations in the POLG1 gene. This gene is involved in the replication of mitochondrial DNA (mtDNA). Although the mechanism is unknown, mutations in the POLG gene often result in fewer copies of mtDNA (mtDNA depletion), particularly in muscle, brain, or liver cells. MtDNA depletion causes a decrease in cellular energy, which could account for the signs and symptoms of MEMSA.
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) is inherited in an autosomal recessive pattern, meaning both copies of the gene in each cell must have mutations for the disorder to manifest. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
- Treatment for MEMSA (myoclonic epilepsy, myopathy, and sensory ataxia) focuses on managing symptoms, addressing complications, and improving quality of life. Specifically, anticonvulsants like levetiracetam, valproic acid, and zonisamide can be used to control myoclonic seizures. For mitochondrial myopathies, CoQ10 supplementation may be helpful in some cases, and exercise programs can improve both biochemical and clinical outcomes.
- Ataxia neuropathy spectrum (ANS)
- Ataxia neuropathy spectrum (ANS) encompasses a range of disorders characterized by both problems with coordination and balance (ataxia) and neuropathy (nerve damage) 24. As the name implies, people with ataxia neuropathy spectrum typically have problems with coordination and balance (ataxia) and disturbances in nerve function (neuropathy). The neuropathy (nerve damage) can be classified as sensory, motor, or a combination of the two (mixed). Sensory neuropathy (sensory nerve damage) causes numbness, tingling, or pain in the arms and legs, and motor neuropathy (motor nerve damage) refers to disturbance in the nerves used for muscle movement.
- Most people with ataxia neuropathy spectrum also have severe brain dysfunction (encephalopathy) and seizures. Some affected individuals have weakness of the external muscles of the eye (ophthalmoplegia), which leads to drooping eyelids (ptosis). Other signs and symptoms can include involuntary muscle twitches (myoclonus), liver disease, depression, migraine headaches, or blindness.
- Ataxia neuropathy spectrum now includes the conditions previously called mitochondrial recessive ataxia syndrome (MIRAS) and sensory ataxia neuropathy dysarthria and ophthalmoplegia (SANDO).
- The most common POLG gene variant in ataxia neuropathy spectrum is the same as that in Alpers-Huttenlocher syndrome (described above), Ala467Thr. It is unclear how the same Ala467Thr mutation can lead to different disorders. Rarely, ataxia neuropathy spectrum (ANS) is caused by the TWNK gene. The TWNK gene provides instructions for making a protein called Twinkle. Mutated polymerase gamma (pol γ) or mutated Twinkle reduce mitochondrial DNA (mtDNA) replication. Although the mechanisms are unknown, mutations in the POLG gene often result in fewer copies of mtDNA (mtDNA depletion), and mutations in the TWNK gene often result in deletions of large regions of mtDNA (mtDNA deletion). MtDNA depletion or deletion occurs most commonly in muscle, brain, or liver cells. MtDNA depletion causes a decrease in cellular energy, which could account for the signs and symptoms of ataxia neuropathy spectrum. It is unclear what role mtDNA deletions play in the signs and symptoms of the condition.
- Mutations in the POLG gene cause a form of the condition that is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
- Mutations in the TWNK gene cause a form of the condition that is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
- As in other POLG-related disorders, people with ataxia neuropathy spectrum typically have mitochondrial DNA (mtDNA) depletion in the tissues affected by the condition, such as the brain. MtDNA depletion decreases the amount of energy available to the cell due to reduced oxidative phosphorylation, which may account for the signs and symptoms of ataxia neuropathy spectrum.
- Ataxia neuropathy spectrum (ANS) treatment focuses on addressing the symptoms and improving quality of life, which may include physical therapy, occupational therapy, speech therapy, and medication to manage specific symptoms.
- Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO)
- Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) is a very rare mitochondrial disease that is part of the ataxia neuropathy spectrum (ANS) typically associated with mutations in the POLG1 gene. Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) primarily affects the brain, muscles, nerves, and eyes 25, 4. Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) usually presents during adulthood; the mean age of onset is 32 years, although it can also be found in younger patients 26. The symptoms caused by sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) and their severity vary widely among individuals with the disorder. While the disorder worsens over time, its progression is slower and milder than other related mitochondrial disorders 4. There is no cure for SANDO. Available treatments focus on symptom management and improving an individual’s quality of life.
- Mitochondrial recessive ataxia syndrome (MIRAS)
- Mitochondrial recessive ataxia syndrome (MIRAS) is a rare mitochondrial genetic disorder characterized by progressive loss of muscle coordination and balance (ataxia), along with other neurological symptoms like seizures, hearing loss, and cognitive decline. It’s caused by mutations in the POLG1 gene, which is responsible for maintaining and repairing mitochondrial DNA (mtDNA). Mitochondrial recessive ataxia syndrome (MIRAS) is an autosomal recessive condition, meaning individuals need to inherit two copies of the mutated POLG1 gene, one from each parent, to develop the disorder.
- MIRAS can present with a variety of symptoms, including:
- Ataxia: Progressive loss of coordination and balance, affecting gait, fine motor skills, and speech.
- Neuropathy (nerve damage): Sensory or mixed sensory-motor neuropathy, causing numbness, tingling, or pain in the limbs.
- Epilepsy: Seizures can occur at any stage of the disease.
- Ophthalmoplegia: Progressive weakness or paralysis of the eye muscles, leading to double vision or drooping eyelids.
- Dysarthria: Difficulty with speech due to muscle weakness in the mouth and tongue.
- Cognitive impairment: Intellectual disability, memory problems, or cognitive regression can occur.
- Movement disorders: Involuntary movements like myoclonus (sudden, brief muscle spasms) can be present.
- Other symptoms: Fatigue, hearing loss, psychiatric symptoms, and liver disease are also possible.
- There is no cure for mitochondrial recessive ataxia syndrome (MIRAS), and treatment focuses on managing symptoms and improving quality of life.
- The prognosis can vary, and some individuals with mitochondrial recessive ataxia syndrome (MIRAS) live fairly normal lives, while others may experience more severe complications.
- Progressive external ophthalmoplegia (PEO and PEO+) also called chronic progressive external ophthalmoplegia (CPEO)
- Progressive external ophthalmoplegia (PEO) is a condition characterized by weakness or paralysis of the eye muscles that control eye movement and eyelids, leading to drooping eyelids (ptosis) and difficulty moving the eyes 27. Progressive external ophthalmoplegia (PEO) typically progresses slowly over time, with symptoms worsening gradually.
- Progressive external ophthalmoplegia (PEO) typically appears in adults between ages 18 and 40 and slowly worsens over time. The first sign of progressive external ophthalmoplegia is typically drooping eyelids (ptosis), which can affect one or both eyelids. As drooping eyelid (ptosis) worsens, affected individuals may use the forehead muscles to try to lift the eyelids, or they may lift up their chin in order to see. Another characteristic feature of progressive external ophthalmoplegia is weakness or paralysis of the muscles that move the eye (ophthalmoplegia). Affected individuals have to turn their head to see in different directions, especially as the ophthalmoplegia worsens. People with progressive external ophthalmoplegia may also have general weakness of the muscles used for movement (myopathy), particularly those in the neck, arms, or legs. The weakness may be especially noticeable during exercise (exercise intolerance). Muscle weakness may also cause difficulty swallowing (dysphagia).
- Although muscle weakness is the primary symptom of progressive external ophthalmoplegia (PEO), this condition can be accompanied by other signs and symptoms. In these instances, the condition is referred to as progressive external ophthalmoplegia plus (PEO+). Additional signs and symptoms can include hearing loss caused by nerve damage in the inner ear (sensorineural hearing loss), weakness and loss of sensation in the limbs due to nerve damage (neuropathy), impaired muscle coordination (ataxia), a pattern of movement abnormalities known as parkinsonism, and depression.
- Progressive external ophthalmoplegia (PEO) can result from mutations in one of several different genes. In some cases, mutations in the POLG, TWNK, RRM2B, and SLC25A4 genes. These genes are critical for the production and maintenance of mitochondrial DNA (mtDNA). Less commonly, mutations that change single nucleotides in genes found in mitochondrial DNA (mtDNA), such as the MT-TL1 gene, cause progressive external ophthalmoplegia. These mutations occur in genes that provide instructions for making molecules called transfer RNAs. Transfer RNAs help assemble protein building blocks (amino acids) into functioning proteins. The transfer RNAs associated with progressive external ophthalmoplegia are present in mitochondria and help assemble the proteins that carry out the steps of oxidative phosphorylation.
- There are at least 67 POLG gene variants that cause progressive external ophthalmoplegia (PEO). Most POLG gene variants change single amino acids in the alpha subunit of polymerase gamma (pol γ), which decreases the efficiency of mitochondrial DNA (mtDNA) replication. As in another POLG-related disorder, Alpers-Huttenlocher syndrome (described above), the most common POLG gene variant in progressive external ophthalmoplegia (PEO) is Ala467Thr. It is unclear how the same variants can lead to different disorders. Researchers have not determined how deletions of mitochondrial DNA (mtDNA) lead to the specific signs and symptoms of progressive external ophthalmoplegia (PEO), although the features of the condition are probably related to impaired oxidative phosphorylation 5. It has been suggested that eye muscles are commonly affected by mitochondrial defects because they are especially dependent on oxidative phosphorylation for energy.
- Progressive external ophthalmoplegia can have different inheritance patterns depending on the gene involved.
- When the nuclear genes POLG, TWNK, RRM2B, or SLC25A4 are involved, progressive external ophthalmoplegia is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. This progressive external ophthalmoplegia (PEO) is better known as Autosomal Recessive Progressive External Ophthalmoplegia (arPEO).
- Certain mutations in the POLG or RRM2B gene can also cause a form of progressive external ophthalmoplegia (PEO) that is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. This progressive external ophthalmoplegia (PEO) is better known as Autosomal Dominant Progressive External Ophthalmoplegia (adPEO).
- When progressive external ophthalmoplegia (PEO) is caused by mutations in the MT-TL1 gene and other mitochondrial transfer RNA genes, it is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mitochondrial DNA (mtDNA). Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mitochondrial DNA (mtDNA) mutations only from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
- Single, large deletions of mitochondrial DNA (mtDNA) are typically not inherited but occur during the formation of a mother’s egg cells or in early development of the embryo. Individuals with these mutations usually have no history of the disorder in their family.
- There is no cure for progressive external ophthalmoplegia (PEO), but treatment focuses on managing symptoms and improving quality of life. Progressive external ophthalmoplegia (PEO) treatment may include physical therapy, medications to address specific symptoms like ptosis, and support for other affected organ systems.
A referral to a medical facility with physicians who specialize in POLG diseases is critical to making the diagnosis as diagnosis of mitochondrial disorders is still a major challenge 21, 1. To diagnose POLG your doctor may perform or recommend the following 4:
- Genetic testing for POLG gene mutation(s)
- Brain-imaging such as computed tomography (CT) scan or magnetic resonance imaging (MRI) to look for changes in the brain associated with POLG disease
- Electroencephalogram (EEG or ‘brain wave’) testing
Because POLG disease is an inherited disorder, affected families should also receive genetic counseling.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Treatment for POLG disease varies significantly based on the specific type of condition and the signs and symptoms present in each person 28. Available treatments for POLG disorder focus on symptom management and quality of life. These may include medications for seizures (epilepsy); occupational, physical, and speech therapy; nutritional support; respiratory support; and standard treatment of liver failure, movement abnormalities, sleep disorders, vision, and hearing issues 4. Medical research has yet to uncover a cure for POLG disease.
The following treatments can help manage the symptoms of POLG disease and improve life for affected individuals and their families 4:
- Anticonvulsant medications for seizures (although valproate must be avoided). The use of valproate, a common anticonvulsant medication, may result in irreversible liver failure.
- Pain medication and muscle relaxants for comfort
- Small frequent meals or a feeding tube for nutritional support may be helpful and the ketogenic diet is sometimes used to help control seizures
- Physical therapy for declining motor skills and muscle strength
- Surgery or special glasses to correct drooping eyelids
- Speech therapy for slurred speech
- Breathing tube and/or artificial ventilation for respiratory failure
- CPAP or BiPAP for temporary cessation of breathing (apnea)
- Physical and occupational therapy for motor involvement
Figure 1. Mitochondria cell
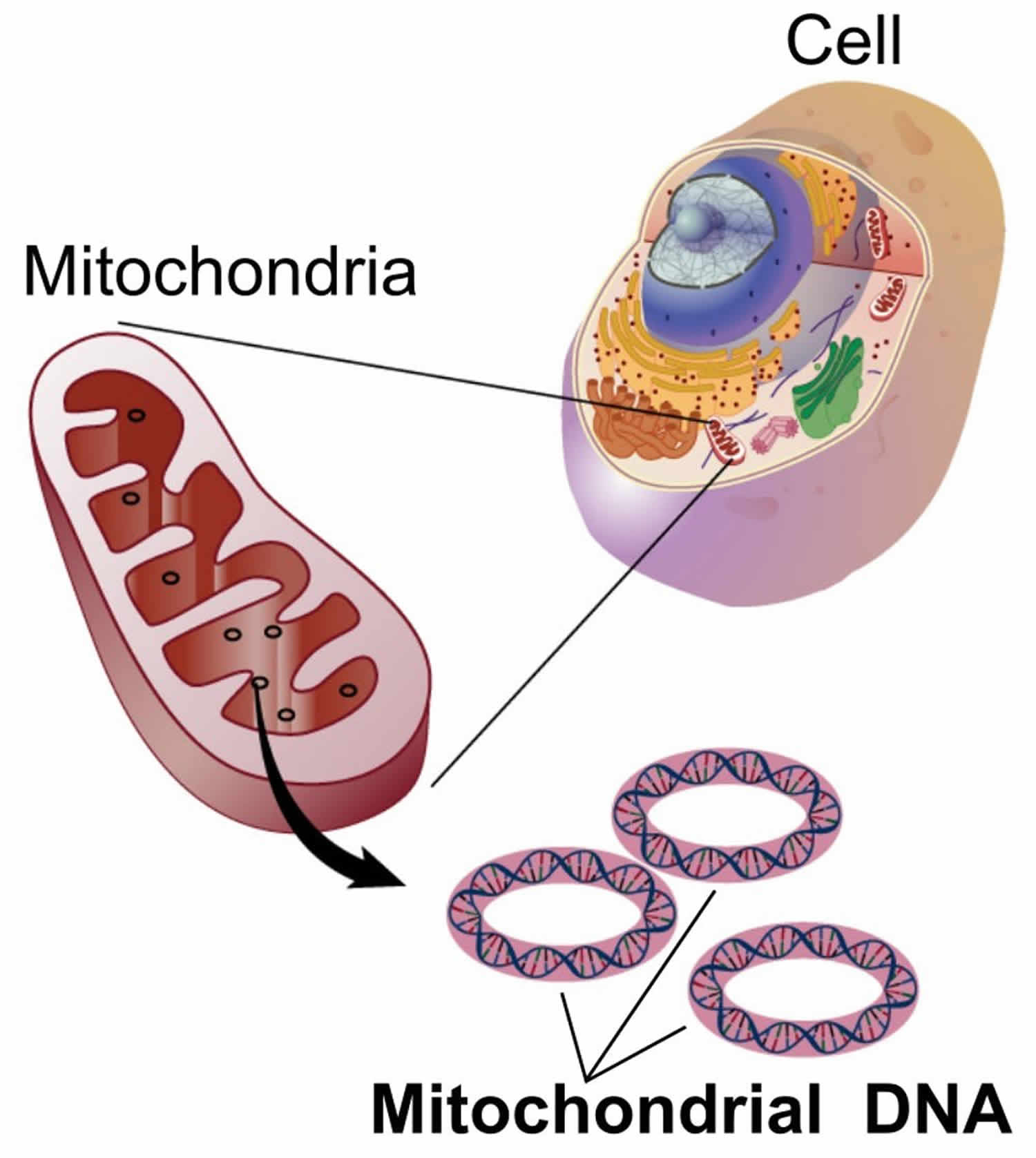
Figure 2. POLG gene mutations
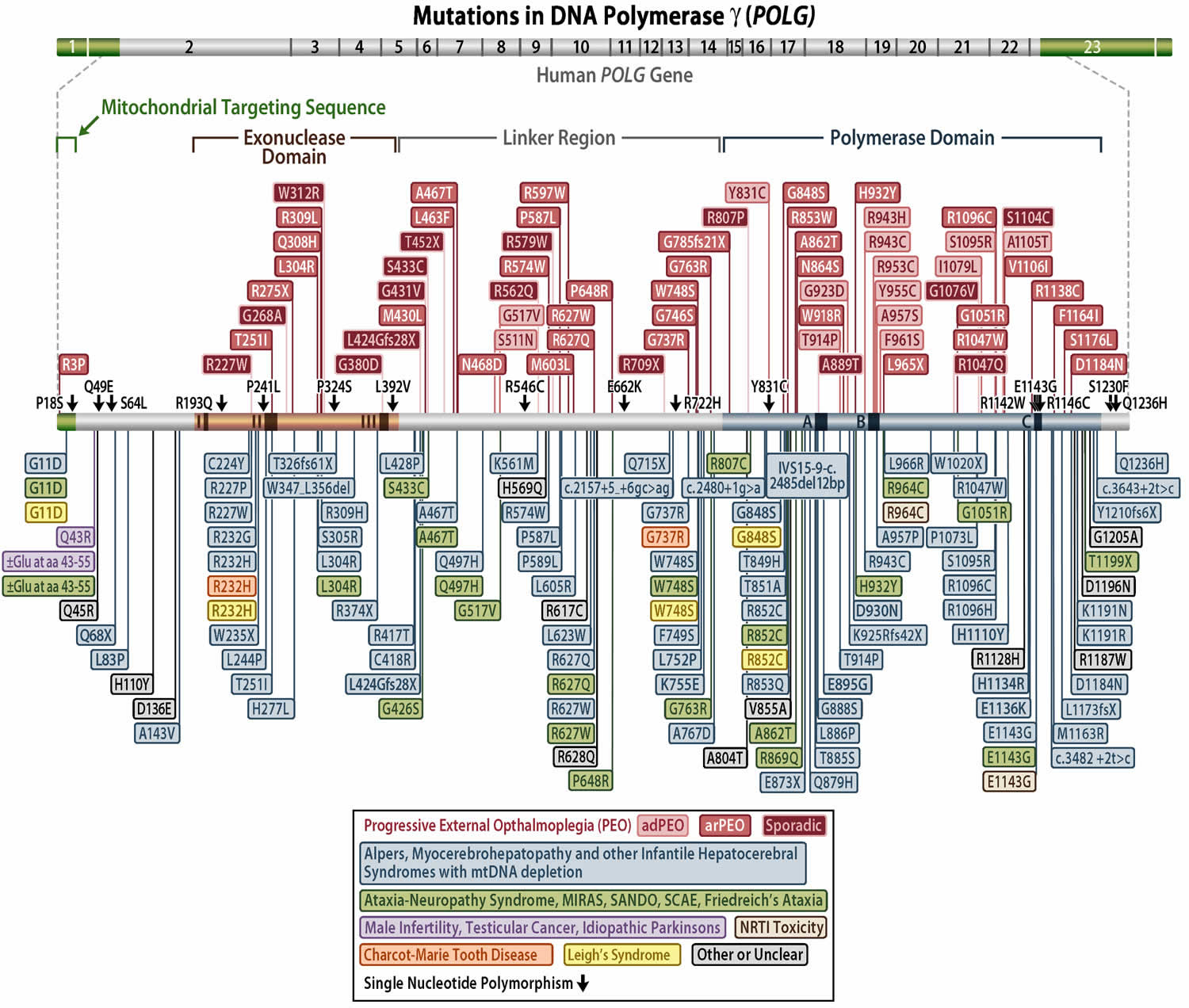
Figure 3. POLG related disorders
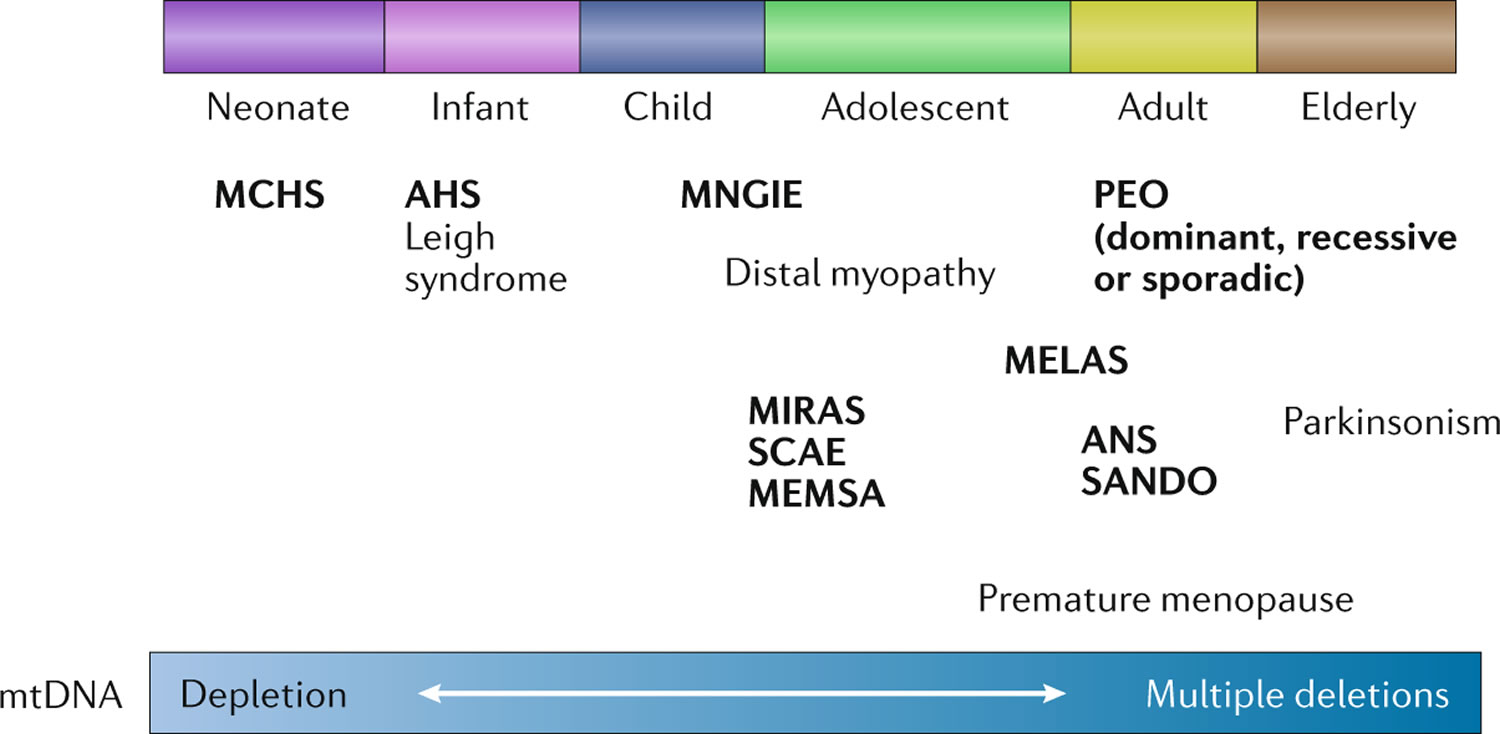
Figure 4. POLG disease diagnostic algorithm
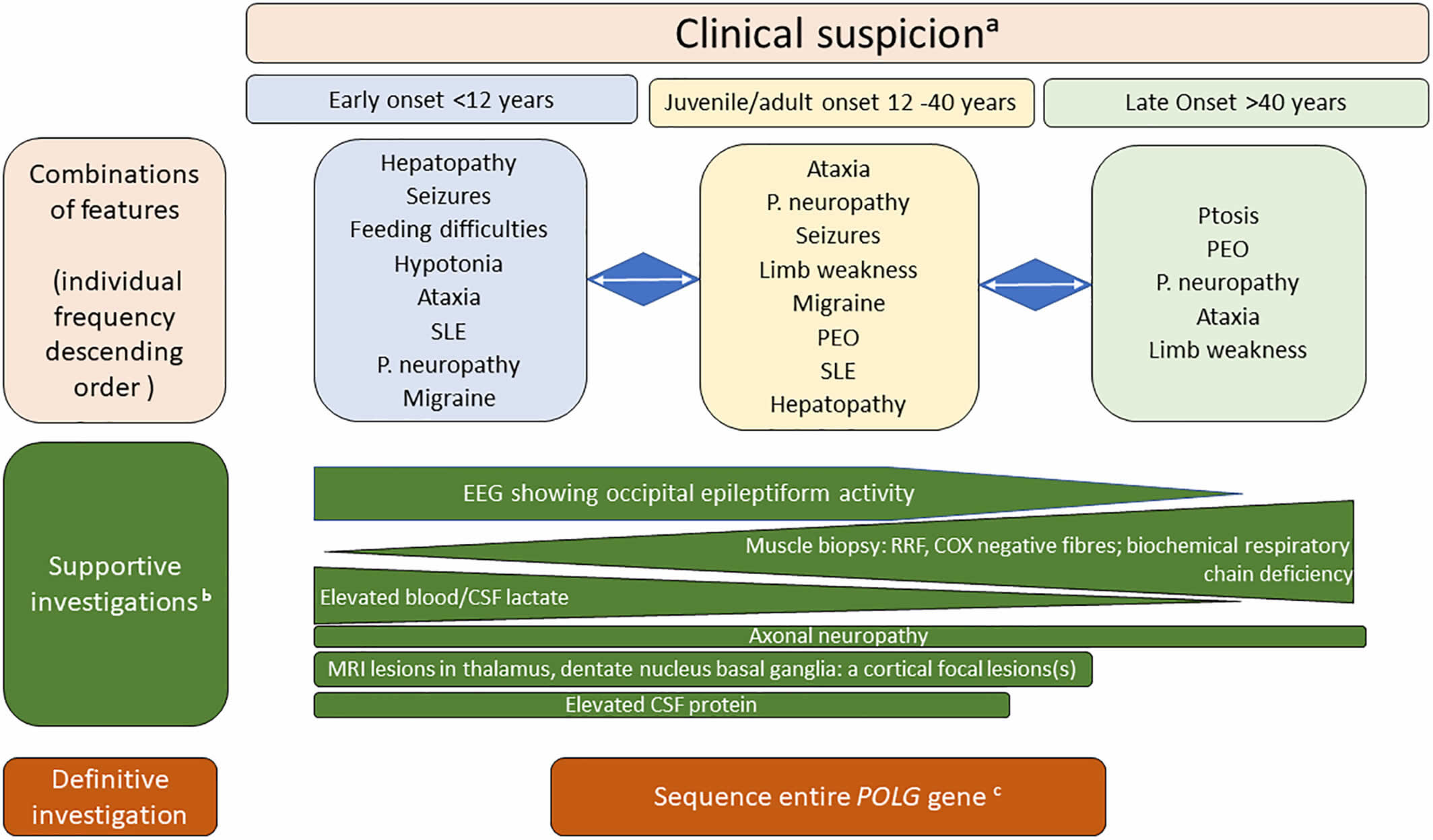
Footnotes: Diagnosing POLG disease; clinical suspicion and relevant investigations according to the age of onset. POLG clinical features form a continuum and age plays a role in which features predominate. Age groups and clinical patterns will dictate which investigations are appropriate and useful. For example, in the older age category, PEO and ataxia dominate the clinical spectrum and in these cases one can choose either to screen the known genes or to take a muscle biopsy which give both structural clues (COX negative fibers) and the possibility to examine mtDNA for multiple deletions. Typical occipital epilepsy occurs in the younger two categories and it is in these that brain MRI imaging also provides important clues. Peripheral neuropathy occurs in all age groups. In earlier studies, it was shown that elevated CSF protein can be helpful, for example in a child with epilepsy and focal MRI changes it can be an important indicator of poor prognosis.
a Direct POLG gene sequence analysis is recommended to confirm the diagnosis in a case of strong clinical suspicion.
b Absence of these findings does not exclude the diagnosis of POLG disease.
c Targeted variant analysis for the most common variants (p. Ala467Thr and p.Trp748Ser) can be performed first in juvenile and late onset disease, whole POLG gene sequence analysis is recommended for all early onset disease and those with strong clinical suspicion of POLG disease regardless of the age of onset.
Abbreviations: CSF = cerebrospinal fluid; RRF = ragged-red fibres; PEO = progressive external ophthalmoplegia; P. neuropathy = peripheral neuropathy; SLE = stroke- like episodes
[Source 1 ]Figure 5. POLG disease survival and life expectancy
What is mitochondria?
You have mitochondria present in every cell of your body except red blood cells. Mitochondria are membrane-bound cell organelles (mitochondrion, singular) within your cells, often called the “powerhouses” of the cell, in which a process called oxidative phosphorylation converts the energy from food you eat into a form called adenosine triphosphate (ATP) that your cells can use 15. Mitochondria use oxygen to break down glucose and other nutrients, releasing energy that is then stored in ATP (adenosine triphosphate). Your mitochondria also contain their own DNA known as mitochondrial DNA (mtDNA), which is essential for the normal function of these structures and is different from the DNA in your other cells nucleus.
The mitochondria in the cells throughout your body are responsible for creating more than 90% of the energy needed by your body to sustain life and support organ function. When mitochondria fail, less and less energy is generated within the cell. Cell injury and even cell death follow. If this process is repeated throughout the body, whole organ systems begin to fail – people get sick, and even die. The parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Chemical energy produced by the mitochondria is stored in a small molecule called adenosine triphosphate (ATP). Mitochondria contain their own small chromosomes. Generally, mitochondria, and therefore mitochondrial DNA, are inherited only from the mother. Problems with mitochondria, the structures that produce energy for all cells, have been linked to the development of Parkinson’s disease.
Mitochondria play a fundamental role in cell physiology; mitochondria organelles are involved in a variety of processes, including bioenergetics, various metabolic pathways, including crucial anabolic and catabolic reactions, such as ATP (adenosine triphosphate) synthesis, the tricarboxylic acid cycle (citric acid cycle or Kreb cycle), and biosynthetic processes, and govern fundamental cellular actions, including proliferation, immunity, and autophagy. Mitochondrial damage and malfunction have been related to the pathogenesis of a large number of human pathologies, such as mitochondrial diseases, neurodegenerative diseases, cancer, cardiovascular diseases, metabolic disorders, and aging. The participation of mitochondria in the redox equilibrium and redox signaling of the cell is also pivotal. Modification of the redox state and increased reactive oxygen species (ROS) production within mitochondria have major consequences for both mitochondrial and extramitochondrial processes and, ultimately, modulate fundamental cellular phenomena such as autophagy and apoptosis.
In people with mitochondrial disease, the parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected 30. Based upon recent epidemiological studies, mitochondrial disorders affect at least 1 in 8000 of the general population 31. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
What is mitochondrial disease?
Mitochondrial diseases can affect almost any part of your body, including the cells of your:
- Brain.
- Nerves.
- Muscles.
- Kidneys.
- Heart.
- Liver.
- Eyes.
- Ears.
- Pancreas.
Figure 6. Mitochondrial diseases
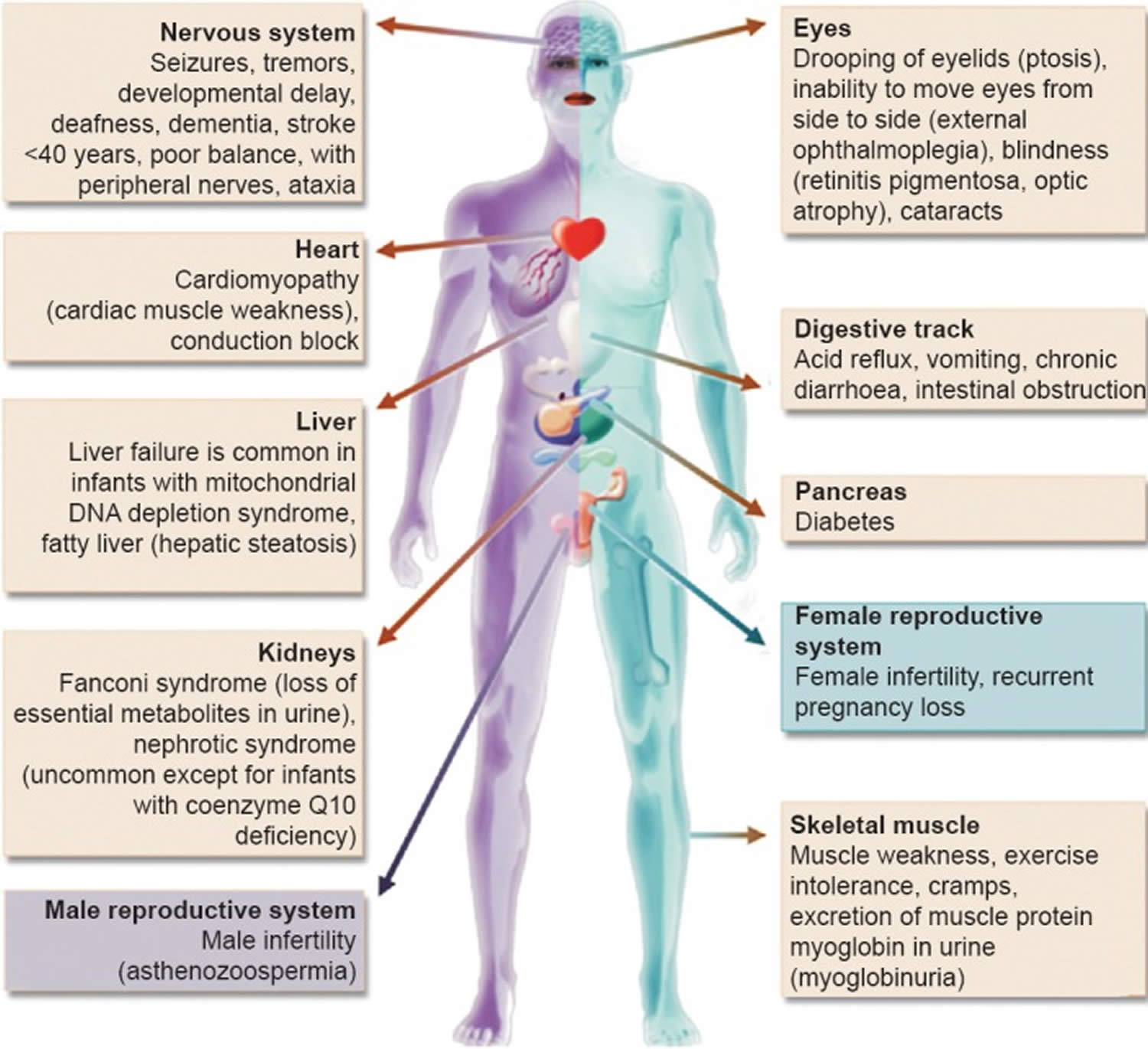
Footnote: Clinical features and the organs affected by mitochondrial diseases.
[Source 32 ]Figure 7. Mitochondrial diseases signs and symptoms
Are mitochondrial diseases difficult to diagnose?
Yes. Because mitochondrial diseases affect so many different organs and tissues of your body, and you may have many different symptoms, mitochondrial diseases can be difficult to diagnose. There’s no single laboratory test that can diagnose a mitochondrial disease. This is why a referral to a medical facility with physicians who specialize in these diseases is critical to making the diagnosis.
What causes POLG
POLG disease is caused by inherited mutations in the POLG gene 4, 6, 2. The POLG gene provides instructions for making the active piece, called the alpha subunit, of a protein called polymerase gamma (pol γ). To be most effective, the alpha subunit attaches to two copies of another protein called the beta subunit to form polymerase gamma (pol γ). Polymerase gamma (pol γ) is a DNA polymerase, which is a type of enzyme that “reads” sequences of your DNA and uses them as templates to produce new DNA. DNA polymerases are important for copying (replicating) cells’ genetic material. DNA polymerases also play critical roles in DNA repair.
Polymerase gamma (pol γ) functions in your mitochondria and is responsible for mitochondrial DNA (mtDNA) maintenance 12, 14. Mitochondria are structures within your cells, often called the “powerhouses” of the cell, in which a process called oxidative phosphorylation converts the energy from food you eat into a form called ATP (adenosine triphosphate) that your cells can use 15. Mitochondria use oxygen to break down glucose and other nutrients, releasing energy that is then stored in ATP. Your mitochondria also contain their own DNA known as mitochondrial DNA (mtDNA), which is essential for the normal function of these structures and is different from the DNA in your other cells nucleus. Polymerase gamma (pol γ) is the only DNA polymerase that is active in mitochondria and that can replicate mitochondrial DNA (mtDNA). This mitochondrial DNA (mtDNA) encodes for some proteins and RNAs necessary for mitochondrial function. Mutations in POLG gene compromise the functioning of the polymerase gamma (pol γ) enzyme responsible for mitochondrial DNA (mtDNA) maintenance. Poor DNA maintenance reduces the amount of mitochondrial DNA (mtDNA) present in the cell and/or introduces mutations in the mitochondrial DNA (mtDNA) 16.
Beyond energy production, your mitochondria also play other crucial roles, including regulating cell death (apoptosis) and contributing to the production of various molecules like proteins, DNA, RNA, lipids, hormones and neurotransmitters and calcium storage reservoirs, which are important for various cellular functions like muscle contraction and neurotransmitter release.
Currently, mutations in the POLG gene are known to cause over 200 disorders and they include 33, 4, 6, 5:
- Alpers-Huttenlocher syndrome also known as Alpers syndrome
- Alpers-Huttenlocher syndrome is characterized by seizures, loss of mental (dementia) and movement abilities (psychomotor regression), spasticity, blindness, and liver disease 17. The liver disease in Alpers-Huttenlocher syndrome can be brought on or made worse by valproic acid and sodium divalproate (divalproex), a common treatment for seizures 18.
- Alpers-Huttenlocher syndrome typically becomes apparent in children between ages 2 and 4. People with Alpers-Huttenlocher syndrome usually have 3 characteristic features: recurrent seizures that do not improve with treatment (intractable epilepsy), loss of mental and movement abilities (psychomotor regression), and liver disease 17.
- People with Alpers-Huttenlocher syndrome usually have additional signs and symptoms. Most have problems with coordination and balance (ataxia) and disturbances in nerve function (neuropathy). Neuropathy (nerve damage) can lead to abnormal or absent reflexes (areflexia). In addition, affected individuals may develop weak muscle tone (hypotonia) that worsens until they lose the ability to control their muscles and movement. Some people with Alpers-Huttenlocher syndrome lose the ability to walk, sit, or feed themselves. Other movement-related symptoms in affected individuals can include involuntary muscle twitches (myoclonus), uncontrollable movements of the limbs (choreoathetosis), or a pattern of movement abnormalities known as parkinsonism.
- People with Alpers-Huttenlocher syndrome may also have other brain-related signs and symptoms. Migraine headaches, often with visual sensations or auras, are common. Additionally, people with Alpers-Huttenlocher syndrome may have decreased brain function that is demonstrated as sleepiness, inability to concentrate, irritability, or loss of language skills or memory. Some people with the condition may lose their eyesight or hearing. People with Alpers-Huttenlocher syndrome can survive from a few months to more than 10 years after the condition first appears.
- The most common POLG gene mutation in Alpers-Huttenlocher syndrome replaces the amino acid alanine with the amino acid threonine at position 467 written as Ala467Thr or A467T 5. This POLG gene Ala467Thr mutation blocks the ability of the alpha subunit to attach to the beta subunits and reduces polymerase gamma (pol γ) ability to synthesize DNA. The Ala467Thr variant is also common in other POLG-related disorders. The different conditions may be determined, in part, by the variant in the other copy of POLG, but there are still some variant combinations that can cause more than one of the disorders. It is unclear how the same variant can lead to different conditions. Although the mechanism is unknown, many people with Alpers-Huttenlocher syndrome have fewer copies of mtDNA (mtDNA depletion). This abnormality is seen only in the tissues affected by the disease. MtDNA depletion leads to impaired oxidative phosphorylation and a decrease in cellular energy. These impairments affect tissues whose cells do not divide continually, such as brain, muscle, and liver. These tissues are most affected because they are more dependent on oxidative phosphorylation for energy, and impaired cells in these tissues are not generally replaced by new cells. The lack of energy supplies in these tissues could account for the signs and symptoms of Alpers-Huttenlocher syndrome.
- Alpers-Huttenlocher syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
- Alpers-Huttenlocher syndrome treatment is limited to symptom management and supportive care and family education should be addressed as soon as the family is able to absorb the diagnosis.
- Childhood Myocerebrohepatopathy Syndrome (MCHS)
- Childhood Myocerebrohepatopathy Syndrome (MCHS) is a severe usually fatal POLG-related disorder that affects the muscles (myo-), brain (cerebro-), and liver (hepato-) 19. Childhood Myocerebrohepatopathy Syndrome (MCHS) key 3 symptoms are myopathy (muscle diseases that affect skeletal muscles that control voluntary movements in the body) or low muscle tone (hypotonia), developmental delay or severe brain dysfunction (encephalopathy), and liver dysfunction. Childhood Myocerebrohepatopathy Syndrome (MCHS) typically presents in the first few months to 3 years of life, characterized by muscle weakness, developmental delay, and liver dysfunction. Other signs and symptoms of Childhood Myocerebrohepatopathy Syndrome (MCHS) include toxic buildup of lactic acid in the body (lactic acidosis), unable to gain weight and grow at the expected rate (failure to thrive), a form of kidney disease called renal tubular acidosis, inflammation of the pancreas (pancreatitis), recurrent episodes of nausea and vomiting (cyclic vomiting) and hearing loss 19. Seizures occur in about 75% of affected individuals. Liver failure typically occurs before the age of 1 year, and the condition is usually fatal with a median age of death in one study of 15.8 months (range: 1 to 184.6 months) 4. Major causes of death include liver failure, sepsis, and status epilepticus (abnormally prolonged seizure or a series of seizures without a period of recovery between them) 20, 21, 12.
- While Childhood Myocerebrohepatopathy Syndrome (MCHS) can present similarly to Alpers Syndrome or Alpers-Huttenlocher syndrome, which also involves liver failure and developmental delay, Childhood Myocerebrohepatopathy Syndrome (MCHS) generally lacks the specific liver histopathological features seen in Alpers-Huttenlocher syndrome. Additionally, Childhood Myocerebrohepatopathy Syndrome (MCHS) seizures are less common and severe, and the liver failure typically occurs before the age of 1 year 22.
- Many variants in the POLG gene can cause Childhood Myocerebrohepatopathy Syndrome (MCHS). Most of these POLG gene variants change single amino acids in the alpha subunit of polymerase gamma (pol γ). These variants reduce the activity of polymerase gamma (pol γ), decreasing mtDNA replication. As in other POLG-related disorders, people with Childhood Myocerebrohepatopathy Syndrome (MCHS) typically have mtDNA depletion in muscle, brain, or liver tissue. MtDNA depletion impairs oxidative phosphorylation in these tissues and decreases the energy available to the cells, which may cause the signs and symptoms of Childhood Myocerebrohepatopathy Syndrome (MCHS).
- Childhood Myocerebrohepatopathy Syndrome (MCHS) is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
- Childhood myocerebrohepatopathy syndrome (MCHS) treatment focuses on managing symptoms like seizures, lactic acidosis, and liver failure and providing supportive care.
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) also known as spinocerebellar ataxia with epilepsy (SCAE)
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) is a genetic disorder characterized by ataxia (lack of coordination), myoclonus (muscle jerks), and myopathy (muscle weakness), as well as seizures 23.
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) signs and symptoms typically appear during young adulthood. The first symptom of MEMSA is usually cerebellar ataxia, which refers to problems with coordination and balance due to defects in the part of the brain that is involved in coordinating movement (cerebellum). Recurrent seizures (epilepsy) usually develop later, often in combination with uncontrollable muscle jerks (myoclonus). The seizures usually begin in the right arm and spread to become generalized throughout the body. Additionally, affected individuals may have severe brain dysfunction (encephalopathy) or muscle weakness (myopathy). The myopathy can affect muscles close to the center of the body (proximal), such as the muscles of the hips, thighs, upper arms, or neck, or muscles farther away from the center of the body (distal), such as the muscles of the hands or feet. The myopathy may be especially noticeable during exercise (exercise intolerance).
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) is primarily caused by mutations in the POLG1 gene. This gene is involved in the replication of mitochondrial DNA (mtDNA). Although the mechanism is unknown, mutations in the POLG gene often result in fewer copies of mtDNA (mtDNA depletion), particularly in muscle, brain, or liver cells. MtDNA depletion causes a decrease in cellular energy, which could account for the signs and symptoms of MEMSA.
- Myoclonic epilepsy myopathy sensory ataxia (MEMSA) is inherited in an autosomal recessive pattern, meaning both copies of the gene in each cell must have mutations for the disorder to manifest. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
- Treatment for MEMSA (myoclonic epilepsy, myopathy, and sensory ataxia) focuses on managing symptoms, addressing complications, and improving quality of life. Specifically, anticonvulsants like levetiracetam, valproic acid, and zonisamide can be used to control myoclonic seizures. For mitochondrial myopathies, CoQ10 supplementation may be helpful in some cases, and exercise programs can improve both biochemical and clinical outcomes.
- Ataxia neuropathy spectrum (ANS)
- Ataxia neuropathy spectrum (ANS) encompasses a range of disorders characterized by both problems with coordination and balance (ataxia) and neuropathy (nerve damage) 24. As the name implies, people with ataxia neuropathy spectrum typically have problems with coordination and balance (ataxia) and disturbances in nerve function (neuropathy). The neuropathy (nerve damage) can be classified as sensory, motor, or a combination of the two (mixed). Sensory neuropathy (sensory nerve damage) causes numbness, tingling, or pain in the arms and legs, and motor neuropathy (motor nerve damage) refers to disturbance in the nerves used for muscle movement.
- Most people with ataxia neuropathy spectrum also have severe brain dysfunction (encephalopathy) and seizures. Some affected individuals have weakness of the external muscles of the eye (ophthalmoplegia), which leads to drooping eyelids (ptosis). Other signs and symptoms can include involuntary muscle twitches (myoclonus), liver disease, depression, migraine headaches, or blindness.
- Ataxia neuropathy spectrum now includes the conditions previously called mitochondrial recessive ataxia syndrome (MIRAS) and sensory ataxia neuropathy dysarthria and ophthalmoplegia (SANDO).
- The most common POLG gene variant in ataxia neuropathy spectrum is the same as that in Alpers-Huttenlocher syndrome (described above), Ala467Thr. It is unclear how the same Ala467Thr mutation can lead to different disorders. Rarely, ataxia neuropathy spectrum (ANS) is caused by the TWNK gene. The TWNK gene provides instructions for making a protein called Twinkle. Mutated polymerase gamma (pol γ) or mutated Twinkle reduce mtDNA replication. Although the mechanisms are unknown, mutations in the POLG gene often result in fewer copies of mtDNA (mtDNA depletion), and mutations in the TWNK gene often result in deletions of large regions of mtDNA (mtDNA deletion). MtDNA depletion or deletion occurs most commonly in muscle, brain, or liver cells. MtDNA depletion causes a decrease in cellular energy, which could account for the signs and symptoms of ataxia neuropathy spectrum. It is unclear what role mtDNA deletions play in the signs and symptoms of the condition.
- Mutations in the POLG gene cause a form of the condition that is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
- Mutations in the TWNK gene cause a form of the condition that is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
- As in other POLG-related disorders, people with ataxia neuropathy spectrum typically have mtDNA depletion in the tissues affected by the condition, such as the brain. MtDNA depletion decreases the amount of energy available to the cell due to reduced oxidative phosphorylation, which may account for the signs and symptoms of ataxia neuropathy spectrum.
- Ataxia neuropathy spectrum (ANS) treatment focuses on addressing the symptoms and improving quality of life, which may include physical therapy, occupational therapy, speech therapy, and medication to manage specific symptoms.
- Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO)
- Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) is a very rare mitochondrial disease that is part of the ataxia neuropathy spectrum (ANS) typically associated with mutations in the POLG1 gene. Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) primarily affects the brain, muscles, nerves, and eyes 25, 4. Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) usually presents during adulthood; the mean age of onset is 32 years, although it can also be found in younger patients 26. The symptoms caused by sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) and their severity vary widely among individuals with the disorder. While the disorder worsens over time, its progression is slower and milder than other related mitochondrial disorders 4. There is no cure for SANDO. Available treatments focus on symptom management and improving an individual’s quality of life.
- Mitochondrial recessive ataxia syndrome (MIRAS)
- Mitochondrial recessive ataxia syndrome (MIRAS) is a rare mitochondrial genetic disorder characterized by progressive loss of muscle coordination and balance (ataxia), along with other neurological symptoms like seizures, hearing loss, and cognitive decline. It’s caused by mutations in the POLG1 gene, which is responsible for maintaining and repairing mitochondrial DNA (mtDNA). Mitochondrial recessive ataxia syndrome (MIRAS) is an autosomal recessive condition, meaning individuals need to inherit two copies of the mutated POLG1 gene, one from each parent, to develop the disorder.
- MIRAS can present with a variety of symptoms, including:
- Ataxia: Progressive loss of coordination and balance, affecting gait, fine motor skills, and speech.
- Neuropathy (nerve damage): Sensory or mixed sensory-motor neuropathy, causing numbness, tingling, or pain in the limbs.
- Epilepsy: Seizures can occur at any stage of the disease.
- Ophthalmoplegia: Progressive weakness or paralysis of the eye muscles, leading to double vision or drooping eyelids.
- Dysarthria: Difficulty with speech due to muscle weakness in the mouth and tongue.
- Cognitive impairment: Intellectual disability, memory problems, or cognitive regression can occur.
- Movement disorders: Involuntary movements like myoclonus (sudden, brief muscle spasms) can be present.
- Other symptoms: Fatigue, hearing loss, psychiatric symptoms, and liver disease are also possible.
- There is no cure for mitochondrial recessive ataxia syndrome (MIRAS), and treatment focuses on managing symptoms and improving quality of life.
- The prognosis can vary, and some individuals with mitochondrial recessive ataxia syndrome (MIRAS) live fairly normal lives, while others may experience more severe complications.
- Progressive external ophthalmoplegia (PEO and PEO+) also called chronic progressive external ophthalmoplegia (CPEO)
- Progressive external ophthalmoplegia (PEO) is a condition characterized by weakness or paralysis of the eye muscles that control eye movement and eyelids, leading to drooping eyelids (ptosis) and difficulty moving the eyes 27. Progressive external ophthalmoplegia (PEO) typically progresses slowly over time, with symptoms worsening gradually.
- Progressive external ophthalmoplegia (PEO) typically appears in adults between ages 18 and 40 and slowly worsens over time. The first sign of progressive external ophthalmoplegia is typically drooping eyelids (ptosis), which can affect one or both eyelids. As drooping eyelid (ptosis) worsens, affected individuals may use the forehead muscles to try to lift the eyelids, or they may lift up their chin in order to see. Another characteristic feature of progressive external ophthalmoplegia is weakness or paralysis of the muscles that move the eye (ophthalmoplegia). Affected individuals have to turn their head to see in different directions, especially as the ophthalmoplegia worsens. People with progressive external ophthalmoplegia may also have general weakness of the muscles used for movement (myopathy), particularly those in the neck, arms, or legs. The weakness may be especially noticeable during exercise (exercise intolerance). Muscle weakness may also cause difficulty swallowing (dysphagia).
- Although muscle weakness is the primary symptom of progressive external ophthalmoplegia (PEO), this condition can be accompanied by other signs and symptoms. In these instances, the condition is referred to as progressive external ophthalmoplegia plus (PEO+). Additional signs and symptoms can include hearing loss caused by nerve damage in the inner ear (sensorineural hearing loss), weakness and loss of sensation in the limbs due to nerve damage (neuropathy), impaired muscle coordination (ataxia), a pattern of movement abnormalities known as parkinsonism, and depression.
- Progressive external ophthalmoplegia (PEO) can result from mutations in one of several different genes. In some cases, mutations in the POLG, TWNK, RRM2B, and SLC25A4 genes. These genes are critical for the production and maintenance of mtDNA. Less commonly, mutations that change single nucleotides in genes found in mtDNA, such as the MT-TL1 gene, cause progressive external ophthalmoplegia. These mutations occur in genes that provide instructions for making molecules called transfer RNAs. Transfer RNAs help assemble protein building blocks (amino acids) into functioning proteins. The transfer RNAs associated with progressive external ophthalmoplegia are present in mitochondria and help assemble the proteins that carry out the steps of oxidative phosphorylation.
- There are at least 67 POLG gene variants that cause progressive external ophthalmoplegia (PEO). Most POLG gene variants change single amino acids in the alpha subunit of polymerase gamma (pol γ), which decreases the efficiency of mitochondrial DNA (mtDNA) replication. As in another POLG-related disorder, Alpers-Huttenlocher syndrome (described above), the most common POLG gene variant in progressive external ophthalmoplegia (PEO) is Ala467Thr. It is unclear how the same variants can lead to different disorders. Researchers have not determined how deletions of mtDNA lead to the specific signs and symptoms of progressive external ophthalmoplegia (PEO), although the features of the condition are probably related to impaired oxidative phosphorylation 5. It has been suggested that eye muscles are commonly affected by mitochondrial defects because they are especially dependent on oxidative phosphorylation for energy.
- Progressive external ophthalmoplegia can have different inheritance patterns depending on the gene involved.
- When the nuclear genes POLG, TWNK, RRM2B, or SLC25A4 are involved, progressive external ophthalmoplegia is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. This progressive external ophthalmoplegia (PEO) is better known as Autosomal Recessive Progressive External Ophthalmoplegia (arPEO).
- Certain mutations in the POLG or RRM2B gene can also cause a form of progressive external ophthalmoplegia (PEO) that is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. This progressive external ophthalmoplegia (PEO) is better known as Autosomal Dominant Progressive External Ophthalmoplegia (adPEO).
- When progressive external ophthalmoplegia (PEO) is caused by mutations in the MT-TL1 gene and other mitochondrial transfer RNA genes, it is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA mutations only from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
- Single, large deletions of mtDNA are typically not inherited but occur during the formation of a mother’s egg cells or in early development of the embryo. Individuals with these mutations usually have no history of the disorder in their family.
- There is no cure for progressive external ophthalmoplegia (PEO), but treatment focuses on managing symptoms and improving quality of life. Progressive external ophthalmoplegia (PEO) treatment may include physical therapy, medications to address specific symptoms like ptosis, and support for other affected organ systems.
POLG gene mutations have been shown to be associated with Charcot-Marie-Tooth 34, 35, Leigh syndrome (a rare inherited neurodegenerative disorder that typically manifests in infancy, characterized by progressive loss of neurological function) 36, 37, and a mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like illness (a rare, autosomal recessive mitochondrial disease characterized by progressive gastrointestinal dysmotility, cachexia, ptosis/ophthalmoplegia, leukoencephalopathy, and peripheral neuropathy) 38.
Little is known about what exactly causes the clinical appearance of POLG disease and what external factors contribute to the development of symptoms and their severity.
POLG signs and symptoms
The primary symptoms of POLG disease depend on the age the signs and symptoms first emerge 6.
For individuals with POLG Early-Onset disease prior to age 12 years, the main symptoms are 6:
- Seizures
- Cognitive regression (intellectual disability)
- Motor impairment
- Cortical visual loss
- Feeding difficulties
- Liver dysfunction
- This group includes Alpers-Huttenlocher syndrome and Childhood Myocerebrohepatopathy Syndrome (MCHS) and is associated with the worst prognosis.
For individuals with Juvenile or Adult-Onset POLG disease between age 12 and 40, the primary symptoms are 6:
- Seizures
- Impaired balance and coordination (ataxia)
- Peripheral nerves damaged (peripheral neuropathy)
- This group includes myoclonic epilepsy myopathy sensory ataxia (MEMSA) or spinocerebellar ataxia with epilepsy (SCAE) and ataxia neuropathy spectrum (ANS) and generally has a better prognosis than the early-onset disease group.
For individuals with Late-Onset POLG disease with onset after age 40 years, the key symptoms are 6:
- Drooping eyelids (ptosis)
- Paralysis of the muscles that control eye movement also called progressive external ophthalmoplegia (PEO)
- Impaired coordination (ataxia)
- Myopathy
- Parkinsonism
- This group includes autosomal recessive progressive external ophthalmoplegia (arPEO), autosomal dominant progressive external ophthalmoplegia (adPEO), and progressive external ophthalmoplegia plus (PEO+) and has the best prognosis overall.
POLG disease can also affect almost any organ. As such, symptoms may also include 4:
- Poor muscle tone (hypotonia)
- Developmental delay
- Movement disorder
- Weakness of the limbs
- Depression
- Anxiety
- Headache
- Hearing loss
- Vision loss
- Slurred speech (dysarthria)
- Respiratory failure
- Temporary cessation of breathing (apnea).
Alpers-Huttenlocher Syndrome
Alpers-Huttenlocher Syndrome also known as Alpers syndrome, one of the most severe phenotypic manifestations in the POLG-related spectrum, is characterized by progressive and severe encephalopathy with recurrent seizures that do not improve with treatment (intractable epilepsy), loss of mental and movement abilities (psychomotor regression), and liver failure 17. The liver disease in Alpers-Huttenlocher syndrome can be brought on or made worse by valproic acid and sodium divalproate (divalproex), a common treatment for seizures 18. Alpers-Huttenlocher syndrome typically becomes apparent in children between ages 2 and 4. Onset is usually between ages two and four years but ranges overall from one month to 36 years. People with Alpers-Huttenlocher syndrome can survive from a few months to more than 10 years after the condition first appears. The typical life expectancy from onset of first symptoms ranges from three months to 12 years. Alpers-Huttenlocher syndrome treatment is limited to symptom management and supportive care and family education should be addressed as soon as the family is able to absorb the diagnosis.
Seizures are the first sign of Alpers-Huttenlocher Syndrome in about 50% of affected children. Seizures may be simple focal, primary generalized, or myoclonic. The most common early seizure types are partial seizures and secondary generalized tonic-clonic seizures. In some children, the first seizure presents with status epilepticus. EEG findings include high-amplitude slow activity with smaller polyspikes or intermittent continuous spike-wave activity 39.
In some instances, the initial seizure type is epilepsia partialis continua (EPC), a classic motor seizure type that involves only one portion of the body (e.g., a limb) with constant and repetitive myoclonic jerking, continuing for hours or days with or without dramatic effects on consciousness. Epilepsia partialis continua (EPC) is not always apparent as an abnormality on EEG and can be mistaken for a conversion reaction. EEG may be normal or show only focal slowing of the background rhythm.
Over time, seizures can evolve into a complex epileptic disorder such as focal status epilepticus, epilepsia partialis continua (EPC), or multifocal myoclonic epilepsy 40, 41, 39.
In some children, seizures are initially controllable with standard dosages of anti-seizure medications (anti-epileptic medications); in others, seizures, such as epilepsia partialis continua (EPC), are refractory from the onset. Over time, seizures become increasingly resistant to anti-seizure medications.
People with Alpers-Huttenlocher syndrome usually have additional signs and symptoms. Most have problems with coordination and balance (ataxia) and disturbances in nerve function (neuropathy). Neuropathy (nerve damage) can lead to abnormal or absent reflexes (areflexia). In addition, affected individuals may develop weak muscle tone (hypotonia) that worsens until they lose the ability to control their muscles and movement. Some people with Alpers-Huttenlocher syndrome lose the ability to walk, sit, or feed themselves. Other movement-related symptoms in affected individuals can include involuntary muscle twitches (myoclonus), uncontrollable movements of the limbs (choreoathetosis), or a pattern of movement abnormalities known as parkinsonism 40. Alpers-Huttenlocher syndrome parkinsonism may temporarily respond to levodopa 42, 43.
People with Alpers-Huttenlocher syndrome may also have other brain-related signs and symptoms. Migraine headaches, often with visual sensations or visual auras that reflect early occipital lobe dysfunction, are common 44, 41, 6. Additionally, people with Alpers-Huttenlocher syndrome may have decreased brain function that is demonstrated as sleepiness, inability to concentrate, irritability, or loss of language skills or memory. Some people with the condition may lose their eyesight or hearing. Stroke and stroke-like episodes may occur as well 40.
Neuropathy and ataxia develop in all persons with Alpers-Huttenlocher Syndrome unless the disease process is so rapid that it results in early death. All neurologic signs and symptoms, including ataxia and nystagmus, may worsen during infections or with other physiologic stressors.
Areflexia (resulting from neuropathy) and hypotonia (possibly the result of generalized weakness as part of systemic illness or pyramidal or extrapyramidal dysfunction) are often both present early in the disease course.
Episodic psychomotor regression is variably present at the time of initial consideration of the diagnosis. The major motor manifestation is a progressive spastic paraparesis resulting from progressive loss of cortical neuronal function. Progressive spasticity occurs universally, has variable onset, and evolves over months to years.
Loss of cognitive function occurs throughout the course of the disease, but the time of onset and rate of progression are variable. Significant sudden or rapid regression is often seen during infectious illnesses. The clinical manifestations may include somnolence, loss of concentration, loss of language skills (both receptive and expressive), irritability with loss of normal emotional responses, and memory deficits. In addition to cognitive impairment caused by refractory epilepsy, high dosages of anti-seizure medications (anti-epileptic medications) can lead to significant cognitive dysfunction. Therefore, the degree of cognitive dysfunction is often difficult to assess due to frequent seizures and high therapeutic doses of anti-seizure medications (anti-epileptic medications).
Vision loss leading to blindness may appear months to years after the onset of other neurologic manifestations. Retinopathy may also play a less important role in vision loss 44, 6. Hearing loss is variable 44, 40.
Liver involvement can progress rapidly to end-stage liver failure within a few months, although this is highly variable. End-stage liver disease is often heralded by hypoalbuminemia and prolonged coagulation time, followed shortly thereafter by fasting hypoglycemia and hyperammonemia. Rapid-onset liver failure has been described when valproic acid (Depakene®) and sodium divalproate (divalproex) (Depakote®) have been used to treat seizures, although the introduction of other anti-seizure medications (anti-epileptic medications), including phenytoin, may also play a role in onset of hepatic failure.
While Alpers-Huttenlocher Syndrome is usually fatal, age of onset, rate of neurologic deterioration, presence of liver failure, and age of death vary among affected individuals 45, 46, 20, 47, 48, 6. Children with Alpers-Huttenlocher Syndrome appear healthy at birth and may develop normally over the first few weeks to years of life. Some have variable degrees of developmental delay prior to the initial recognition of neurodegeneration.
Alpers-Huttenlocher Syndrome disease progression is variable in timing and rapidity. The rate of neurodegeneration varies and is marked by periods of stability. Loss of neurologic function results in dementia, spastic quadriparesis from corticospinal tract involvement, visual loss, and death.
Neuroimaging
Brain CT or MRI may be normal early in the course of Alpers-Huttenlocher Syndrome . As the illness evolves, neuroimaging shows gliosis initially more pronounced in the occipital lobe regions and generalized cortical atrophy. Restricted diffusion unilaterally in the pulvinar and occipital region is described in the acute phase. FLAIR and T2-weighted sequence images demonstrate high signal intensity in deep gray matter nuclei, especially in the thalamus and cerebellum 49. Progressive cerebellar atrophy can occur in addition to cortical atrophy. The pons, midbrain, and globus pallidum can also be involved. Lesions described in the inferior olivary nuclei may also be a part of Alpers-Huttenlocher Syndrome and are associated with palatal myoclonus. Brain magnetic resonance spectroscopy (MRS) typically shows reduced N-acetylaspartate, normal creatine, and lactate.
Histopathologic abnormalities
- Brain. The gross appearance of the brain varies from normal to severe atrophy, depending on the state of disease progression. Central nervous system regions affected in Alpers-Huttenlocher Syndrome are the same as those affected by Leigh syndrome but typically evolve in the reverse order. For example, in Alpers-Huttenlocher Syndrome , gliosis is most severe and occurs earliest in the cerebral cortex, followed by the cerebellum, basal ganglia, and brain stem. Involved regions demonstrate neuronal degeneration, characteristic spongiform or microcystic degeneration, and – as seen in Leigh syndrome – gliosis, necrosis, and capillary proliferation. The cortical ribbon shows patchy lesions, but the calcarine cortex, which is characteristically involved early in the course of the disease, is usually narrowed, granular, and discolored. Microscopic abnormalities throughout the cerebral cortex evolve as the disease progresses. Early in the course of the disease, spongiosis, astrocytosis, and neuronal loss are prevalent in the superficial cortex. Later, the deeper laminae are affected. In the most advanced stages, the entire cortex becomes a thin dense gliotic scar. Usually, the striate cortex is the most affected part of the brain, followed by the thalamus, hippocampus, and cerebellum. These pathologic features differ from those resulting from hypoxic injury, recurrent seizures, or other causes of hepatic failure.
- Liver. Liver histology may demonstrate macro- and microvesicular steatosis, centrilobular necrosis, disorganization of the normal lobular architecture, hepatocyte loss with or without bridging fibrosis or cirrhosis, regenerative nodules, bile duct proliferation, or mitochondrial proliferation with a vivid eosinophilic cytoplasm. Florid cirrhosis occurs late in the disease. This pathology differs from that seen in chemically induced or toxic hepatopathies.
POLG gene mutation
The most common POLG gene mutation in Alpers-Huttenlocher syndrome replaces the amino acid alanine with the amino acid threonine at position 467 written as Ala467Thr or A467T 5. This POLG gene Ala467Thr mutation blocks the ability of the alpha subunit to attach to the beta subunits and reduces polymerase gamma (pol γ) ability to synthesize DNA. The Ala467Thr variant is also common in other POLG-related disorders. The different conditions may be determined, in part, by the variant in the other copy of POLG, but there are still some variant combinations that can cause more than one of the disorders. It is unclear how the same variant can lead to different conditions. Although the mechanism is unknown, many people with Alpers-Huttenlocher syndrome have fewer copies of mtDNA (mtDNA depletion). This abnormality is seen only in the tissues affected by the disease. MtDNA depletion leads to impaired oxidative phosphorylation and a decrease in cellular energy. These impairments affect tissues whose cells do not divide continually, such as brain, muscle, and liver. These tissues are most affected because they are more dependent on oxidative phosphorylation for energy, and impaired cells in these tissues are not generally replaced by new cells. The lack of energy supplies in these tissues could account for the signs and symptoms of Alpers-Huttenlocher syndrome.
Alpers-Huttenlocher syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Childhood Myocerebrohepatopathy Syndrome (MCHS)
Childhood Myocerebrohepatopathy Syndrome also called Childhood Myocerebrohepatopathy Spectrum (MCHS) is a severe usually fatal POLG-related disorder that affects the muscles (myo-), brain (cerebro-), and liver (hepato-) 19. Childhood Myocerebrohepatopathy Syndrome (MCHS) key 3 symptoms are muscle diseases that affect skeletal muscles that control voluntary movements in the body (myopathy) or low muscle tone (hypotonia), developmental delay or severe brain dysfunction (encephalopathy), and liver dysfunction. Childhood Myocerebrohepatopathy Syndrome (MCHS) typically presents in the first few months to 3 years of life (a median age of 4.7 months), characterized by muscle weakness (myopathy), or low muscle tone (hypotonia), developmental delay, and liver dysfunction. Other signs and symptoms of childhood myocerebrohepatopathy spectrum (MCHS) include toxic buildup of lactic acid in the body (lactic acidosis), unable to gain weight and grow at the expected rate (failure to thrive), a form of kidney disease called renal tubular acidosis, inflammation of the pancreas (pancreatitis), recurrent episodes of nausea and vomiting (cyclic vomiting) and hearing loss 19. Seizures occur in about 75% of affected individuals. Liver failure typically occurs before the age of 1 year, and the condition is usually fatal with a median age of death in one study of 15.8 months (range: 1 to 184.6 months) 4. Major causes of death include liver failure, sepsis, and status epilepticus (abnormally prolonged seizure or a series of seizures without a period of recovery between them) 20, 21, 12.
Childhood Myocerebrohepatopathy Syndrome (MCHS) can present very similarly to Alpers Syndrome or Alpers-Huttenlocher syndrome, which also involves liver failure and developmental delay. However, childhood myocerebrohepatopathy syndrome (MCHS) generally lacks the specific liver histopathological features seen in Alpers-Huttenlocher syndrome. Additionally, childhood myocerebrohepatopathy syndrome (MCHS) seizures are less common and severe, and the liver failure typically occurs before the age of 1 year 22.
Childhood myocerebrohepatopathy syndrome (MCHS) treatment focuses on managing symptoms like seizures, lactic acidosis, and liver failure and providing supportive care.
POLG gene mutation
Many variants in the POLG gene can cause Childhood Myocerebrohepatopathy Syndrome (MCHS). Most of these POLG gene variants change single amino acids in the alpha subunit of polymerase gamma (pol γ). These variants reduce the activity of polymerase gamma (pol γ), decreasing mtDNA replication. As in other POLG-related disorders, people with Childhood Myocerebrohepatopathy Syndrome (MCHS) typically have mtDNA depletion in muscle, brain, or liver tissue. MtDNA depletion impairs oxidative phosphorylation in these tissues and decreases the energy available to the cells, which may cause the signs and symptoms of Childhood Myocerebrohepatopathy Syndrome (MCHS).
Childhood Myocerebrohepatopathy Syndrome (MCHS) is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Ataxia Neuropathy Spectrum (ANS)
Ataxia neuropathy spectrum (ANS) encompasses a range of disorders characterized by both problems with coordination and balance (ataxia) and neuropathy (nerve damage) 24. As the name implies, people with ataxia neuropathy spectrum typically have problems with coordination and balance (ataxia) and disturbances in nerve function (neuropathy). The neuropathy (nerve damage) can be classified as sensory, motor, or a combination of the two (mixed). Sensory nerve damage (sensory neuropathy) causes numbness, tingling, or pain in the arms and legs, and can be severe enough to contribute to ataxia – so-called sensory ataxia and motor nerve damage (motor neuropathy) refers to disturbance in the nerves used for muscle movement. About 25% of affected individuals have cramps, but clinical myopathy is less common and, if present, is not the main component of issues pertaining to gait dysfunction or balance difficulties.
Most people but not all affected individuals with ataxia neuropathy spectrum also have severe brain dysfunction (encephalopathy) and seizures. The encephalopathy is similar to that seen in Alpers-Huttenlocher syndrome but tends to be more slowly progressive and can even be mild. Some affected individuals have weakness of the external muscles of the eye (ophthalmoplegia), which leads to drooping eyelids (ptosis). Other signs and symptoms can include involuntary muscle twitches (myoclonus), liver disease, depression, migraine headaches, or blindness 20. Liver disease range from no dysfunction, to elevated enzymes and mild synthetic dysfunction, to liver failure in some cases 20, 41. Psychiatric illness including depression is common. Headaches, generally migraines, are also common and may precede other symptoms by many years.
Ataxia neuropathy spectrum now includes the conditions previously called mitochondrial recessive ataxia syndrome (MIRAS) and sensory ataxia neuropathy dysarthria and ophthalmoplegia (SANDO) 50.
As in other POLG-related disorders, people with ataxia neuropathy spectrum typically have mtDNA depletion in the tissues affected by the condition, such as the brain. MtDNA depletion decreases the amount of energy available to the cell due to reduced oxidative phosphorylation, which may account for the signs and symptoms of ataxia neuropathy spectrum. Although muscle pathology may show COX-negative fibers, there may be no pathologic findings.
Ataxia neuropathy spectrum (ANS) treatment focuses on addressing the symptoms and improving quality of life, which may include physical therapy, occupational therapy, speech therapy, and medication to manage specific symptoms.
POLG gene mutation
The most common POLG gene variant in ataxia neuropathy spectrum is the same as that in Alpers-Huttenlocher syndrome (described above), Ala467Thr. It is unclear how the same Ala467Thr mutation can lead to different disorders. Rarely, ataxia neuropathy spectrum (ANS) is caused by the TWNK gene. The TWNK gene provides instructions for making a protein called Twinkle. Mutated polymerase gamma (pol γ) or mutated Twinkle reduce mtDNA replication. Although the mechanisms are unknown, mutations in the POLG gene often result in fewer copies of mtDNA (mtDNA depletion), and mutations in the TWNK gene often result in deletions of large regions of mtDNA (mtDNA deletion). MtDNA depletion or deletion occurs most commonly in muscle, brain, or liver cells. MtDNA depletion causes a decrease in cellular energy, which could account for the signs and symptoms of ataxia neuropathy spectrum. It is unclear what role mtDNA deletions play in the signs and symptoms of the condition.
Mutations in the POLG gene cause a form of the condition that is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Mutations in the TWNK gene cause a form of the condition that is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Progressive external ophthalmoplegia (PEO) or chronic progressive external ophthalmoplegia (CPEO)
Progressive external ophthalmoplegia (PEO) is a condition characterized by weakness or paralysis of the eye muscles that control eye movement and eyelids, leading to drooping eyelids (ptosis) and difficulty moving the eyes 27. Progressive external ophthalmoplegia (PEO) typically progresses slowly over time, with symptoms worsening gradually. Progressive external ophthalmoplegia (PEO) typically appears in adults between ages 18 and 40 and slowly worsens over time. The first sign of progressive external ophthalmoplegia is typically drooping eyelids (ptosis), which can affect one or both eyelids. As drooping eyelid (ptosis) worsens, affected individuals may use the forehead muscles to try to lift the eyelids, or they may lift up their chin in order to see. Another characteristic feature of progressive external ophthalmoplegia is weakness or paralysis of the muscles that move the eye (ophthalmoplegia). Affected individuals have to turn their head to see in different directions, especially as the ophthalmoplegia worsens. People with progressive external ophthalmoplegia may also have general weakness of the muscles used for movement (myopathy), particularly those in the neck, arms, or legs. The weakness may be especially noticeable during exercise (exercise intolerance). Muscle weakness may also cause difficulty swallowing (dysphagia).
Although muscle weakness is the primary symptom of progressive external ophthalmoplegia (PEO), this condition can be accompanied by other signs and symptoms. In these instances, the condition is referred to as progressive external ophthalmoplegia plus (PEO+). Additional signs and symptoms can include hearing loss caused by nerve damage in the inner ear (sensorineural hearing loss), weakness and loss of sensation in the limbs due to nerve damage (neuropathy), impaired muscle coordination (ataxia), a pattern of movement abnormalities known as parkinsonism, and depression.
There is no cure for progressive external ophthalmoplegia (PEO), but treatment focuses on managing symptoms and improving quality of life. Progressive external ophthalmoplegia (PEO) treatment may include physical therapy, medications to address specific symptoms like ptosis, and support for other affected organ systems.
Progressive external ophthalmoplegia gene mutation
Progressive external ophthalmoplegia (PEO) can result from mutations in one of several different genes. In some cases, mutations in the POLG, TWNK, RRM2B, and SLC25A4 genes. These genes are critical for the production and maintenance of mtDNA. Less commonly, mutations that change single nucleotides in genes found in mtDNA, such as the MT-TL1 gene, cause progressive external ophthalmoplegia. These mutations occur in genes that provide instructions for making molecules called transfer RNAs. Transfer RNAs help assemble protein building blocks (amino acids) into functioning proteins. The transfer RNAs associated with progressive external ophthalmoplegia are present in mitochondria and help assemble the proteins that carry out the steps of oxidative phosphorylation.
There are at least 67 POLG gene variants that cause progressive external ophthalmoplegia (PEO). Most POLG gene variants change single amino acids in the alpha subunit of polymerase gamma (pol γ), which decreases the efficiency of mitochondrial DNA (mtDNA) replication. As in another POLG-related disorder, Alpers-Huttenlocher syndrome (described above), the most common POLG gene variant in progressive external ophthalmoplegia (PEO) is Ala467Thr. It is unclear how the same variants can lead to different disorders. Researchers have not determined how deletions of mtDNA lead to the specific signs and symptoms of progressive external ophthalmoplegia (PEO), although the features of the condition are probably related to impaired oxidative phosphorylation 5. It has been suggested that eye muscles are commonly affected by mitochondrial defects because they are especially dependent on oxidative phosphorylation for energy.
Progressive external ophthalmoplegia inheritance pattern
Progressive external ophthalmoplegia can have different inheritance patterns depending on the gene involved.
- When the nuclear genes POLG, TWNK, RRM2B, or SLC25A4 are involved, progressive external ophthalmoplegia is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. This progressive external ophthalmoplegia (PEO) is better known as Autosomal Recessive Progressive External Ophthalmoplegia (arPEO).
- Certain mutations in the POLG or RRM2B gene can also cause a form of progressive external ophthalmoplegia (PEO) that is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. This progressive external ophthalmoplegia (PEO) is better known as Autosomal Dominant Progressive External Ophthalmoplegia (adPEO).
- When progressive external ophthalmoplegia (PEO) is caused by mutations in the MT-TL1 gene and other mitochondrial transfer RNA genes, it is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA mutations only from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
- Single, large deletions of mtDNA are typically not inherited but occur during the formation of a mother’s egg cells or in early development of the embryo. Individuals with these mutations usually have no history of the disorder in their family.
Autosomal Recessive Progressive External Ophthalmoplegia (arPEO)
Progressive PEO without systemic involvement is the hallmark of Autosomal Recessive Progressive External Ophthalmoplegia (arPEO). Caution needs to be exercised, however, when making the diagnosis of Autosomal Recessive Progressive External Ophthalmoplegia (arPEO), as some POLG genetic mutations associated with Autosomal Recessive Progressive External Ophthalmoplegia (arPEO) are also associated with ataxia neuropathy spectrum (ANS) and other POLG-related disorders with systemic involvement. Therefore, many individuals who have no other clinical findings at the time of diagnosis with isolated Autosomal Recessive Progressive External Ophthalmoplegia (arPEO) develop other signs and symptoms of POLG-related disorders over subsequent years or decades 51, 52, 53.
Autosomal Dominant Progressive External Ophthalmoplegia (adPEO)
The universal signs and symptoms of this adult-onset progressive external ophthalmoplegia (PEO) is progressive weakness of the extraocular eye muscles resulting in ptosis and strabismus 51. A generalized myopathy is present in most affected individuals, leading to early fatigue and exercise intolerance. Some affected individuals have variable degrees of sensorineural hearing loss, axonal neuropathy, ataxia, depression, parkinsonism, hypogonadism, and cataracts 42, 54. Cardiomyopathy (heart muscle disease) and gastrointestinal dysmotility are less common.
POLG diagnosis
A referral to a medical facility with physicians who specialize in POLG diseases is critical to making the diagnosis as diagnosis of mitochondrial disorders is still a major challenge 21.
To diagnose POLG your doctor may perform or recommend the following 4:
- Genetic testing for POLG gene mutation(s)
- Brain-imaging such as computed tomography (CT) scan or magnetic resonance imaging (MRI) to look for changes in the brain associated with POLG disease
- Electroencephalogram (EEG or ‘brain wave’) testing for seizures and related brain issues.
Because POLG disease is an inherited disorder, affected families should also receive genetic counseling.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
A POLG-related disorder should be suspected in individuals with combinations of the following clinical features and laboratory and neuroimaging findings.
Clinical features
Clinical features form a continuum but vary in their age of onset. Apart from progressive external ophthalmoplegia (PEO) or ptosis, other features could present at any time from infancy to adulthood. The most common clinical features by age of onset are 4:
- Prior to age 12 years (early-onset disease):
- Liver involvement
- Feeding difficulties
- Seizures
- Hypotonia and muscle weakness that can evolve into corticospinal tract dysfunction (spasticity and dystonia)
- Between age 12 and 40 years (juvenile/adult-onset disease):
- Ataxia
- Peripheral neuropathy
- Seizures
- Stroke-like episodes
- PEO (in individuals with longer survival)
- After age 40 years (late-onset disease):
- Ptosis
- PEO
- Ataxia
- Muscle weakness
- Other features
- Developmental delay, especially in childhood-onset disease
- Movement disorder (e.g., myoclonus, dysarthria, choreoathetosis, parkinsonism)
- Myopathy (e.g., proximal > distal limb weakness with fatigue and exercise intolerance)
- Episodic psychomotor regression
- Psychiatric illness (e.g., depression, mood disorder), more commonly reported in adult-onset phenotypes
- Endocrinopathy (e.g., premature ovarian failure)
Laboratory findings
- Elevated serum lactate in serum and cerebrospinal fluid (CSF) is common throughout the spectrum of phenotypes but is more common in early-onset disease. However, normal values do not eliminate the likelihood of a POLG-related disorder.
- CSF protein levels are generally elevated in individuals with Alpers-Huttenlocher syndrome and other POLG-related disorders, but absence of this finding does not exclude a POLG-related disorder.
- Evidence of liver dysfunction or failure can be present, which may occur following exposure to certain anti-seizure medications. This could result in elevation of liver enzymes (alanine transaminase [ALT], aspartate transaminase [AST], and gamma-glutamyl transferase [GGT]) as well as synthetic liver dysfunction, causing hypoglycemia, hyperammonemia, elevated glutamine, hyperbilirubinemia, prolonged bleeding times (international normalized ratio [INR], prothrombin time [PT], partial thromboplastin time [PTT]), hypoalbuminemia, and low cholesterol levels.
- Respiratory chain defect and/or a defect of mitochondrial DNA (mtDNA) (depletion or multiple deletions) can be present. This could result in respiratory chain dysfunction, identified by either enzymatic assays or polarographic assays. Depletion of mtDNA can be measured by comparing mtDNA to nuclear DNA content in an affected tissue (e.g., liver). Normal respiratory chain function or absence of mtDNA depletion does not rule out a POLG-related disorder.
- In muscle biopsy samples, ragged-red fibers, COX-negative fibers, excessive lipid deposits, and abnormal respiratory chain activities can be present. However, biochemical findings on muscle biopsy can be normal.
Neuroimaging features
- Brain computerized tomography (CT) or magnetic resonance imaging (MRI) may be normal early in the course of Alpers-Huttenlocher syndrome.
- As Alpers-Huttenlocher syndrome evolves, neuroimaging shows gliosis (initially more pronounced in occipital lobe regions) and generalized brain atrophy. These findings are also reported in some individuals with adult-onset POLG-related disorders.
- Cortical focal lesions manifesting as T2/FLAIR hyperintensities in cortical and subcortical areas can be seen. These findings are typical in Alpers-Huttenlocher syndrome but have also been reported in sensory ataxia neuropathy dysarthria and ophthalmoplegia (SANDO) and ataxia neuropathy spectrum (ANS) 55, 56.
Other diagnostic studies
- Abnormal EEG activity over the occipital lobes in individuals with epilepsy
- Abnormal nerve conduction studies (NCVs)
POLG treatment
There is no cure for POLG disease. POLG disease treatment varies significantly based on the specific type of condition and the signs and symptoms present in each person 28. Available treatments for POLG disorder focus on symptom management and quality of life. These may include medications for seizures (epilepsy); occupational, physical, and speech therapy; nutritional support; respiratory support; and standard treatment of liver failure, movement abnormalities, sleep disorders, vision, and hearing issues 4.
The following treatments can help manage the symptoms of POLG disease and improve life for affected individuals and their families 4:
- Anticonvulsant medications for seizures (although valproate must be avoided). The use of valproate, a common anticonvulsant medication, may result in irreversible liver failure.
- Pain medication and muscle relaxants for comfort
- Small frequent meals or a feeding tube for nutritional support may be helpful and the ketogenic diet is sometimes used to help control seizures
- Physical therapy for declining motor skills and muscle strength
- Surgery or special glasses to correct drooping eyelids
- Speech therapy for slurred speech
- Breathing tube and/or artificial ventilation for respiratory failure
- CPAP or BiPAP for temporary cessation of breathing (apnea)
- Physical and occupational therapy for motor involvement.
- The use of folinic acid should be considered 57.
- The use of levoarginine has been reported to be helpful in reducing the frequency and severity of the strokes associated with MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) syndrome, and can be considered for use in persons with POLG-related disorders, especially if deficiency in the plasma or cerebrospinal fluid arginine concentration is confirmed 58.
- The use of levocarnitine should be reserved for individuals with reduced free carnitine levels in the blood, and the levels should be monitored 59.
- Creatine monohydrate, coenzyme Q10, B vitamins, and antioxidants such as alpha-lipoic acid, vitamin E, and vitamin C have been used as mitochondrial supplements based on limited case reports and small series but with a lack of objective evidence based on randomized controlled trials. Use of all in POLG-related disorders is reasonable given the general lack of toxicity but is not mandatory 59, 60, 61, 62, 63.
- Vitamin and cofactor therapy with the intent to fortify mitochondrial function may be offered, yet there is insufficient evidence demonstrating objective benefit in cohorts of persons. There have not been formal studies of the use of these vitamins and cofactors in Alpers-Huttenlocher syndrome or other POLG-related disorders 64, 65, 59.
The use of other treatments for refractory epilepsy, such as corticotropin or prednisone, ketogenic diet, and intravenous immunoglobulin G (IVIg), are unproven in the treatment of Alpers-Huttenlocher syndrome 4.
Table 1. Recommended Evaluations Following Diagnosis of POLG Related Disorders
| System | Evaluation | Comment |
|---|---|---|
| Neurologic | Neurologic evaluation |
|
| Ataxia | Orthopedics / physical medicine & rehabilitation / physical therapy (PT) & occupational therapy (OT) eval | To include assessment of:
|
| Development | Developmental assessment |
|
| Neurobehavioral/ Psychiatric | Neuropsychiatric evaluation |
|
| Depression screen |
| |
| Musculoskeletal | Orthopedics / physical medicine & rehabilitation / physical therapy (PT) & occupational therapy (OT) evaluation | To include assessment of:
|
| Gastrointestinal/ Feeding | Gastroenterology / nutrition / feeding team evaluation |
|
| Eyes | Ophthalmologic evaluation |
|
| Hearing | Audiologic evaluation |
|
| ENT/Mouth | ENT evaluation |
|
| Cardiovascular | Cardiac eval |
|
| Respiratory | Pulmonary eval |
|
| Endocrine | Screen for pancreatic, thyroid, and adrenal function. |
|
| Pregnancy | High-risk obstetrical care |
|
| Genetic counseling | By genetics professionals 4 |
|
| Family support & resources | By clinicians, wider care team, and family support organizations | Assessment of family and social structure to determine need for:
|
Footnotes:
1 In some instances, the first neuroimaging study may be normal, but with certain phenotypes, such as Alpers-Huttenlocher syndrome, changes may be seen in a relatively short amount of time.
2 AST elevation, and to a lesser extent ALT elevation, may be due to muscle disease. Therefore, simultaneously obtaining a serum CK level helps differentiate between liver & muscle involvement, which can both be seen in individuals with POLG-related disorders.
3 Parikh et al 28
4 Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Table 2. POLG Treatment Options
| Manifestation/Concern | Treatment | Considerations/Other |
|---|---|---|
| Developmental delay / Intellectual disability / Neurobehavioral issues | See Developmental Delay / Intellectual Disability Management Issues below. | Occupational therapy (OT), physical therapy (PT), and speech therapy (ST) are indicated in Alpers-Huttenlocher syndrome to maintain or improve neurologic function for as long as possible and for comfort care. |
| Epilepsy | Standardized treatment w/ASM by experienced neurologist |
|
| Movement disorders | Standard treatment by an experienced neurologist |
|
| Spasticity | Orthopedics / physical medicine & rehab / physical therapy (PT) & occupational therapy (OT) incl stretching to help avoid contractures & falls |
|
| Inadequate weight gain & linear growth (children) or unexplained weight loss (all ages) | Feeding therapy Gastrostomy tube placement may be required for persistent feeding issues. |
|
| GI & bowel dysfunction | Standard treatment by GI specialist for feeding or nutritional issues |
|
| Monitor for constipation. |
| |
| Liver failure | Standard treatment |
|
| Eyes | Standardized treatment by ophthalmologist |
|
| Ptosis |
| |
| Low vision services |
| |
| Cerebral visual impairment | No specific treatment |
|
| Hearing | Hearing aids may be helpful per otolaryngologist |
|
| Respiratory | Standard treatment by pulmonologist for respiratory issues |
|
| Family/Community |
|
|
Developmental Delay and Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay or intellectual disability in the United States; standard recommendations may vary from country to country 4.
- Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
- Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
- All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- Individualized education plan (IEP) services:
- An individualized education plan (IEP) provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child’s IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child’s access to academic material. Beyond that, private supportive therapies based on the affected individual’s needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- Individualized education plan (IEP) services:
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. Developmental Disabilities Administration (DDA) is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction Management
Gross motor dysfunction
Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation). Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers). For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, Botox®, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction
Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues
Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An augmentative and alternative communication (AAC) evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. Augmentative and alternative communication (AAC) devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Neurobehavioral and Psychiatric Management
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). Applied behavior analysis (ABA) therapy is targeted to the individual child’s behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder (ADHD), when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Evaluations by a multidisciplinary team of health care providers based on clinical findings; routine evaluation of growth, nutrition, oral intake, and respiratory status; monitoring of liver enzymes every three months or as clinically indicated; monitoring of epilepsy with repeat liver function tests after introduction of any new anti-seizure medication 4.
Table 3. Recommended Surveillance
| System/Concern | Evaluation | Frequency |
|---|---|---|
| Neurologic |
| At each visit |
| Development |
| |
| Neurobehavioral/ Psychiatric |
| |
| Musculoskeletal |
| |
| Feeding |
| |
| Liver function & metabolic status |
| Every 3 months, or as clinically indicated |
| Annually or as clinically indicated | |
| ||
| Gastrointestinal |
| At each visit or as clinically indicated |
| Respiratory |
| At each visit |
| As clinically indicated or every 2-3 yers | |
| Auditory |
| As clinically indicated |
| Ophthalmologic involvement |
| Per treating ophthalmologist(s) |
| Per treating clinicians | |
| Cardiovascular |
| At each visit |
| Endocrine |
| Upon critical illness or as clinically indicated |
| Family/Community |
| At each visit |
Agents to avoid
Valproic acid (Depakene®) and sodium divalproate (divalproex) (Depakote®) because of the risk of precipitating and/or accelerating liver disease 18, 4.
Liver transplantation
Liver transplantation is not advised in children with Alpers-Huttenlocher syndrome because transplanting the liver does not alter the rapid progression of the brain disease 69. However, liver transplantation in adults who have an acceptable quality of life may be of benefit.
- In one report, one of two individuals undergoing liver transplantation survived 41. In another report of solid organ transplantation in primary mitochondrial disease, of six individuals with POLG-related disease, two survived without complications 70.
- In another report, a woman underwent liver transplantation at age 19 years, eight years after experiencing fulminant hepatic failure following onset of valproate therapy. Molecular genetic testing seven years after her liver transplantation confirmed the diagnosis of a POLG-related disorder; her phenotype fit best with sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) 71, 70.
POLG prognosis
Your doctor can give you the best advice on your life expectancy after a POLG disease diagnosis. Your prognosis (outlook) depends on the age your symptoms first appeared, your affected organs and your general health. The signs and symptoms of POLG disease can appear anywhere between infancy and late adulthood and can vary widely among affected individuals 4:
- In individuals with early-onset disease prior to age 12 years, liver involvement, feeding difficulties, seizures (epilepsy), low muscle tone (hypotonia), and muscle weakness are the most common clinical features. This group has the worst prognosis.
- In the juvenile or adult-onset form with onset between the age 12 to 40 years, disease is typically characterized by peripheral nerves damaged (peripheral neuropathy), impaired balance and coordination (ataxia), seizures, stroke-like episodes, and, in individuals with longer survival, progressive external ophthalmoplegia (PEO). Progressive external ophthalmoplegia (PEO) is a condition characterized by weakness of the eye muscles that control eye movement, leading to progressive inability to move the eyes and eyelids, often with drooping eyelids (ptosis). This group generally has a better prognosis than the early-onset group.
- Late-onset disease with onset after age 40 years is characterized by drooping eyelids (ptosis) and progressive external ophthalmoplegia (PEO), with additional features such as peripheral nerves damaged (peripheral neuropathy), impaired balance and coordination (ataxia), and muscle weakness. This group overall has the best prognosis.
Some affected children and adults have the same expected lifespan as someone who doesn’t have this condition. Others might experience drastic changes in their health over a very short period of time. Some may have occasional flare-ups of symptoms throughout their lives. Although there’s no cure for POLG-related disorders, research is ongoing to learn more.
In a survival analysis involving 155 patients, 61 were alive at the time of data analysis and one had been lost to follow-up. Median age at death was 7.4 years (range 1 month to 91 years) 1. The main cause of death was liver failure (32%, n = 30/93), followed by infection/sepsis (20%, n = 19/93), multi-organ failure (19%, n = 18/93), status epilepticus (14%, n = 13/93), one suicidal death. The cause of death was unknown in 13% (n = 12/93) of the individuals 1.
Further analysis showed that median survival time from disease onset to death was 19 months (range 0.5 to 600 months) for those with disease onset prior to the age 12 years, 151 months (range 4 to 487 months) for those with disease onset between 12 and 40 years, and 191 months (range 17 to 336 months) for those with disease onset after the age of 40 years 1.
The presence of epilepsy was associated with significantly worse survival, and the median survival time from seizure onset to death was 37 months (range less than 1 to 487 months) 1. Survival analysis also showed that patients with pathogenic compound heterozygous POLG variants had significantly worse survival compared to those with pathogenic homozygous variants, regardless of specific variant types 1. Further analysis showed that survival after the onset of seizures in those with early onset disease was significantly worse than those who developed seizures as part of juvenile or adult onset disease 1. Furthermore, patients who developed liver dysfunction showed a significantly worse survival than those without liver impairment 1.
- Hikmat O, Naess K, Engvall M, et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J Inherit Metab Dis. 2020; 43: 726–736. https://doi.org/10.1002/jimd.12211[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Saneto RP, Naviaux RK. Polymerase gamma disease through the ages. Dev Disabil Res Rev. 2010;16(2):163-74. doi: 10.1002/ddrr.105[↩][↩][↩][↩]
- Rahman, S., Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat Rev Neurol 15, 40–52 (2019). https://doi.org/10.1038/s41582-018-0101-0[↩][↩]
- Cohen BH, Chinnery PF, Copeland WC. POLG-Related Disorders. 2010 Mar 16 [Updated 2024 Feb 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26471[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- POLG gene. https://medlineplus.gov/genetics/gene/polg[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Hikmat O, Naess K, Engvall M, Klingenberg C, Rasmussen M, Tallaksen CM, Brodtkorb E, Ostergaard E, de Coo IFM, Pias-Peleteiro L, Isohanni P, Uusimaa J, Darin N, Rahman S, Bindoff LA. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J Inherit Metab Dis. 2020 Jul;43(4):726-736. doi: 10.1002/jimd.12211[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Çakmak Durmaz Ç, Langerscheidt F, Mantey I, Xia X, Zempel H. Knockdown of POLG Mimics the Neuronal Pathology of Polymerase-γ Spectrum Disorders in Human Neurons. Cells. 2025 Mar 22;14(7):480. doi: 10.3390/cells14070480[↩]
- Zempel H., Sadzot B., Haag N. Treatment Avenues for the Juvenile and Adult Onset Mitochondriopathies Alpers Syndrome, Ataxia Neuropathy Spectrum, MEMSA and PEO Caused by Polymerase-Gamma Mutations Ala467Thr and Trp748Ser. SM J. Neurol. Neurosci. 2017;3:1013[↩]
- Fadic R., Johns D. Clinical Spectrum of Mitochondrial Diseases. Semin. Neurol. 1996;16:11–20. doi: 10.1055/s-2008-1040954[↩]
- Finsterer J. Mitochondriopathies. Eur. J. Neurol. 2004;11:163–186. doi: 10.1046/j.1351-5101.2003.00728.x[↩]
- Hance N., Ekstrand M.I., Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum. Mol. Genet. 2005;14:1775–1783. doi: 10.1093/hmg/ddi184[↩]
- Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. 2019 Jan;15(1):40-52. doi: 10.1038/s41582-018-0101-0[↩][↩][↩][↩][↩][↩]
- Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. 2019;15(1):40-52. doi:10.1038/s41582-018-0101-0[↩]
- Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in Mitochondrial DNA Replication and Repair. Chemical Reviews. 2006;106:383–405. doi: 10.1021/cr040463d[↩][↩]
- Kukat C, Wurm CA, Spahr H, Falkenberg M, Larsson NG, Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A. 2011;108:13534–9. doi: 10.1073/pnas.1109263108[↩][↩][↩]
- Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012 Jan-Feb;47(1):64-74. doi: 10.3109/10409238.2011.632763[↩][↩]
- Alpers-Huttenlocher syndrome. https://medlineplus.gov/genetics/condition/alpers-huttenlocher-syndrome[↩][↩][↩][↩][↩]
- Saneto RP, Lee IC, Koenig MK, Bao X, Weng SW, Naviaux RK, Wong LJ. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure. 2010 Apr;19(3):140-6. doi: 10.1016/j.seizure.2010.01.002[↩][↩][↩][↩]
- Childhood myocerebrohepatopathy spectrum. https://medlineplus.gov/genetics/condition/childhood-myocerebrohepatopathy-spectrum[↩][↩][↩][↩][↩][↩]
- Wong LJ, Naviaux RK, Brunetti-Pierri N, et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008 Sep;29(9):E150-72. doi: 10.1002/humu.20824[↩][↩][↩][↩][↩][↩]
- Hikmat O, Tzoulis C, Chong WK, Chentouf L, Klingenberg C, Fratter C, Carr LJ, Prabhakar P, Kumaraguru N, Gissen P, Cross JH, Jacques TS, Taanman JW, Bindoff LA, Rahman S. The clinical spectrum and natural history of early-onset diseases due to DNA polymerase gamma mutations. Genet Med. 2017 Nov;19(11):1217-1225. doi: 10.1038/gim.2017.35. Epub 2017 Apr 27. Erratum in: Genet Med. 2019 Apr;21(4):1027. doi: 10.1038/s41436-018-0098-1[↩][↩][↩][↩][↩]
- Hikmat O, Tzoulis C, Chong WK, Chentouf L, Klingenberg C, Fratter C, Carr LJ, Prabhakar P, Kumaraguru N, Gissen P, Cross JH, Jacques TS, Taanman JW, Bindoff LA, Rahman S. The clinical spectrum and natural history of early-onset diseases due to DNA polymerase gamma mutations. Genet Med. 2017 Nov;19(11):1217-1225. doi: 10.1038/gim.2017.35. Erratum in: Genet Med. 2019 Apr;21(4):1027. doi: 10.1038/s41436-018-0098-1[↩][↩][↩]
- Myoclonic epilepsy myopathy sensory ataxia. https://medlineplus.gov/genetics/condition/myoclonic-epilepsy-myopathy-sensory-ataxia/[↩][↩]
- Ataxia neuropathy spectrum. https://medlineplus.gov/genetics/condition/ataxia-neuropathy-spectrum[↩][↩][↩]
- Fadic R, Russell JA, Vedanarayanan VV, Lehar M, Kuncl RW, Johns DR. Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology. 1997;49(1):239-245. doi:10.1212/wnl.49.1.239[↩][↩]
- Li LX, Jiang LT, Pan YG, et al. Clinical and Molecular Features of POLG-Related Sensory Ataxic Neuropathy with Dysarthria and Ophthalmoparesis. J Mol Neurosci. 2021;71(12):2462-2467. doi:10.1007/s12031-021-01831-9[↩][↩]
- Progressive external ophthalmoplegia. https://medlineplus.gov/genetics/condition/progressive-external-ophthalmoplegia[↩][↩][↩]
- Parikh S, Goldstein A, Karaa A, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017 Dec;19(12):10.1038/gim.2017.107. doi: 10.1038/gim.2017.107[↩][↩][↩][↩]
- https://tools.niehs.nih.gov/polg/_assets/images/POLG_mutation.png[↩]
- Schapira AH. Mitochondrial disease. Lancet 2006;368(9529):70‐82.[↩]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG. Prevalence of mitochondrial DNA disease in adults. Annals of Neurology 2008;63(1):35‐9.[↩]
- Khan NA, Govindaraj P, Meena AK, Thangaraj K. Mitochondrial disorders: challenges in diagnosis & treatment. Indian J Med Res. 2015 Jan;141(1):13-26. doi: 10.4103/0971-5916.154489[↩][↩]
- Human DNA Polymerase Gamma Mutation Database https://tools.niehs.nih.gov/polg/index.cfm/main/home/limitresults/false[↩]
- Harrower T, Stewart JD, Hudson G, Houlden H, Warner G, O’Donovan DG, Findlay LJ, Taylor RW, De Silva R, Chinnery PF. POLG1 mutations manifesting as autosomal recessive axonal Charcot-Marie-Tooth disease. Arch Neurol. 2008 Jan;65(1):133-6. doi: 10.1001/archneurol.2007.4[↩]
- Phillips J, Courel S, Rebelo AP, Bis-Brewer DM, Bardakjian T, Dankwa L, Hamedani AG, Züchner S, Scherer SS. POLG mutations presenting as Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2019 Jun;24(2):213-218. doi: 10.1111/jns.12313[↩]
- Naess K, Freyer C, Bruhn H, Wibom R, Malm G, Nennesmo I, von Döbeln U, Larsson NG. MtDNA mutations are a common cause of severe disease phenotypes in children with Leigh syndrome. Biochim Biophys Acta. 2009 May;1787(5):484-90. doi: 10.1016/j.bbabio.2008.11.014[↩]
- Taanman JW, Rahman S, Pagnamenta AT, Morris AA, Bitner-Glindzicz M, Wolf NI, Leonard JV, Clayton PT, Schapira AH. Analysis of mutant DNA polymerase gamma in patients with mitochondrial DNA depletion. Hum Mutat. 2009 Feb;30(2):248-54. doi: 10.1002/humu.20852[↩]
- Tang S, Dimberg EL, Milone M, Wong LJ. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol. 2012 May;259(5):862-8. doi: 10.1007/s00415-011-6268-6[↩]
- Hikmat O, Eichele T, Tzoulis C, Bindoff LA. Understanding the Epilepsy in POLG Related Disease. Int J Mol Sci. 2017 Aug 24;18(9):1845. doi: 10.3390/ijms18091845[↩][↩][↩]
- Horvath R, Hudson G, Ferrari G, Fütterer N, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain. 2006 Jul;129(Pt 7):1674-84. doi: 10.1093/brain/awl088[↩][↩][↩][↩]
- Tzoulis C, Engelsen BA, Telstad W, Aasly J, Zeviani M, Winterthun S, Ferrari G, Aarseth JH, Bindoff LA. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain. 2006 Jul;129(Pt 7):1685-92. doi: 10.1093/brain/awl097[↩][↩][↩][↩]
- Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004 Sep 4-10;364(9437):875-82. doi: 10.1016/S0140-6736(04)16983-3[↩][↩]
- Mancuso M, Filosto M, Oh SJ, DiMauro S. A novel polymerase gamma mutation in a family with ophthalmoplegia, neuropathy, and Parkinsonism. Arch Neurol. 2004 Nov;61(11):1777-9. doi: 10.1001/archneur.61.11.1777[↩]
- Hakonen AH, Heiskanen S, Juvonen V, et al. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005 Sep;77(3):430-41. doi: 10.1086/444548[↩][↩][↩]
- Davidzon G, Greene P, Mancuso M, Klos KJ, Ahlskog JE, Hirano M, DiMauro S. Early-onset familial parkinsonism due to POLG mutations. Ann Neurol. 2006 May;59(5):859-62. doi: 10.1002/ana.20831[↩]
- Nguyen KV, Sharief FS, Chan SS, Copeland WC, Naviaux RK. Molecular diagnosis of Alpers syndrome. J Hepatol. 2006 Jul;45(1):108-16. doi: 10.1016/j.jhep.2005.12.026[↩]
- Cohen BH, Naviaux RK. The clinical diagnosis of POLG disease and other mitochondrial DNA depletion disorders. Methods. 2010 Aug;51(4):364-73. doi: 10.1016/j.ymeth.2010.05.008[↩]
- Saneto RP, Cohen BH, Copeland WC, Naviaux RK. Alpers-Huttenlocher syndrome. Pediatr Neurol. 2013 Mar;48(3):167-78. doi: 10.1016/j.pediatrneurol.2012.09.014[↩]
- Alves CAPF, Gonçalves FG, Grieb D, Lucato LT, Goldstein AC, Zuccoli G. Neuroimaging of Mitochondrial Cytopathies. Top Magn Reson Imaging. 2018 Aug;27(4):219-240. doi: 10.1097/RMR.0000000000000173[↩]
- Fadic R, Russell JA, Vedanarayanan VV, Lehar M, Kuncl RW, Johns DR. Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology. 1997 Jul;49(1):239-45. doi: 10.1212/wnl.49.1.239[↩]
- Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001 Jul;28(3):211-2. doi: 10.1038/90034[↩][↩]
- Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, Papadimitriou A, Spelbrink H, Silvestri L, Casari G, Comi GP, Zeviani M. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol. 2002 Aug;52(2):211-9. doi: 10.1002/ana.10278[↩]
- Van Goethem G, Martin JJ, Dermaut B, Löfgren A, Wibail A, Ververken D, Tack P, Dehaene I, Van Zandijcke M, Moonen M, Ceuterick C, De Jonghe P, Van Broeckhoven C. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord. 2003 Feb;13(2):133-42. doi: 10.1016/s0960-8966(02)00216-x[↩]
- Pagnamenta AT, Taanman JW, Wilson CJ, Anderson NE, Marotta R, Duncan AJ, Bitner-Glindzicz M, Taylor RW, Laskowski A, Thorburn DR, Rahman S. Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum Reprod. 2006 Oct;21(10):2467-73. doi: 10.1093/humrep/del076[↩]
- Parada-Garza JD, López-Valencia G, Miranda-García LA, Pérez-García G, Ruiz-Sandoval JL. MRI findings in SANDO variety of the ataxia-neuropathy spectrum with a novel mutation in POLG (c.3287G>T): A case report. Neuromuscul Disord. 2020 Jul;30(7):590-592. doi: 10.1016/j.nmd.2020.04.008[↩]
- García-Cabo C, Carvajal-García P, Fernández-Vega I, Peña-Suárez J, Mateos-Marcos V, Suárez-Santos P, Álvarez-Martínez V, Morís-de la Tassa G. Ataxia y neuropatía sensitiva de inicio en la edad adulta como manifestación clínica de mutaciones en el gen POLG [Adult-onset sensory neuropathy and ataxia as a clinical manifestation of POLG gene mutations]. Rev Neurol. 2023 Feb 1;76(3):75-81. Spanish. doi: 10.33588/rn.7603.2022322[↩]
- Hasselmann O, Blau N, Ramaekers VT, Quadros EV, Sequeira JM, Weissert M. Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol Genet Metab. 2010 Jan;99(1):58-61. doi: 10.1016/j.ymgme.2009.08.005[↩]
- El-Hattab AW, Almannai M, Scaglia F. Arginine and citrulline for the treatment of MELAS syndrome. J Inborn Errors Metab Screen. 2017 Jan;5:10.1177/2326409817697399. doi: 10.1177/2326409817697399[↩]
- Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, Anselm I, Cohen BH, Falk MJ, Greene C, Gropman AL, Haas R, Hirano M, Morgan P, Sims K, Tarnopolsky M, Van Hove JL, Wolfe L, DiMauro S. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015 Sep;17(9):689-701. doi: 10.1038/gim.2014.177[↩][↩][↩]
- Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R; Mitochondrial Medicine Society Clinical Directors Working Group; Clinical Director’s Work Group. Practice patterns of mitochondrial disease physicians in North America. Part 2: treatment, care and management. Mitochondrion. 2013 Nov;13(6):681-7. doi: 10.1016/j.mito.2013.09.003[↩]
- Rodriguez MC, MacDonald JR, Mahoney DJ, Parise G, Beal MF, Tarnopolsky MA. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007 Feb;35(2):235-42. doi: 10.1002/mus.20688[↩]
- Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics. 2008 Oct;5(4):558-68. doi: 10.1016/j.nurt.2008.07.002[↩]
- Gold DR, Cohen BH. Treatment of mitochondrial cytopathies. Semin Neurol. 2001 Sep;21(3):309-25. doi: 10.1055/s-2001-17948[↩]
- Camp KM, Krotoski D, Parisi MA, et al. Nutritional interventions in primary mitochondrial disorders: Developing an evidence base. Mol Genet Metab. 2016 Nov;119(3):187-206. doi: 10.1016/j.ymgme.2016.09.002[↩]
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R, Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol. 2009 Nov;11(6):414-30. doi: 10.1007/s11940-009-0046-0[↩]
- Besse A, Wu P, Bruni F, Donti T, Graham BH, Craigen WJ, McFarland R, Moretti P, Lalani S, Scott KL, Taylor RW, Bonnen PE. The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metab. 2015 Mar 3;21(3):417-27. doi: 10.1016/j.cmet.2015.02.008[↩]
- Mirza NS, Alfirevic A, Jorgensen A, Marson AG, Pirmohamed M. Metabolic acidosis with topiramate and zonisamide: an assessment of its severity and predictors. Pharmacogenet Genomics. 2011 May;21(5):297-302. doi: 10.1097/FPC.0b013e3283441b95[↩]
- Hakonen AH, Heiskanen S, Juvonen V, Lappalainen I, Luoma PT, Rantamaki M, Goethem GV, Lofgren A, Hackman P, Paetau A, Kaakkola S, Majamaa K, Varilo T, Udd B, Kaariainen H, Bindoff LA, Suomalainen A. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005 Sep;77(3):430-41. doi: 10.1086/444548[↩]
- Kelly DA. Liver transplantation: to do or not to do? Pediatr Transplant. 2000 Aug;4(3):170-2. doi: 10.1034/j.1399-3046.2000.00125.x[↩]
- Parikh S, Karaa A, Goldstein A, Ng YS, Gorman G, Feigenbaum A, Christodoulou J, Haas R, Tarnopolsky M, Cohen BK, Dimmock D, Feyma T, Koenig MK, Mundy H, Niyazov D, Saneto RP, Wainwright MS, Wusthoff C, McFarland R, Scaglia F. Solid organ transplantation in primary mitochondrial disease: Proceed with caution. Mol Genet Metab. 2016 Jul;118(3):178-184. doi: 10.1016/j.ymgme.2016.04.009[↩][↩]
- Wong LJ, Naviaux RK, Brunetti-Pierri N, Zhang Q, Schmitt ES, Truong C, Milone M, Cohen BH, Wical B, Ganesh J, Basinger AA, Burton BK, Swoboda K, Gilbert DL, Vanderver A, Saneto RP, Maranda B, Arnold G, Abdenur JE, Waters PJ, Copeland WC. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008 Sep;29(9):E150-72. doi: 10.1002/humu.20824[↩]





{kind=link}