Achondrogenesis
Achondrogenesis also called achondrogenesis syndrome, is a group of severe disorders that are present from birth and affect the development of cartilage and bone 1. Infants with achondrogenesis usually have a small body, extremely short arms and legs (micromelia), other skeletal abnormalities, and underdeveloped lungs 2. As a result of serious health problems, infants with achondrogenesis usually die before birth, are stillborn, or die soon after birth from respiratory failure. However, some infants have lived for a short time with intensive medical support.
Researchers have described at least three forms of achondrogenesis, designated as type 1A (Houston-Harris type), type 1B (Fraccaro type) and type 2 (Langer-Saldino type). The types are distinguished by their signs and symptoms, inheritance pattern, and genetic cause. However, types 1A and 1B are often hard to tell apart without genetic testing.
Achondrogenesis types 1A and 1B are rare genetic disorders; their incidence is unknown. Combined, achondrogenesis type 2 and hypochondrogenesis (a similar skeletal disorder) occur in 1 in 40,000 to 60,000 newborns 1.
Achondrogenesis is usually diagnosed during pregnancy by ultrasound and genetic testing is used to distinguish between the three types 2. Type 1A and 1B achondrogenesis are both inherited in an autosomal recessive pattern. Type 1A is caused by mutations in the TRIP11 gene 3. Type 1B is caused by mutations in the SLC26A2 gene 4. Type 2 achondrogenesis is caused by new (de novo) mutations in the COL2A1 gene 5. Because of the severity of this condition, most infants with achondrogenesis die before or shortly after birth 1.
Figure 1. Achondrogenesis
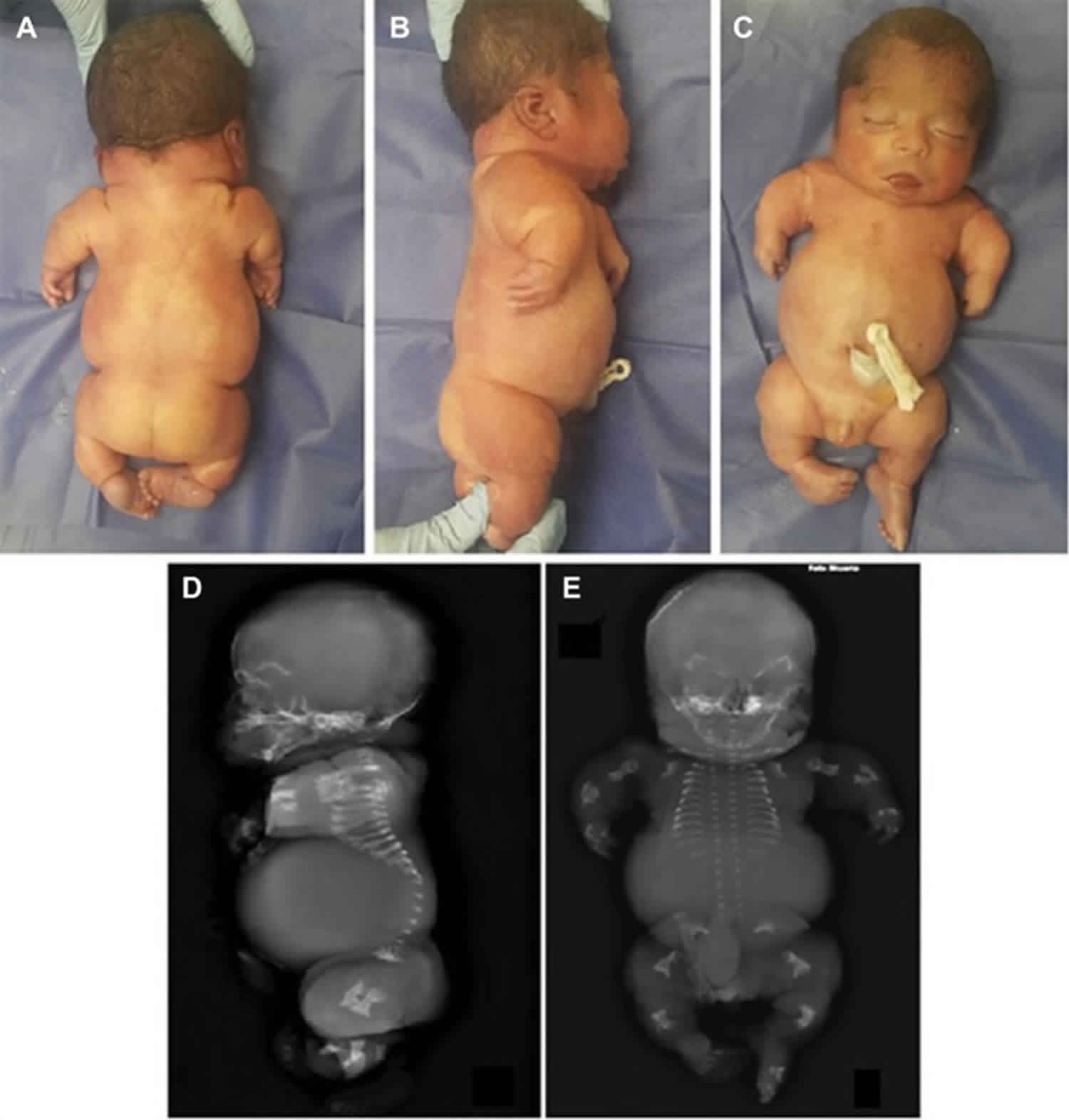
Footnote: (A–C) Posterior, lateral, and frontal views. Patient phenotype shows flat face, short nose, protruding tongue and eyes, low-set ears, narrow bell-shaped thorax, and severe micromelia. (D, E) Postmortem X-ray. Lateral and frontal views showing deficient mineralization of the calvaria and vertebral bodies, unossified sacrum, hypoplastic thorax, and markedly short and beaded ribs with flared and spurred ends.
[Source 6 ]Achondrogenesis type 1A
Achondrogenesis type 1A, which is also called the Houston-Harris type, is the least well understood of the three forms. Affected infants have extremely short limbs, a narrow chest, short ribs that fracture easily, and a lack of normal bone formation (ossification) in the skull, spine, and pelvis.
Some of the common clinical features of achondrogenesis type 1A include:
- low birth weight
- skeletal abnormalities
- very short limbs
- short neck
- incomplete ossification of bones – particularly the skull
- congenital cardiac anomalies
- patent ductus arteriosus
- atrial septal defects
- ventricular septal defects
- breathing problems
- small chest
- underdeveloped lungs
- protruberant abdomen
Achondrogenesis type 1B
Achondrogenesis type 1B, also known as the Parenti-Fraccaro type, is characterized by extremely short limbs, a narrow chest, and a prominent, rounded abdomen. The fingers and toes are short and the feet may turn inward and upward (clubfeet). The fetuses frequently present in breech position. Affected infants frequently have a narrow thorax with protuberant abdomen with soft out-pouching around the belly-button (an umbilical hernia) or near the groin (an inguinal hernia) and the characteristics include flat face, severe micromelia with short stubby fingers and toes. Stillbirth is common.
Achondrogenesis type 2
Infants with achondrogenesis type 2, which is sometimes called the Langer-Saldino type, have short arms and legs, a narrow chest with short ribs, and underdeveloped lungs. This condition is also associated with a lack of ossification in the spine and pelvis. Distinctive facial features include a prominent forehead, a small chin, microtia (under developed ears) and, in some cases, an opening in the roof of the mouth (a cleft palate). The abdomen is enlarged, and affected infants often have a condition called hydrops fetalis, in which excess fluid builds up in the body before birth.
Majority of cases are stillborn or die within few hours after birth. The neonate is extremely short stature and present with large calvarium, large fontanels, hypertelorism, depressed nasal bridge, anteverted nostrils, hypertrophied tongue, micrognathia and cystic hygroma can be present
Achondrogenesis causes
Mutations in the TRIP11, SLC26A2, and COL2A1 genes cause achondrogenesis type 1A, type 1B, and type 2, respectively 1.
The genetic cause of achondrogenesis type 1A was unknown until recently, when researchers discovered that achondrogenesis type 1A can result from mutations in the TRIP11 gene 6. The TRIP11 gene provides instructions for making a protein called GMAP-210. This protein plays a critical role in the Golgi apparatus, a cell structure in which newly produced proteins are modified so they can carry out their functions. Mutations in the TRIP11 gene prevent the production of functional GMAP-210, which alters the structure and function of the Golgi apparatus. Researchers suspect that cells called chondrocytes in the developing skeleton may be most sensitive to these changes. Chondrocytes give rise to cartilage, a tough, flexible tissue that makes up much of the skeleton during early development. Most cartilage is later converted to bone, except for the cartilage that continues to cover and protect the ends of bones and is present in the nose and external ears. Malfunction of the Golgi apparatus in chondrocytes likely underlies the problems with bone formation in achondrogenesis type 1A.
Achondrogenesis type 1B is the most severe of a spectrum of skeletal disorders caused by mutations in the SLC26A2 gene 4. The SLC26A2 gene provides instructions for making a protein that is essential for the normal development of cartilage and for its conversion to bone. Mutations in the SLC26A2 gene cause the skeletal problems characteristic of achondrogenesis type 1B by disrupting the structure of developing cartilage, which prevents bones from forming properly.
Achondrogenesis type 2 is one of several skeletal disorders that result from mutations in the COL2A1 gene 7. The COL2A1 gene provides instructions for making a protein that forms type II collagen. This type of collagen is found mostly in cartilage and in the clear gel that fills the eyeball (the vitreous). It is essential for the normal development of bones and other connective tissues that form the body’s supportive framework. Mutations in the COL2A1 gene interfere with the assembly of type II collagen molecules, which prevents bones and other connective tissues from developing properly.
Achondrogenesis inheritance pattern
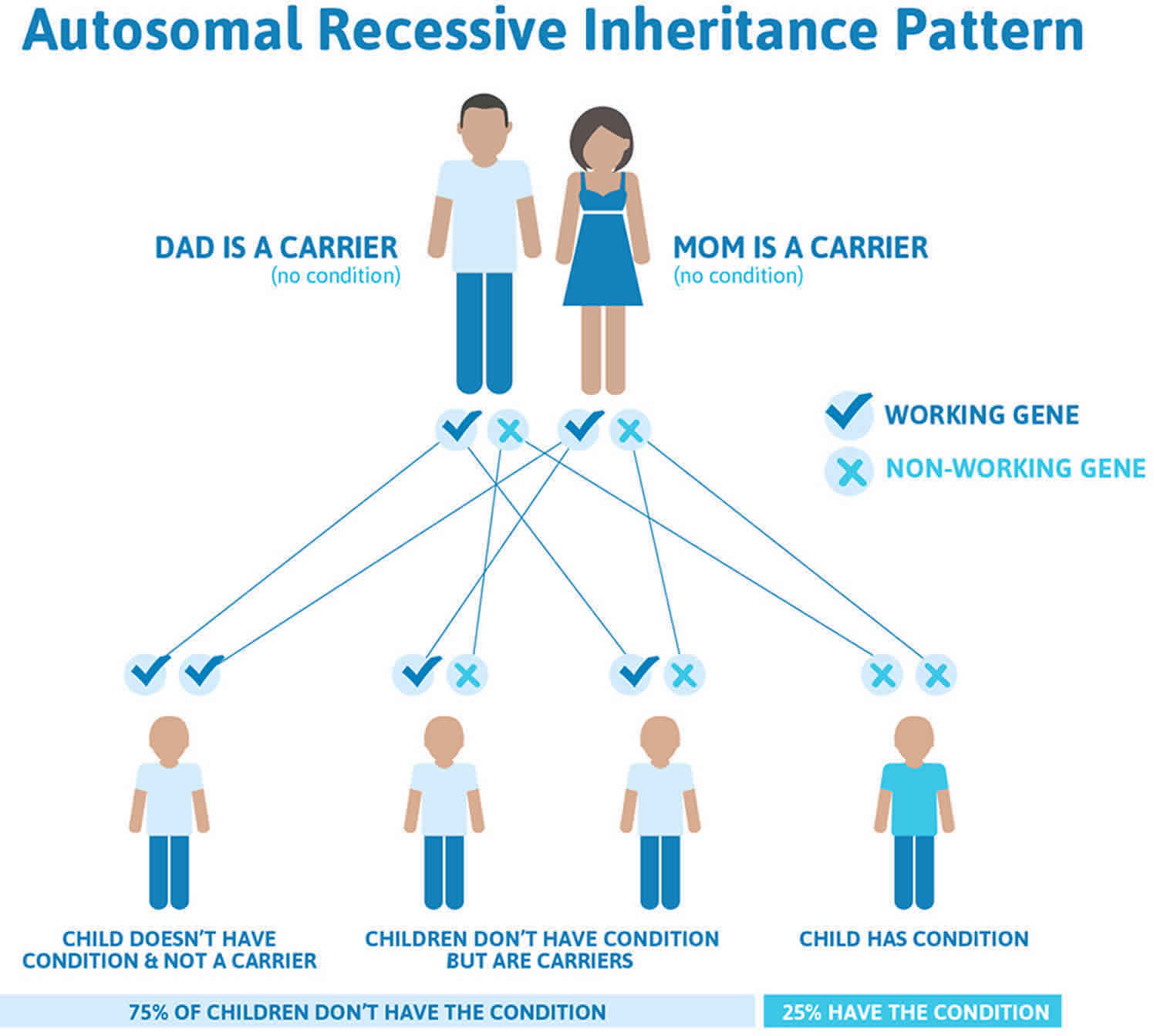
Achondrogenesis type 1A and type 1B both have an autosomal recessive pattern of inheritance, which means both copies of the TRIP11 or SLC26A2 gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene but do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2. Achondrogenesis type 1A and type 1B autosomal recessive inheritance pattern
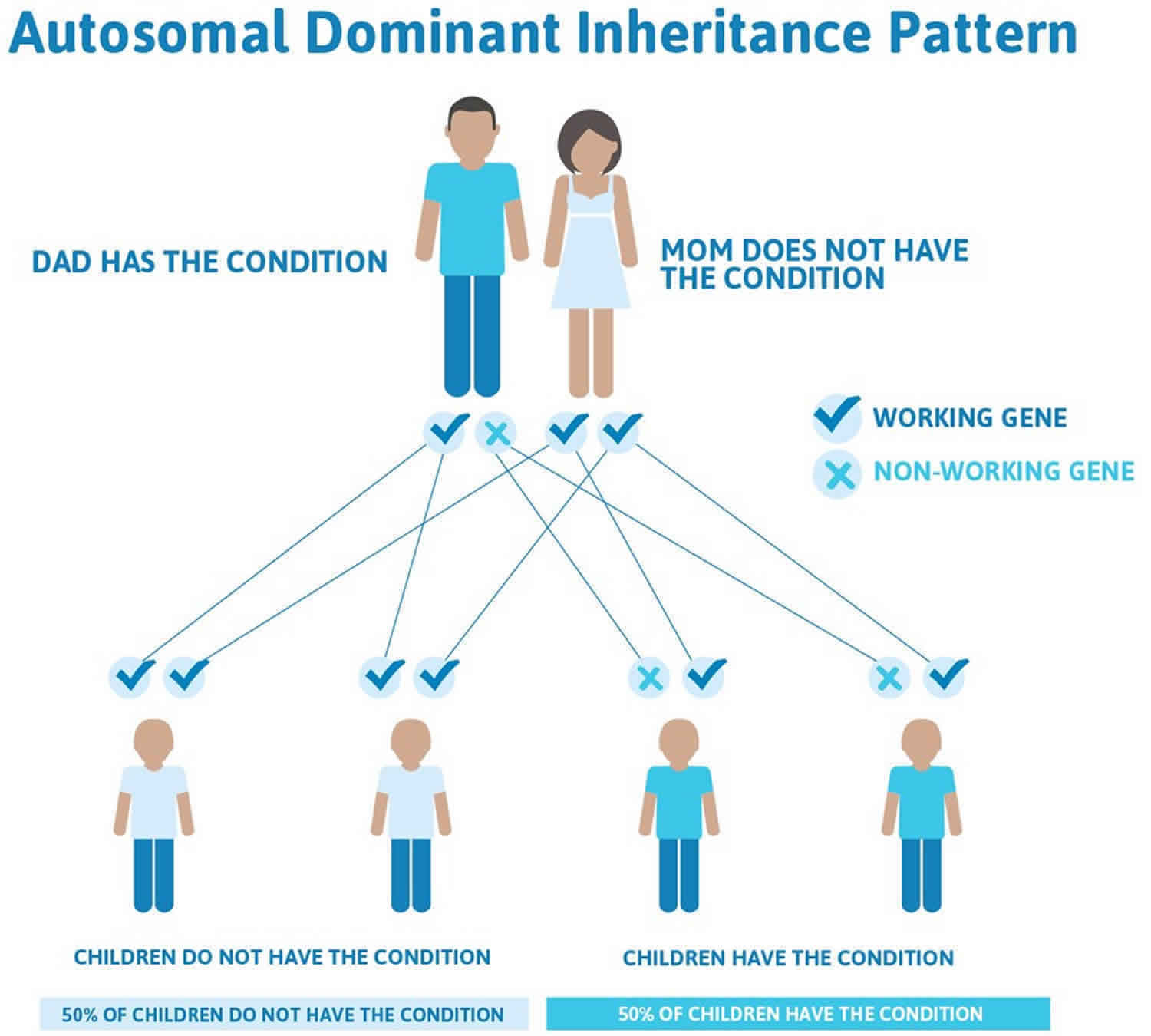
Achondrogenesis type 2 is considered an autosomal dominant disorder because one copy of the altered gene in each cell is sufficient to cause the condition. It is almost always caused by new (de novo) mutation in the COL2A1 gene and typically occurs in people with no history of the disorder in their family 5. There are rare cases of families who have had more than one pregnancy with achondrogenesis type 2 5. These cases are thought to occur because of mosaicism, which means that a portion of the DNA in a person’s cells is different than the DNA in the rest of his/her body. Germline mosaicism occurs then a person’s reproductive cells (sperm or egg) carry a mutation not found in the rest of the cells in the body. Somatic mosaicism occurs when a person has a some body cells that carry a mutation and some body cells that do not carry the mutation. Individuals with germline or somatic mosaicism are generally healthy and do not show signs of the disorder.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 3. Achondrogenesis type 2 autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Achondrogenesis symptoms
Achondrogenesis is characterized by premature birth, abnormal accumulation of fluid in the body (hydrops fetalis), and a head that may be abnormal in shape and less ossified, meaning that they are not hard like regular bone. The head may look disproportionately large, because the body is small. In addition, affected individuals have extremely short arms and legs (micromelia) and ribs, short neck, flat vertebrae and many other bones of the skeleton are not properly developed. In infants born with achondrogenesis the abdomen is prominent and the thoracic cage is small. Other abnormalities are incomplete closure of the roof of the mouth (cleft palate), corneal clouding, and ear deformities. The disorder is life-threatening either before birth or shortly after birth usually due to underdeveloped thorax and small lungs.
All three types of achondrogenesis have similar features, and it can be difficult to tell the types apart based only on signs and symptoms.
Achondrogenesis type IA (Houston-Harris type) is characterized by varying facial abnormalities (flat face, protruding eyes and protruding tongue or only minor facial anomalies), short trunk and limbs, short beaded ribs and thin skull bones (deficient ossification of the skull). Bone formation is abnormal in the spine, pelvis and extremities, but the degree of the severity of skeletal involvement may be variable. However, small thorax leads to underdevelopment of lungs and death soon after birth.
Achondrogenesis type IB (Fraccaro type) is characterized by short trunk and limbs, narrow chest, and prominent abdomen. Affected infants may have a protrusion around the belly-button (umbilical hernia), or near the groin (inguinal hernia), and have short fingers and toes with feet turned inward. The face may be flat, the palate may be cleft and the neck is usually short. In some cases, the soft tissue of the neck may be abnormally thickened. Achondrogenesis type IB is sub-classified as a sulfation disorder, a small group of disorders associated with mutations in the gene SLC26A2. This group includes diastrophic dysplasia and recessive multiple epiphyseal dysplasia, which are milder conditions. It is important to note that one diagnosis does not change to another while the baby is developing, even if the genetic changes are located in the same gene.
Achondrogenesis type 2 (Langer-Saldino type) is characterized by a narrow chest, abnormally small or short bones in the arms and/or legs, thin ribs, flat vertebra or deficient ossification of vertebral bodies, underdeveloped lungs, small chin, cleft palate and club feet. Bone formation is abnormal in the spine and pelvis. Abnormal accumulation of fluid may occur (hydrops fetalis) and the abdomen may be enlarged.
Achondrogenesis diagnosis
Prenatal diagnosis of achondrogenesis by ultrasound is possible after 14-15 weeks gestation. Prenatal diagnosis by chorionic villus sampling (10-12 weeks gestation) or amniocentesis (15-18 weeks gestation) is possible if the specific gene mutations have been identified in a family member.
At birth, achondrogenesis is suspected when the infant has extremely short underdeveloped arms and legs, short ribs and small chest, and short trunk. Achondrogenesis is diagnosed by physical features, X-ray (radiographic) findings and examination of tissue samples under a microscope (histology). Molecular genetic testing of the TRIP11, SLC26A2 and COL2A1 genes can be performed to confirm the diagnosis and determine the type of achondrogenesis.
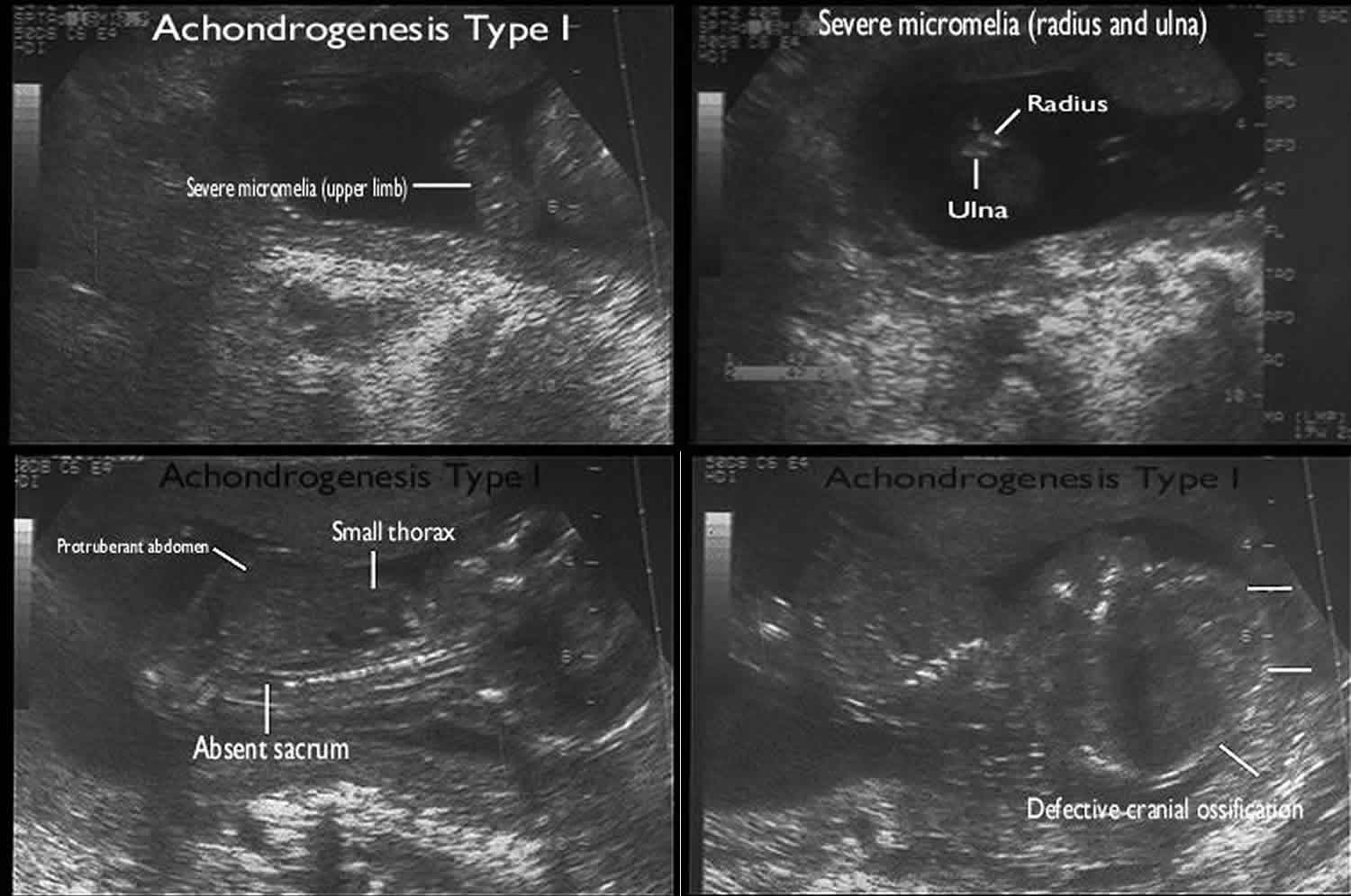
Achondrogenesis ultrasound
Ultrasound diagnosis may be possible after 13 weeks of gestation, where nuchal edema may be evident as an early (though non-specific) sign 8.
The fetal bony structures are often unable to be identified. There may also be extreme micromelia 9. Calvarial ossification may be preserved with the type II subtype which can, in turn, give a floating head appearance.
Additional sonographic findings include:
- micrognathia
- macrocephaly
- frontal bossing
- flat face
- anteverted nares
- long philtrum
- narrow fetal thorax
- lung aplasia/hypoplasia
- rib fractures may be present in type 1A
Other ancillary sonographic features that may be present include:
- polyhydramnios
- development of hydrops fetalis
Figure 4. Achondrogenesis ultrasound
Achondrogenesis treatment
Treatment of achondrogenesis is symptomatic and supportive and involves palliative care, in which physicians attempt to reduce or minimize pain, stress and specific symptoms associated with the disorder. Genetic counseling is recommended for families with an affected child. Psychosocial support for the entire family is essential as well.
Achondrogenesis prognosis
Most infants with achondrogenesis have severely underdeveloped lungs which leads to serious breathing problems and lung failure. Most die before or shortly after birth because of the severity of this disorder.
- Achondrogenesis. https://ghr.nlm.nih.gov/condition/achondrogenesis[↩][↩][↩][↩]
- Achondrogenesis. https://rarediseases.org/rare-diseases/achondrogenesis[↩][↩]
- Vanegas S, Sua LF, Lopez-Tenorio J. Ramirez-Montano D, Pachajoa H.. Achondrogenesis, type 1A: clinical, histologic, molecular and prenatal ultrasound diagnosis.. Appl Clin Genet.. 2018; 11:69-73. https://www.ncbi.nlm.nih.gov/pubmed/29872333[↩]
- Bonafé L, Mittaz-Crettol L, Ballhausen D, et al. Achondrogenesis Type 1B. 2002 Aug 30 [Updated 2013 Nov 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1516[↩][↩]
- Comstock JM, Putnam AR, Sangle N, Lowichik A, Rose N, Optiz JM. Recurrence of Achondrogenesis type 2 in sibs: Additional evidence for germline mosaicism.. Am J Med Genet Part A. 2010; 152A:1822-1824. https://www.ncbi.nlm.nih.gov/pubmed/20583175[↩][↩][↩]
- Vanegas S, Sua LF, López-Tenorio J, Ramírez-Montaño D, Pachajoa H. Achondrogenesis type 1A: clinical, histologic, molecular, and prenatal ultrasound diagnosis. Appl Clin Genet. 2018;11:69–73. Published 2018 May 25. doi:10.2147/TACG.S157235 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5973320[↩][↩]
- Comstock, J. M., Putnam, A. R., Sangle, N. , Lowichik, A. , Rose, N. C. and Opitz, J. M. (2010), Recurrence of achondrogenesis type 2 in sibs: Additional evidence for germline mosaicism. Am. J. Med. Genet., 152A: 1822-1824. doi:10.1002/ajmg.a.33463[↩]
- Achondrogenesis. https://radiopaedia.org/articles/achondrogenesis?lang=us[↩]
- Lee HS, Doh JW, Kim CJ, Chi JG. Achondrogenesis type II (Langer-Saldino achondrogenesis): a case report. J Korean Med Sci. 2000;15(5):604–608. doi:10.3346/jkms.2000.15.5.604 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3054684/pdf/11069003.pdf[↩]
{kind=link}