Axenfeld-Rieger syndrome
Axenfeld-Rieger syndrome (ARS), also called Rieger syndrome, Rieger’s anomaly, Axenfeld and Rieger anomaly or iridogoniodysgenesis with somatic anomalies, is a rare genetic disorder that primarily affects your eyes, but can also affect other parts of your body 1, 2, 3, 4, 5, 6, 7, 8. In current medical terminology, the terms Axenfeld anomaly, Rieger anomaly, and Rieger syndrome are no longer used 9. Instead, these eye findings and associated systemic manifestations are now recognized under a spectrum of disorders called Axenfeld-Riger syndrome (ARS) 7. Axenfeld-Rieger syndrome is characterized by abnormalities in the anterior segment of the eye, such as cornea defects and iris defects, that can lead to glaucoma 6, 7, 8. People with Axenfeld-Rieger syndrome may have an off-center pupil (corectopia) or extra holes in the eyes that can look like multiple pupils (polycoria) (see Figures 2 to 4 below). About 50% of people with Axenfeld-Rieger syndrome develop glaucoma, a condition that increases pressure inside of the eye (intraocular pressure [IOP]). When glaucoma occurs with Axenfeld-Rieger syndrome, it most often develops in late childhood or adolescence, although it can occur as early as infancy. Glaucoma can cause vision loss or blindness. Axenfeld-Rieger syndrome can also cause abnormalities of the cornea, which is the clear front covering of the eye. Even though Axenfeld-Rieger syndrome is primarily an eye disorder, this syndrome can affect other parts of the body. Most people with Axenfeld-Rieger syndrome have distinctive facial features such as widely spaced eyes (hypertelorism), a flattened mid-face with a broad, flat nasal bridge and a prominent forehead 5. Many people with Axenfeld-Rieger syndrome also have issues with their teeth including unusually small teeth (microdontia) or fewer than normal teeth (oligodontia) 5. Some people with Axenfeld-Rieger syndrome have extra folds of skin around their belly button (redundant periumbilical skin) 5. Other, less common features can include heart defects, the opening of the urethra on the underside of the penis (hypospadias), narrowing of the anus (anal stenosis), and abnormalities of the pituitary gland that can result in slow growth 5. Several genes have been associated with Axenfeld-Rieger syndrome – the two known genes are PITX2 and FOXC1 genes 5, 6, 10. Axenfeld-Rieger syndrome has an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder, but it can also occur sporadically 6, 5. Axenfeld-Rieger syndrome has an estimated prevalence of 1 in 50,000 to 1 in 200,000 babies 11, 12, 13, 14. There is no racial or gender preference 9. In most cases, both eyes are affected by the characteristic features of Axenfeld-Rieger syndrome 9.
Researchers have described at least 3 types of Axenfeld-Rieger syndrome. The types, which are numbered 1 through 3, are distinguished by their genetic cause 5:
- Axenfeld-Rieger syndrome type 1 is linked to mutations in the Pituitary Homeobox 2 (PITX2) gene on chromosome 4q25 15. Axenfeld-Rieger syndrome type 1 signs and symptoms include 16:
- Abnormalities in the anterior segment of the eye (e.g., iris hypoplasia, corectopia)
- Dental anomalies (e.g., microdontia, hypodontia)
- Redundant periumbilical skin
- Craniofacial anomalies
- Cardiovascular defects
- Axenfeld-Rieger syndrome type 2 is linked to deletions in the region on chromosome 13q14 whose the molecular basis is yet to be identified 17, 18. Axenfeld-Rieger syndrome type 2 is characterized by abnormalities in the anterior chamber of the eye, developmental delay and other systemic anomalies may be present but less specific compared to type 1 and type 3. Nine of the affected individuals also showed evidence of glaucoma. With the exception of 1 individual, all of the affected persons also demonstrated one or more systemic abnormalities, including dental and hearing defects, mild craniofacial dysmorphism, hydrocephalus, cryptorchidism, fetal lobulation of kidney, congenital heart defect, and congenital hip abnormalities 19. The redundancy of the periumbilical skin, which has been described as a common feature of the Axenfeld-Rieger syndrome, was found in no affected members of this kindred 19. One child of an affected individual died in infancy and was said to have had an abnormal navel, but this could not be documented by medical records 19. Phillips et al. 20 pointed to forkhead (FOXO1A) as an excellent candidate for the site of the mutation in this form of Axenfeld-Rieger syndrome. They stated that if such mutations are found, this would be an example of a gene that can function both as an oncogene (producing rhabdomyosarcoma) and as a ‘teratogene’ (producing RIEG2) 20.
- Axenfeld-Rieger syndrome type 3 is linked to mutations in the Forkhead-Box C1 gene (FOXC1) gene on chromosome 6p25 21, 22. Axenfeld-Rieger syndrome type 3 is characterized anteriorly displaced Schwalbe line, the presence of another ocular anomaly (hypoplasia of iris stroma, corectopia, or iridocorneal adhesions), and nonocular anomalies including maxillary hypoplasia, hypodontia, microdontia, protuberant periumbilical skin, sensorineural hearing loss, and congenital heart or kidney abnormalities 23.
- Some people with Axenfeld-Rieger syndrome do not have identified mutations in the PITX2 or FOXC1 genes 5. In these individuals, the cause of the condition is unknown. Other as-yet-unidentified genes may also cause Axenfeld-Rieger syndrome.
- Genetic heterogeneity in conditions like Axenfeld-Rieger syndrome means that various genes can lead to similar features or phenotypes 9. The inheritance pattern of most cases of Axenfeld-Rieger syndrome is autosomal dominant, although sporadic cases also arise. Axenfeld-Rieger syndrome is typically characterized by high penetrance but exhibits variable expressivity 9.
Your child’s doctor or an eye specialist will diagnose your child with Axenfeld-Rieger syndrome. Diagnosis usually occurs at birth if your child has obvious issues with their eyes or other parts of their body. Diagnosis may also occur after your child develops symptoms.
Most Axenfeld-Rieger syndrome cases are diagnosed during infancy or childhood; however, glaucoma typically occurs in late childhood or adulthood 7, 24.
Your child’s eye specialist (ophthalmologist) will perform an eye exam to look at your child’s eyes (including inside them). They might perform a few tests to diagnose Axenfeld-Rieger syndrome, including:
- A visual acuity test to see if your child’s vision is affected.
- Gonioscopy to check for glaucoma.
- DNA tests and genetic testing to confirm if your child has the mutations that cause Axenfeld-Rieger syndrome.
Axenfeld-Rieger syndrome treatment depends on where your child has symptoms and what (if any) issues their symptoms cause. Talk to your child’s doctor or your child’s eye specialist (ophthalmologist) about which treatments your child will need and how often they need follow-up exams to monitor changes in their eyes and body.
An annual slit lamp examination should be performed, along with gonioscopy, intraocular pressure (IOP) measurements and funduscopy to assess the retinal nerve fiber layer and optic nerve head involvement due to possible glaucoma 13. Autoperimetry (automated measurements of the visual fields) is necessary whenever glaucoma is suspected 13. Should childhood glaucoma develop, drug therapy is recommended before surgery. In case of glaucoma, the goal of treatment is to reach low intraocular pressure (IOP). Surgery is performed if eye drops are not sufficient in lowering intraocular pressure (IOP). Medications that decrease aqueous output (beta-blockers, alpha-agonists and carbonic anhydrase inhibitors) are more beneficial than those affecting outflow. However, alpha-agonists should be used with caution in young children because of possible central nervous system (brain and spinal cord) depression. If surgery is necessary, the procedure of choice is trabeculectomy with the adjunctive use of antimetabolites. If photophobia is present in patients with corectopia and polycoria, contact lenses may be used to cover the holes in the iris 13.
Figure 1. Eye anatomy
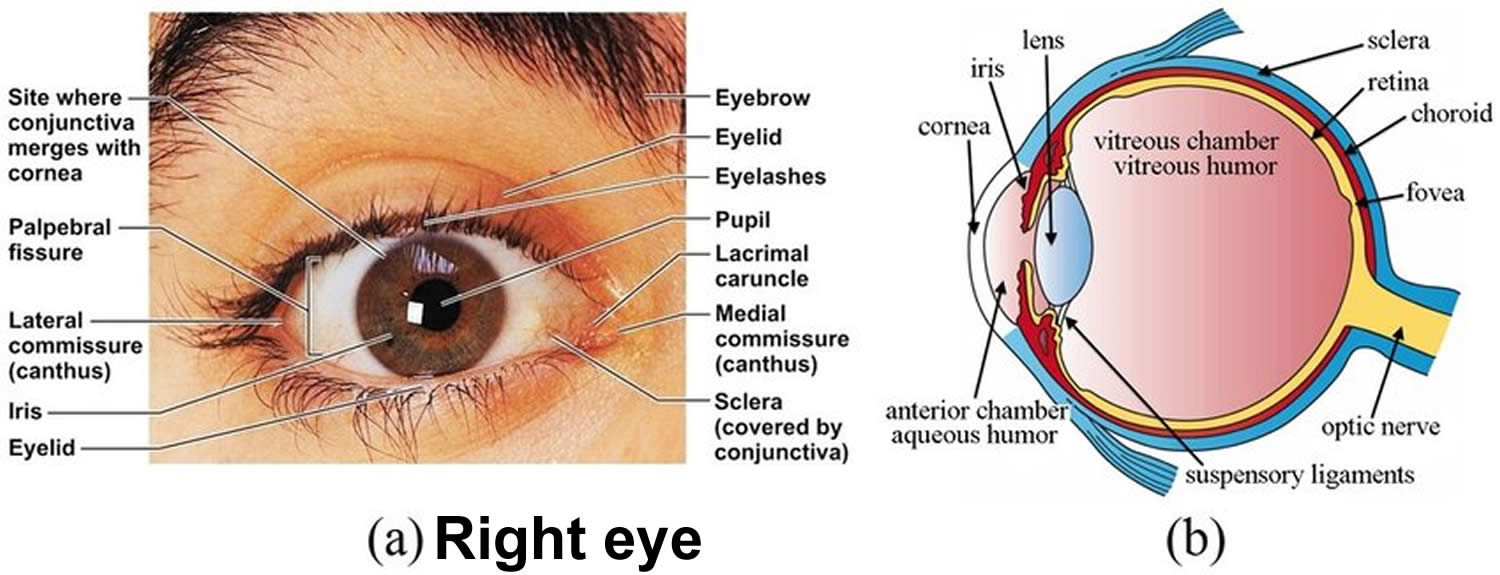
Footnote: External anatomy of right eye, (a) Frontal view, (b) Internal view.
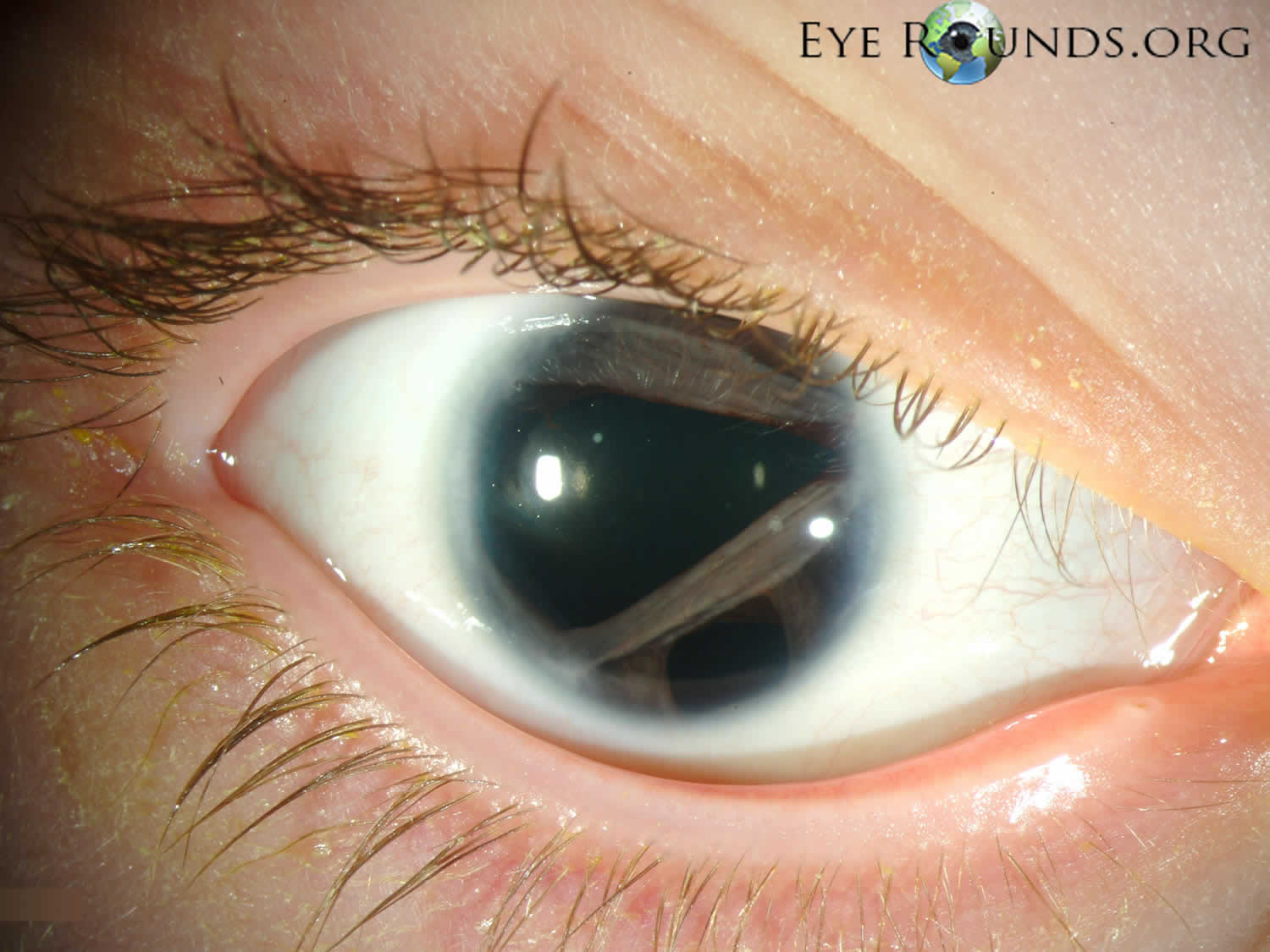
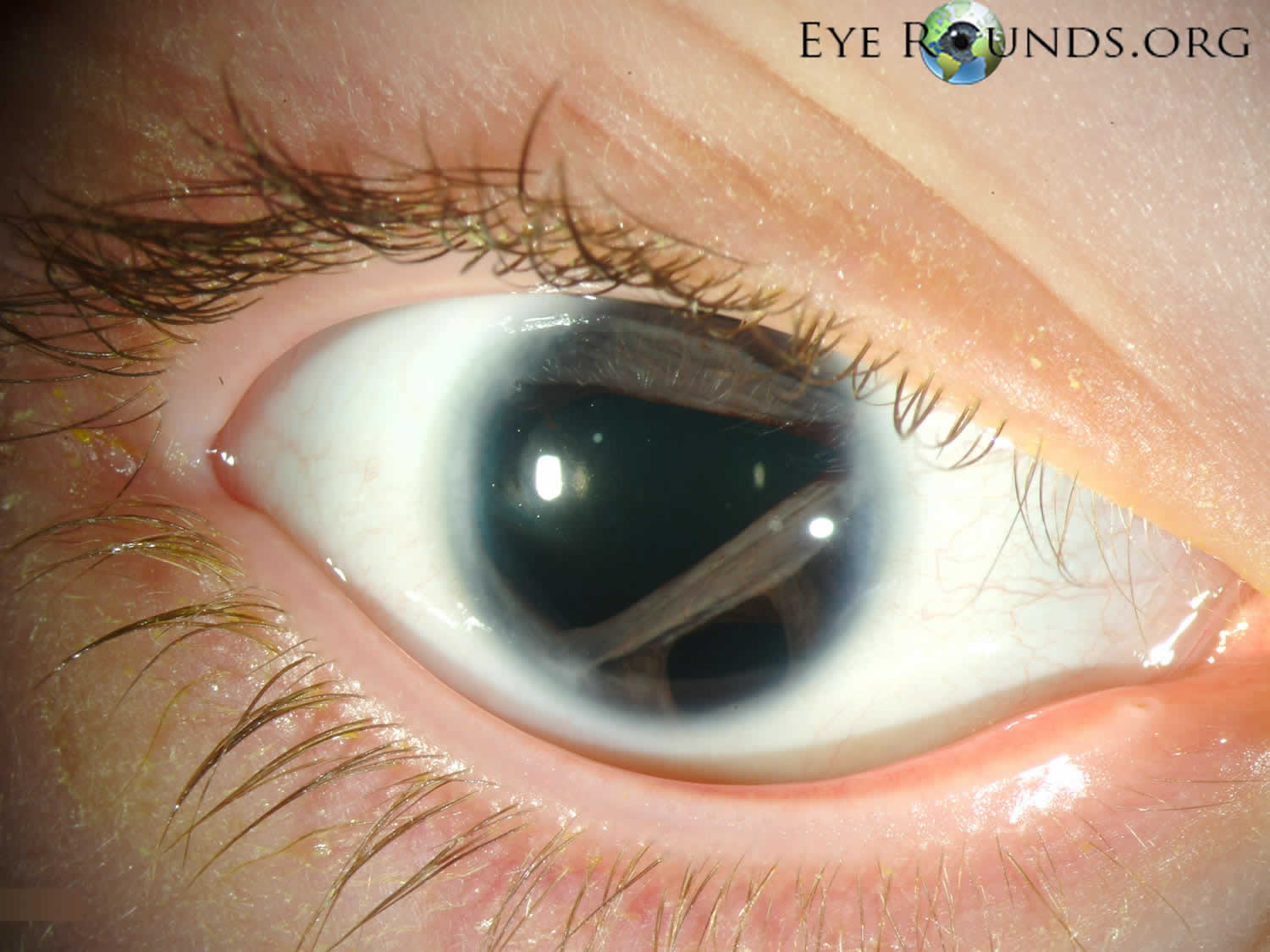
[Source 25 ]Figure 2. Axenfeld-Rieger syndrome eye changes
Footnotes: Eye changes observed in Axenfeld–Rieger syndrome. (a) Corectopia on the right eye (displaced pupil is shown by an arrow); (b) Polycoria (the extra hole on the iris is shown by an arrow); (c) Posterior embryotoxon, the prominent Schwalbe’s line is indicated with arrows; (d) Iris strands bridging the chamber angle as seen on gonioscopy.
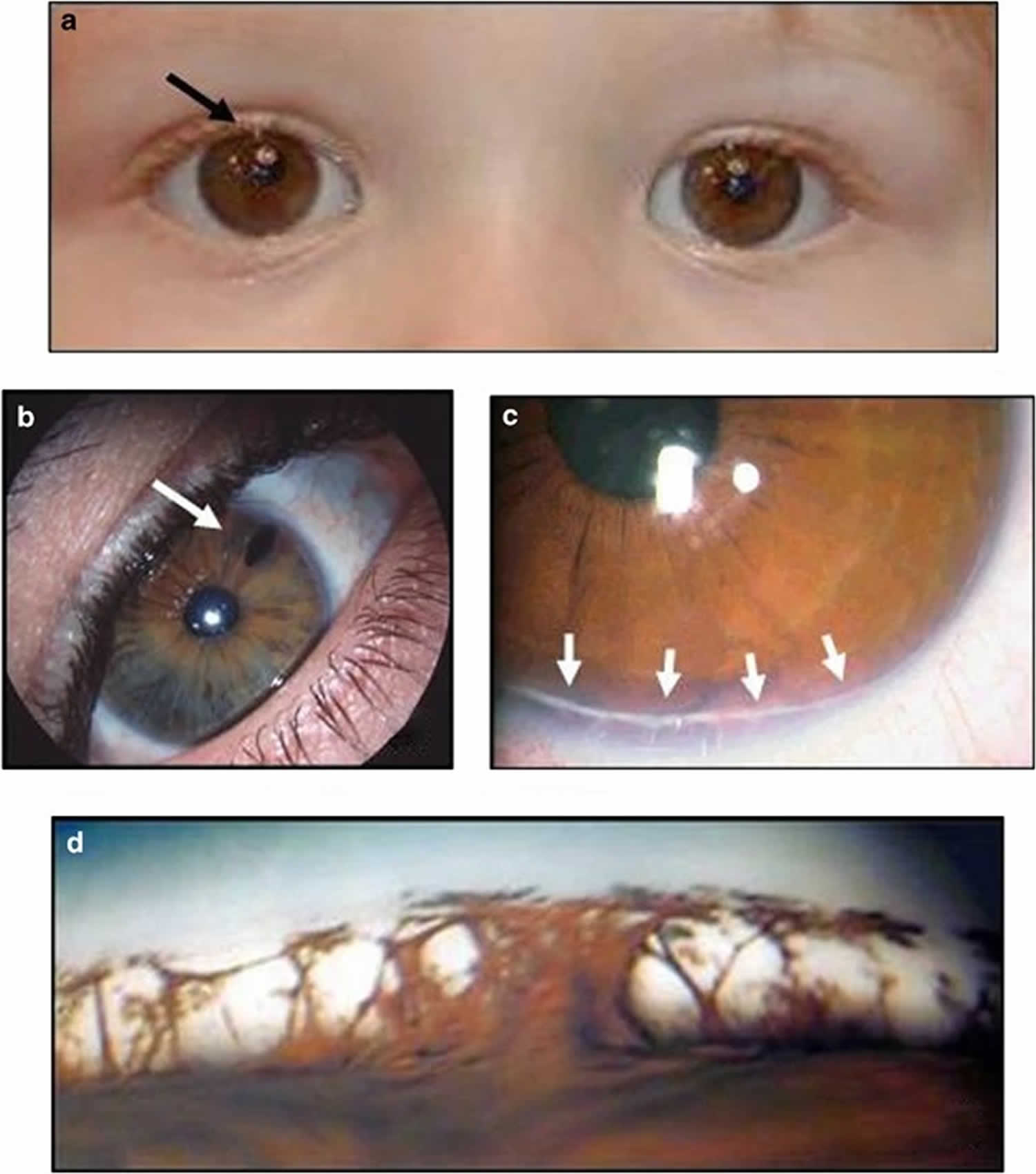
[Source 3 ]Figure 3. Axenfeld-Rieger syndrome in a young boy
Footnotes: Displacement of the pupil (corectopia), multiple holes in the iris (polycoria), and posterior embryotoxon in a young boy with Axenfeld-Rieger syndrome. This patient also has glaucoma, redundant periumbilical skin, maxillary hypoplasia, and teeth anomalies. Posterior embryotoxon is a prominent white line running parallel to the limbus on the endothelial surface of the peripheral cornea. Posterior embryotoxon is present in nearly all patients with Axenfeld-Rieger syndrome, but can also be seen in normal individuals or seen in the presence of other systemic disease such as Alagille’s syndrome 26. It is important to recognize that 8-15% of the normal population may have a subtle, mild form of posterior embryotoxon without other complications, including glaucoma 27. The posterior embryotoxon in Axenfeld-Rieger syndrome may be more dramatic and associated with other anterior segment findings, as described above 28. Posterior embryotoxon is a corneal abnormality that is visible with slit-lamp biomicroscopy as a thin grey-white, arcuate ridge on the inner surface of the cornea, adjacent to the limbus 29. Posterior embryotoxon is an anteriorly displaced Schwalbes line, the junction of Descemets membrane and the uveal trabecular meshwork 29. Histolgically, posterior embryotoxon consists of a central collagen core surrounded by a thin layer of Descemets membrane, and is separated from the anterior chamber by a layer of endothelium.
[Source 30 ]Figure 4. Axenfeld-Rieger syndrome
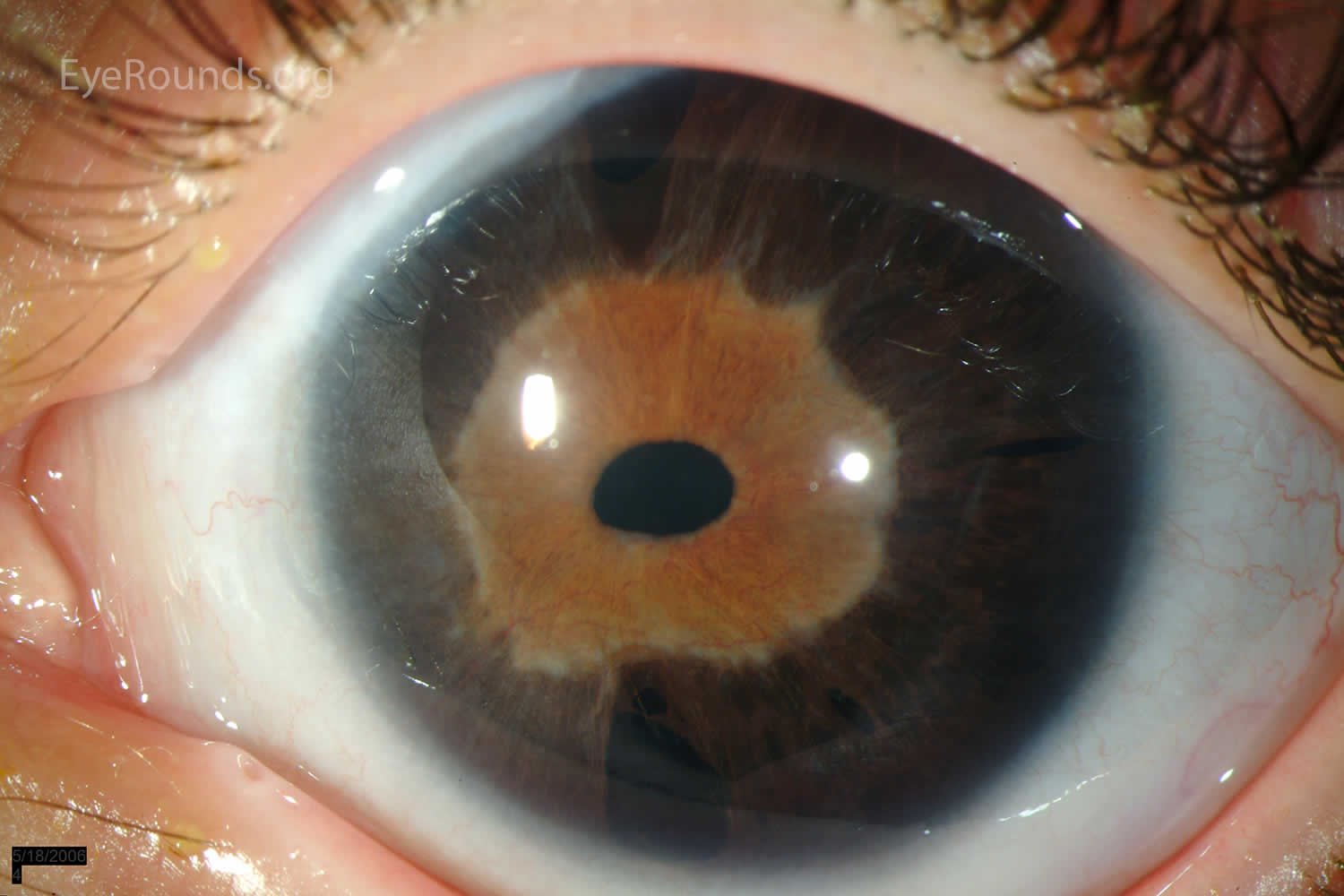
Figure 5. Normal aqueous outflow
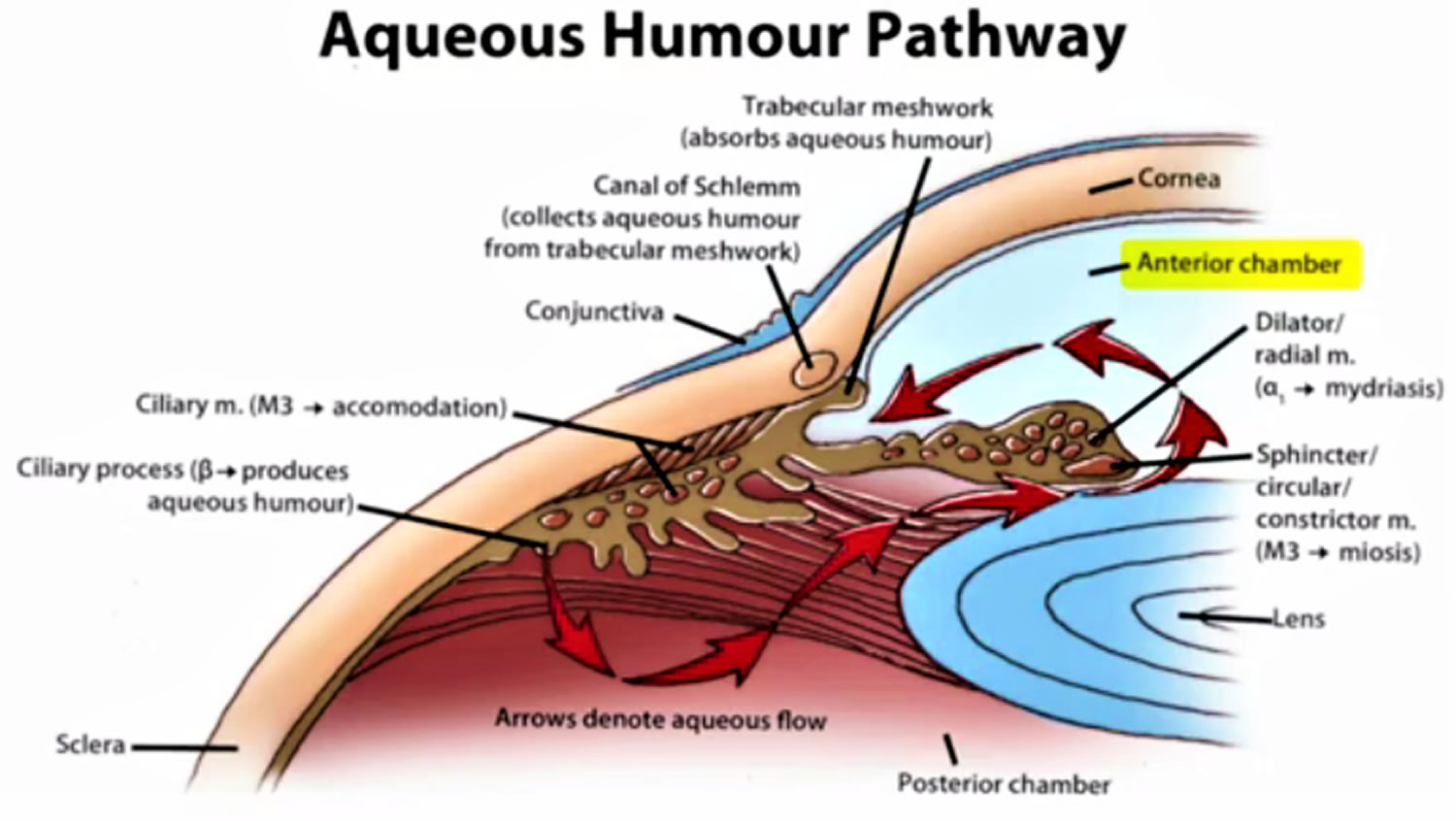
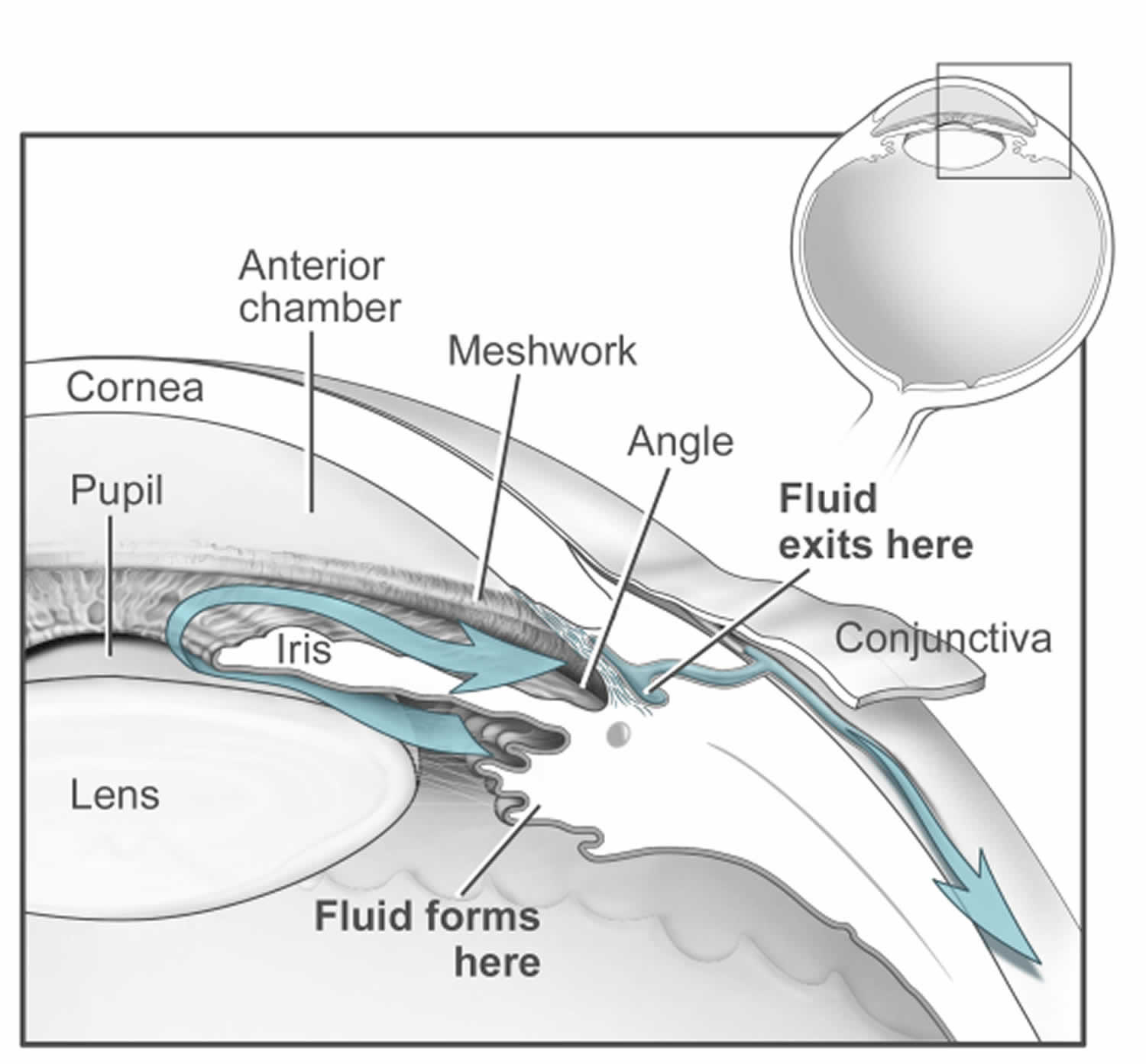
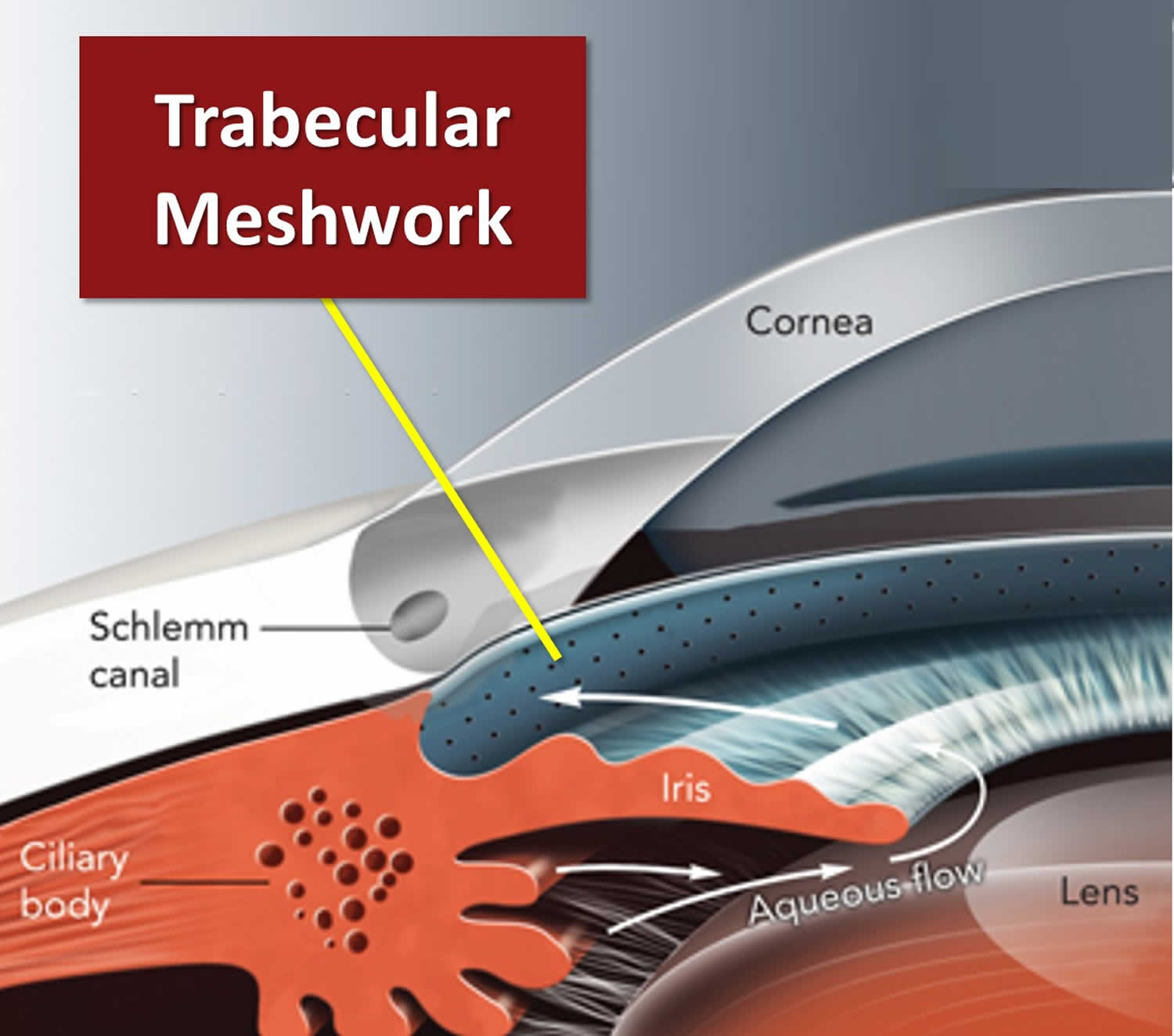
Footnotes: The ciliary body is a structure that sits directly behind the iris (the colored part of your eye). One of ciliary body’s jobs is to create an important fluid called aqueous humor, a fluid that nourishes the cornea and lens. Aqueous humor flows through a specific route into the front of the eye (the anterior chamber). This route allows aqueous humor to send important nutrients and oxygen to other parts of the eye, such as the lens and cornea. The aqueous humor is produced behind the iris, flows into the anterior chamber through the pupil, and exits the eye between the iris and cornea via the trabecular meshwork, a specialized eye tissue located at the chamber angle of the eye next to the cornea 32. In a healthy eye, this is a constant process. The ciliary body is always producing aqueous humor, and 80%-90% aqueous humor is always draining through the trabecular meshwork. The trabecular meshwork is a specialized spongy tissue in the anterior chamber of the eye that regulates the outflow of aqueous humor 32. The trabecular meshwork acts as a filter, controlling how quickly aqueous humor drains out of the eye through a structure called Schlemm’s canal, ultimately maintaining intraocular pressure (IOP). The canal of Schlemm, also known as Schlemm’s canal or the scleral venous sinus, is a circular, lymphatic-like vessel in the eye that drains aqueous humor from the anterior chamber into the episcleral blood vessels. The canal of Schlemm and the trabecular meshwork (TM) play a crucial role in maintaining intraocular pressure (IOP) by facilitating the outflow of aqueous humor. Too much aqueous humor production or obstruction of its outflow causes a rise in intraocular pressure (IOP) that can lead to glaucoma.
[Source 33 ]Figure 6. Posterior embryotoxon (black arrows)
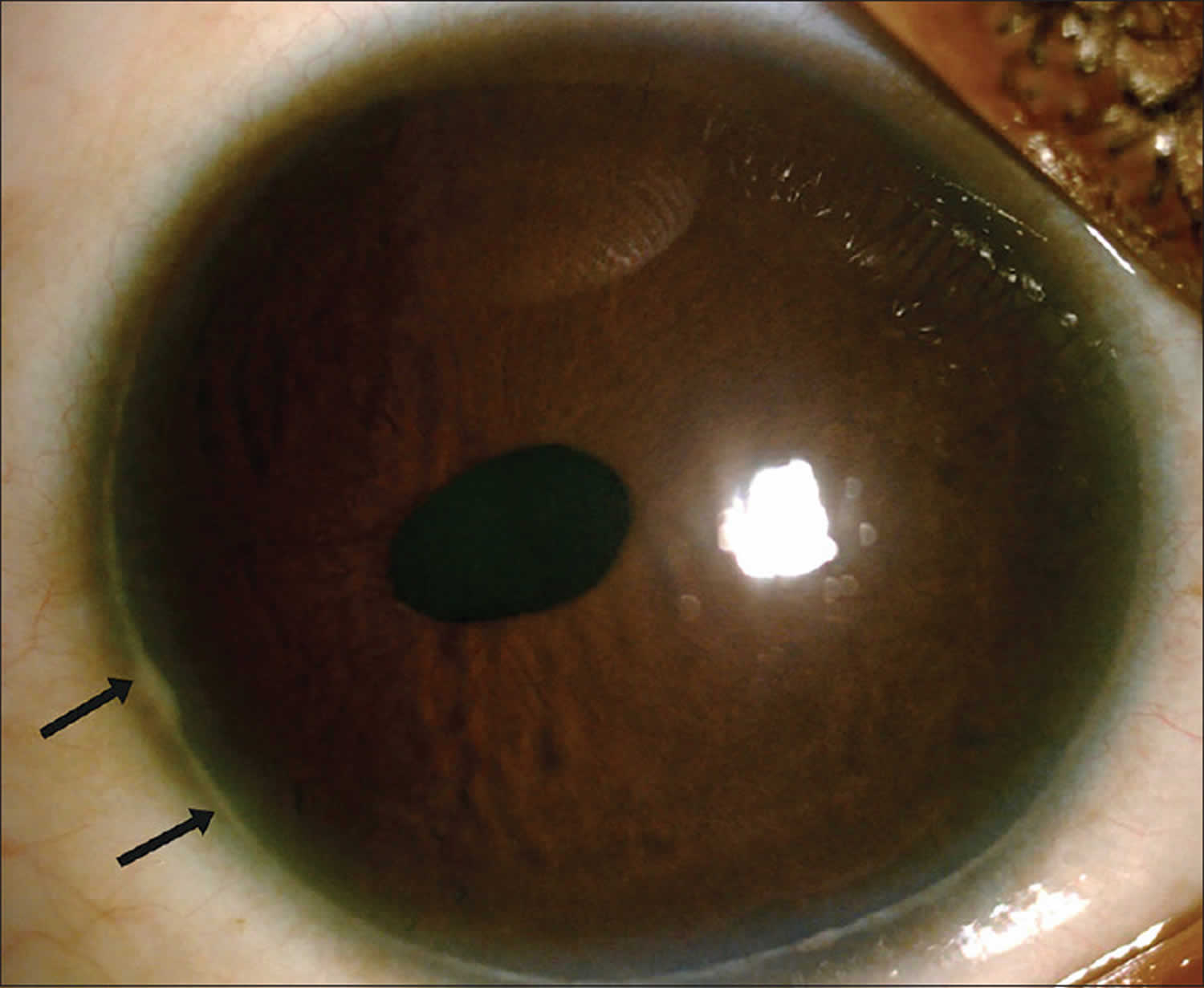
Figure 7. Gonioscopy
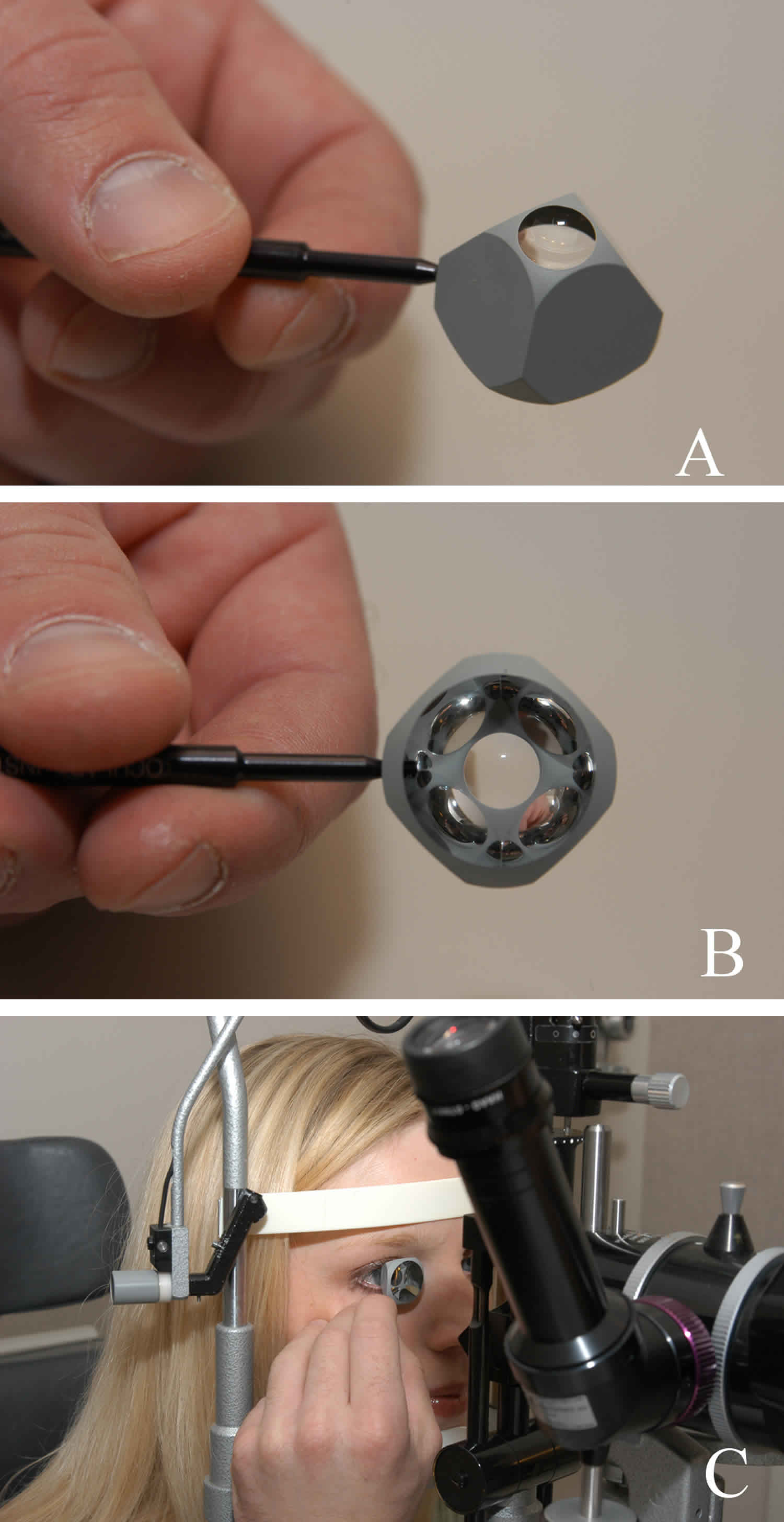
Footnotes: Gonioscopy. (A) and (B) Gonioscopy lens. (C) The gonioscopy lens is gently held against the cornea. Eye doctors look through the gonioscopy lens to see the drainage angle.
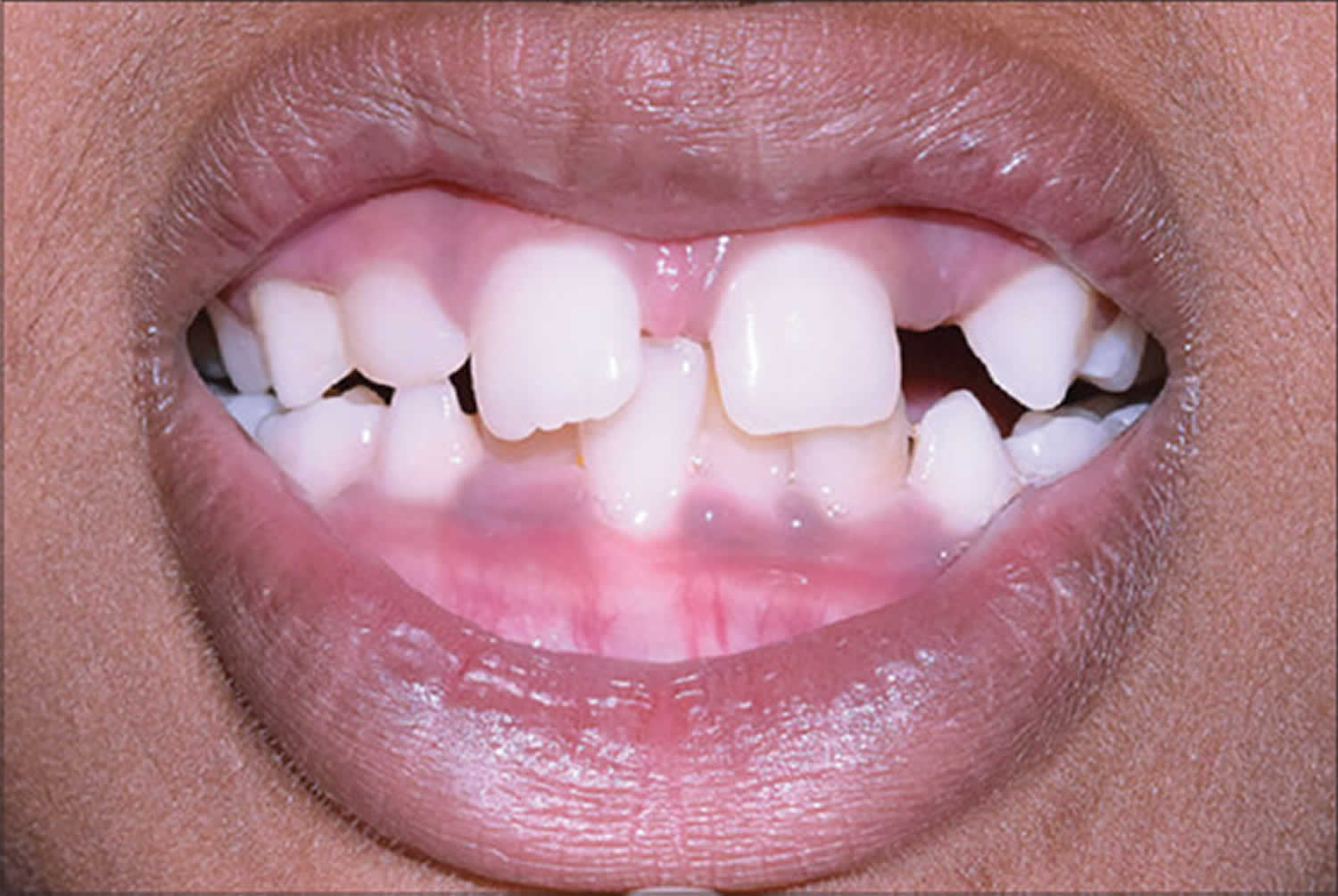
[Source 34 ]Figure 8. Axenfeld–Rieger syndrome dental abnormalities (oligodontia and microdontia)
What is the difference between Axenfeld-Rieger syndrome and aniridia?
Axenfeld-Rieger syndrome and aniridia are both congenital conditions (something you’re born with) that affect babies’ eyes.
Aniridia is the medical term for being born without an iris (the colored part of your eye). Some babies who have aniridia don’t have irises, while others only have part of an iris.
Axenfeld-Rieger syndrome usually causes symptoms in a child’s eyes, but it may not exclusively affect their irises. It most commonly causes issues in the front of the eye (the anterior chamber). It can also cause symptoms in other parts of your child’s body as they grow and develop.
Diagnosis for both conditions usually occurs when a baby is born. Visit your eye care specialist if you notice any changes in your baby’s eyes or vision.
Axenfeld-Rieger syndrome causes
Mutations in PITX2 gene on chromosome 4q25, FOXC1 gene on chromosome 6p25, PAX6 on chromosome 11p13, FOXO1A gene on chromosome 13q14, CYP1B1 gene on chromosome 2p22.2 and
COL4A1, PAX6, FOXE3, CPAMD8, and PXDN genes have been associated with Axenfeld-Rieger syndrome 9, 35, 36, 37, 38, 12, 39, 40, 41, 42, 40, 20. Cases of Axenfeld-Rieger syndrome with mutations in CYP1B1 have also been reported 43. However, approximately 40%–70% of Axenfeld-Rieger syndrome cases result from mutations in at least two known genes, PITX2 and FOXC1 genes 6. The FOXC1 gene, also known as forkhead box 1, has been referred to as forkhead, drosophila, homolog-like 7 (FKHL7), and forkhead-related activator 3 (FREAC3) 44.
Forkhead-Box C1 gene (FOXC1) gene mutations are more commonly associated with isolated ocular findings compared to Pituitary Homeobox 2 gene (PITX2) gene mutations that are associated with defects in other organ systems. However, rates of secondary glaucoma are similar between both mutations 45, 46, 47. FOXC1 mutations are also associated with a more variable eye presentation 45. Systemic manifestations are more commonly associated with PITX2 mutations compared to FOXC1 45, 46. PITX2 mutations have been shown to be more closely related to the development of Axenfeld-Rieger syndrome compared to FOXC1 48. Odontogenic disease, umbilical abnormalities, and Meckel diverticulum are more common in PITX2 mutations, whereas congenital heart disease, feeding difficulties, hearing loss, and skeletal abnormalities are more common in FOXC1 mutations 45.
Pituitary Homeobox 2 gene (PITX2) gene mutations cause Axenfeld-Rieger syndrome type 1, and Forkhead-Box C1 gene (FOXC1) gene mutations cause Axenfeld-Rieger syndrome type 3 5. The gene associated with type 2 is likely located on chromosome 13, but it has not been identified 5. The proteins produced from the PITX2 and FOXC1 genes are transcription factors, which means they attach (bind) to DNA and help control the activity of other genes 5. These transcription factors are active before birth in the developing eye and in other parts of the body. They appear to play important roles in embryonic development, particularly in the formation of structures in the anterior segment of the eye 5.
Mutations in either the PITX2 or FOXC1 gene disrupt the activity of other genes that are needed for normal development 5. Impaired regulation of PITX2 or FOXC1 genes leads to problems in the formation of the anterior segment of the eye and other parts of the body. These developmental abnormalities underlie the characteristic features of Axenfeld-Rieger syndrome. Affected individuals with PITX2 gene mutations are more likely than those with FOXC1 gene mutations to have abnormalities affecting parts of the body other than the eye 5.
Rates of glaucoma within the first 2 years of diagnosis are more frequent for FOXC1 mutations compared to PITX2, where secondary glaucoma may present in later childhood, adolescence, or adulthood 45. However, some people with Axenfeld-Rieger syndrome do not have identified mutations in the PITX2 or FOXC1 genes 5. In these individuals, the cause of the condition is unknown. Other as-yet-unidentified genes may also cause Axenfeld-Rieger syndrome 49..
Defects in differentiation, migration, or arrested development of neural crest cells in the anterior chamber, facial bones, teeth, cardiovascular system, and periumbilical skin are considered to be the etiological basis for the systemic and eye findings characteristic of Axenfeld-Rieger syndrome 16.
Histologically, patients with Axenfeld-Rieger syndrome have been found to have a monolayer of endothelial-like cells with a Descemet-like membrane extending from the cornea, across the anterior chamber and angle structures onto the surface of the iris 16. The membrane is typically found in the quadrant with associated the ectropion uveae/corectopia and the iris atrophy is found in the opposite quadrant 24.
The cause of glaucoma in Axenfeld-Rieger syndrome is thought to be angle dysgenesis or incomplete development of the trabecular meshwork and the Schlemm canal 9. It is believed that an arrest of the development of neural crest cells leads to the persistence of primordial endothelial cells over the angle and the iris, resulting in iridogoniodysgenesis that obstructs aqueous outflow and leads to glaucoma 9.
Axenfeld-Rieger syndrome inheritance pattern
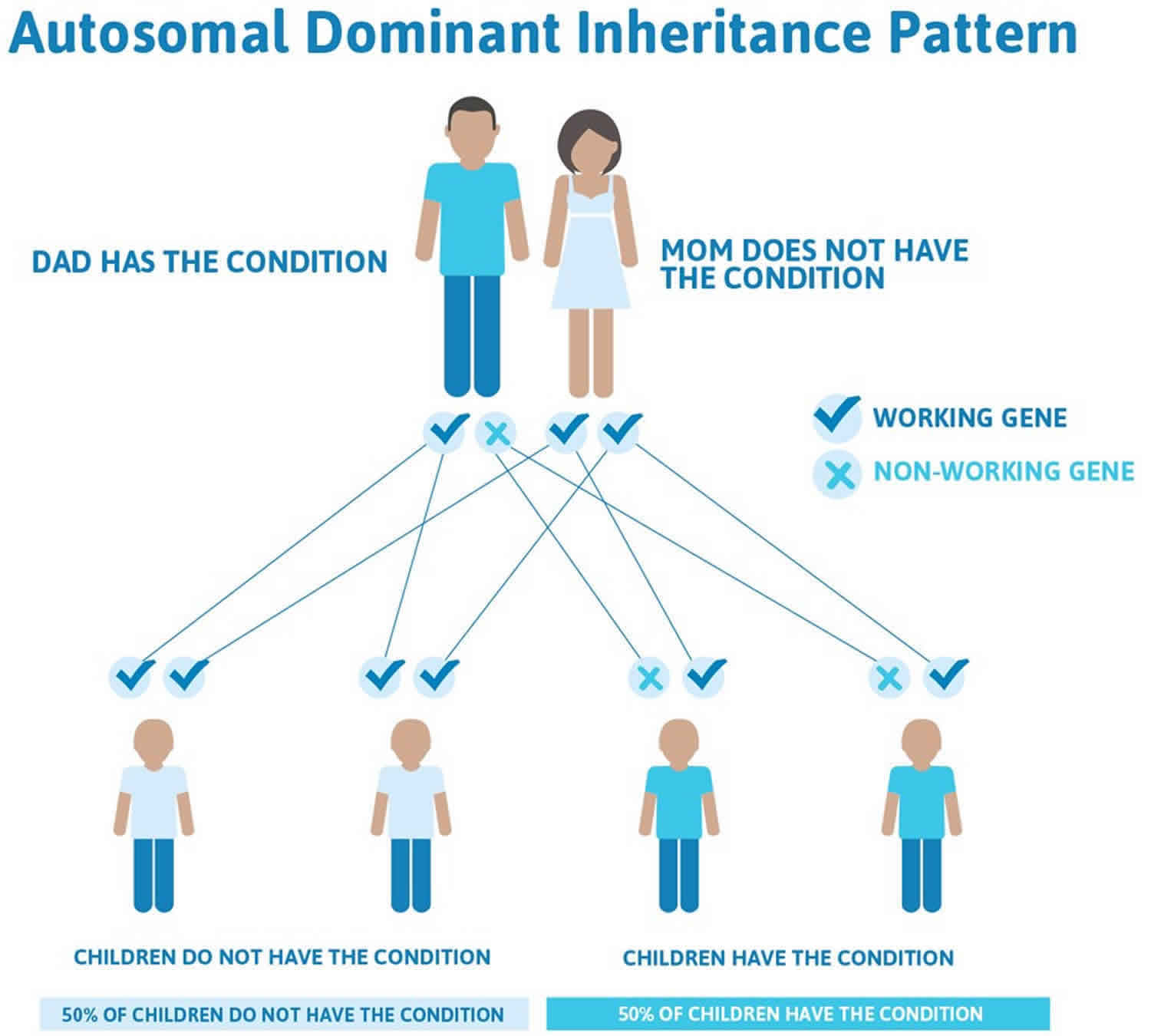
Axenfeld-Rieger syndrome is an autosomal dominant genetic disorder. That means only one copy of the genetic mutation needs to pass from one of a baby’s biological parents to their child for the child to inherit it. This doesn’t mean that if you carry the specific mutation in your genes that your baby will absolutely have Axenfeld-Rieger syndrome. However, there’s a 50% chance they will. Axenfeld-Rieger syndrome can also occur sporadically in 50%–70% of patients 50, 6. People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 9. Axenfeld-Rieger syndrome autosomal dominant inheritance pattern
Axenfeld-Rieger syndrome pathophysiology
Axenfeld-Rieger syndrome is believed to be caused by abnormal neural crest migration during early embryogenesis 9. Important eye structures such as the ciliary body, cornea, and iris stroma rely on adequate neural crest cell migration 9. Transcription factors in different genes strongly regulate these processes 51, 52.
During late fetal development, the primordial endothelium that covers the cornea is expected to undergo resorption; disruption in this process may result in the development of posterior embryotoxon and abnormal insertion of the iris, causing pupillary changes such as pseudopolycoria or ectropion uveae 51, 52. The previously discussed abnormalities in the development of the anterior chamber may affect the development of the Schlemm canal, impairing the outflow of aqueous humor and increasing the risk of glaucoma 51, 52.
Abnormal neural crest migration associated with Axenfeld-Rieger syndrome often affects extraocular tissues, such as vestibuloacoustic ganglion tissue, which may cause hearing loss in some patients with Axenfeld-Rieger syndrome 51, 52. The pathogenesis of Axenfeld-Rieger syndrome involves abnormalities in the differentiation, development, or migration of neural crest cells, contributing to the ocular and systemic features observed in affected individuals. The third wave of neural crest gives rise to the iris stroma 53.
Research suggests abnormalities or disturbances in the third wave of neural crest cell migration can result in Axenfeld-Rieger syndrome 54. It is proposed that the developmental arrest of the tissues derived from neural crest cells in late gestation leads to the retention of the primordial endothelium over the iris and the angle of the anterior chamber 7. This abnormal tissue continues to contract, resulting in some of the progressive changes seen in patients with Axenfeld-Rieger syndrome 7.
Axenfeld-Rieger syndrome signs and symptoms
Axenfeld-Rieger syndrome symptoms can be divided into eye symptoms that affect your child’s eyes and systemic symptoms that affect other parts of your child’s body.
Axenfeld-Rieger syndrome eye symptoms
Eye symptoms of Axenfeld-Rieger syndrome include:
- Thin or underdeveloped irises (corectopia) and multiple holes in the iris (polycoria).
- Off-centered pupils (the black center of your eye), or pupils that are out of their usual place.
- Issues with the cornea (the clear part at the front of your eye) called posterior embryotoxon. Posterior embryotoxon is a prominent white line running parallel to the limbus on the endothelial surface of the peripheral cornea. Posterior embryotoxon is present in nearly all patients with Axenfeld-Rieger syndrome, but can also be seen in normal individuals or seen in the presence of other systemic disease such as Alagille’s syndrome 26. It is important to recognize that 8-15% of the normal population may have a subtle, mild form of posterior embryotoxon without other complications, including glaucoma 27. The posterior embryotoxon in Axenfeld-Rieger syndrome may be more dramatic and associated with other anterior segment findings, as described above 28. Posterior embryotoxon is a corneal abnormality that is visible with slit-lamp biomicroscopy as a thin grey-white, arcuate ridge on the inner surface of the cornea, adjacent to the limbus 29. Posterior embryotoxon is an anteriorly displaced Schwalbes line, the junction of Descemets membrane and the uveal trabecular meshwork 29. Histolgically, posterior embryotoxon consists of a central collagen core surrounded by a thin layer of Descemets membrane, and is separated from the anterior chamber by a layer of endothelium. Shields published a case series of 24 patients with Axenfeld-Rieger syndrome, in which 5 patients had posterior embryotoxon visible only with gonioscopy 7.
Photophobia may be a symptom resulting from the pupillary and iris abnormalities.
Axenfeld-Rieger syndrome will make your child more likely to develop other eye conditions, including:
- Glaucoma. Glaucoma is seen in approximately 50% of the cases with Axenfeld-Rieger syndrome. Development arrest of the neural crest cells with their retention in the anterior chamber drainage angle during fetal development results in incomplete development of the trabecular meshwork or Schlemm canal 16. The extent of iris defects and iris stands in the angle do not correlate well with the severity of glaucoma. However, the high iris insertion appears to be more pronounced in eyes with glaucoma 7. Although glaucoma may present in early infancy, most cases occur during adolescence or early adulthood.
- Cataracts
- Coloboma, a birth defect where part of the eye’s tissue is missing, leading to a gap or hole in the iris, retina, choroid, or optic disc. These gaps can affect vision depending on their size and location, with those affecting the iris often causing a “keyhole” pupil appearance but typically not significantly impairing vision. Colobomas can also be present in other parts of the eye, including the lens and eyelids.
- Macular degeneration
- Strabismus (crossed eyes).
Axenfeld-Rieger syndrome systemic symptoms
Systemic symptoms of Axenfeld-Rieger syndrome are less common than ocular symptoms. If your child has systemic symptoms, they might include 6, 55, 56, 57, 58:
- Issues with their skull. Hypertelorism (wide-set eyes) and a flattened face shape. Axenfeld-Rieger syndrome can also cause babies to have an unusually broad, prominent brow or forehead.
- Teeth symptoms. Babies with Axenfeld-Rieger syndrome are sometimes born with unusually small teeth. They might be missing some teeth too.
- Extra skin. Your child might develop extra folds of skin on their abdomen near their belly button. Male babies sometimes also develop extra skin on the underside of their penis.
- Narrow urethra or anal openings.
- Heart defects.
- Issues with their pituitary gland. Babies born with Axenfeld-Rieger syndrome can sometimes have delayed growth (they take longer to start growing and developing than most children).
Abnormalities of the pituitary gland and other surrounding areas are less common, but more serious findings. Cases of empty sella syndrome 59 and arachnoid cysts have been reported 60. Growth hormone deficiency and short stature have also been described 60.
Axenfeld-Rieger syndrome diagnosis
Your child’s doctor or an eye specialist will diagnose your child with Axenfeld-Rieger syndrome. Diagnosis usually occurs at birth if your child has obvious issues with their eyes or other parts of their body. Diagnosis may also occur after your child develops symptoms.
Your child’s eye specialist (ophthalmologist) will perform an eye exam to look at your child’s eyes (including inside them). They might perform a few tests to diagnose Axenfeld-Rieger syndrome, including:
- A visual acuity test to see if your child’s vision is affected.
- Gonioscopy to check for glaucoma.
- DNA tests and genetic testing to confirm if your child has the mutations that cause Axenfeld-Rieger syndrome.
Physical Examination
The clinical examination must include 61:
- Fixation of light: The patient’s ability to fixate and follow light should be tested with each eye separately. There may be exotropia (where one or both eyes turn outward, away from the nose) due to poor fixation and nystagmus in long-standing cases.
- Sclera: The sclera(e) may appear bluish in color because of high myopia, scleral thinning, and exposure to underlying uveal tissue 62.
- Cornea: Corneal examination might reveal signs of corneal enlargement or buphthalmos. Normal corneal size from birth to 6 months should be between 9.5 to 11.5 mm. A size of greater than 12 mm should raise the suspicion of glaucoma. A corneal diameter of more than 13 mm in any child older than 6 months indicates corneal enlargement. The slit-lamp examination may reveal horizontal or oblique tears and breaks in the Descemet membrane called Haab striae (see Figure 4). Another critical finding is corneal edema. This usually starts as epithelial edema and then gradually involves the deeper layers of the cornea, occasionally causing permanent opacities impairing vision profoundly 63.
- Anterior chamber: The anterior chamber is usually deep.
- Iris: Iridodonesis, ectropion uvea, hypoplasia, or any atrophic patches may be present 64.
- Pupil: The pupil may be oval, dilated, and ischemic.
- Lens: The clinician should evaluate for lenticular opacities or lens subluxation due to excessive stretching of zonules 65.
- Optic disc: This typically demonstrates reversible cupping in the early stages. Later stages may present with an enlarged cup-to-disc ratio or even atrophy 66, 67.
- Intraocular pressure (IOP): Intraocular pressure (IOP) is usually elevated at presentation and can be measured using a pneumotonometer in the outpatient setting 68.
Diagnostic procedures
The main diagnostic test for primary congenital glaucoma is the measurement of the intraocular pressure (IOP), which should be done prior to instilling dilating drops. In a cooperative infant or young child, this measurement can be obtained in the clinic setting with a Perkins applanation tonometer, Tono-pen (a portable Mackay-Marg-type tonometer) and/or Icare rebound tonometer. In older patients, standard Goldmann applanation tonometry can be performed. A pneumotonometer may be useful to confirm intraocular pressure (IOP) during examination under anesthesia or in clinic if available, and may be less influenced by corneal abnormalities. A Schiötz indentation tonometry is not recommended in these patients due to under- or overestimation of intraocular pressure (IOP) in childhood glaucoma 69, 70. For the uncooperative child, an examination under anesthesia should be performed.
Of note, the Icare rebound tonometer has decreased the need for examinations under anesthesia as it does not require a topical anesthetic 71. Two models available in the United States (Icare TAO1i and Icare ic100) require the patient to be upright, while the newest model, recently approved in the US (Icare ic200), allows measurement in a supine patient. The IOP measured by Icare in cooperative, awake children with known or suspected glauacoma, has been shown to be within 3 mmHg of IOP obtained by Goldmann applanation tonometry in 63% and is higher than measured by Goldmann applanation tonometry in 75% of children 72. By contrast, Icare tonometry may under-measure the IOP compared to Tono-pen readings in the setting of corneal edema 73.
Because anesthetic agents variably alter the intraocular pressure (IOP), with most lowering intraocular pressure (IOP), measurements should be obtained as soon as possible after induction of anesthesia and before intubation. If the intraocular pressure (IOP) is actually elevated, it often remains greater than 20 mmHg under anesthesia, which suggests glaucoma. The normal intraocular pressure (IOP) is lower in infants and young children than adults. A newborn has an average intraocular pressure (IOP) of 10-12 mm Hg, increasing to 14 mm Hg by age 7 or 8 years of age. An asymmetric measurement or an elevated intraocular pressure (IOP) measurement in the presence of other clinical signs helps make the diagnosis of glaucoma.
Corneal diameter measurement is another key diagnostic procedure for primary congenital glaucoma. Some providers check horizontal diameters only, while some check horizontal and vertical diameters. If there is pannus or scarring obscuring the limbus, the measurement may not be accurate. In the office a millimeter ruler can be placed above the eyes and if the child is not cooperative, a close-up digital photograph can be taken with the ruler in place, and a measurement can be made from the photo. This is most amenable to horizontal corneal diameter measurement. While under anesthesia, calipers with the tips placed at the limbus 180 degrees apart are used across the widest diameter, and then measured with a graduated ruler to check the measurement. Ideally, the measurement can be estimated to the nearest 0.25 mm 74.
Examination for Haab striae is done with an oblique slit beam with a portable slit lamp if the patient is younger or under anesthesia, or on a regular slit lamp in the clinic if the patient is older. Retroillumination can also be used to identify Haab striae. . In older patients with treated primary congenital glaucoma, corneal endothelial protuberances and hyperproliferation of the Descemet membrane/pre-Descemet’s layer complex have been demonstrated with anterior segment OCT (ASOCT). These may demarcate areas in which the edges of the Descemet membrane have re-approximated during the healing process 63.
If a view through the cornea allows it, gonioscopy is done in clinic if tolerated, ideally a Sussman (or similar) indirect gonioscopy lens as it fits easily between a young child’s small palpebral fissure. Using a gonioscopy lens without a handle may be easier as it allows the examiner to hold open the eyelids while placing the lens. More commonly, for initial diagnosis of primary congenital glaucoma, gonioscopy is performed under under anesthesia with a Koeppe or similar direct gonioscopy lens and portable slit lamp. There are different sized Koeppe lenses to fit different corneal diameters. The Koeppe lens is best handled with a glove to avoid fingerprint smudges. The Koeppe lens cup is filled with balanced salt solution and placed quickly on the eye or placed on the eye and tilted with one edge abutting the sclera while filling the space between the lens and eye with solution. Then a binocular microscope such as the portable slit lamp is angled towards the angle of interest and the lens can be shifted slightly toward the angle to optimize the view.
Gonioscopy in these cases helps guide surgical planning in cases of primary congenital glaucoma, and may also identify other angle abnormalities which might identify other secondary glaucoma types, for example Axenfeld-Rieger anomaly (many irido-corneal attachments with anteriorly placed Schwalbe line). Infants with primary congenital glaucoma usually do not have a visible scleral spur because the peripheral iris inserts into the trabecular meshwork (in contrast to normal infants whose peripheral iris and ciliary body have recessed to the scleral spur or posterior to it). There may also be scalloped edges of the peripheral iris and pale peripheral iris stroma in front of the angle causing a “morning mist” appearance. If there are peripheral anterior synechiae, posterior embryotoxon, or other abnormalities, then the diagnosis is unlikely primary congenital glaucoma. Gonioscopy photographs can be taken by instilling the eye with coupling gel and angling the camera lens (i.e. RetCam) obliquely toward the angle of interest and adjusting the focus until the angle comes into clear view.
Axial length is measured with A-scan ultrasonography, ideally using the immersion and not contact method, either in clinic or under anesthesia. It is best done under anesthesia, during baseline examination to determine if the axial length is greater than normal for the patient’s age, and repeated approximately every 3-4 months to assess if the growth rate is greater than average. Of note, measuring axial length itself is not an indication for examination under anesthesia if a patient is otherwise doing well, and can be performed at intervals when examination under anesthesia is needed for clinical management. Sampaolesi and Kiskis provided linear regressions from data of normal children. Sampaolesi used immersion A-scans and found the normal axial length for a one-month-old lies between 17.25 mm (5th percentile) and 20.25 mm (95th percentile). Sampaolesi also recommended that axial length be measured after dilation with cycloplegic drops 75, 74.
Optic nerve evaluation is performed with either indirect or direct ophthalmoloscopy with attention to the cup-to-disc ratio. In the setting of a small pupil, a magnified view of the nerve can be obtained by using a direct ophthalmoscope through a Koeppe gonioscopy lens on the eye. Fundus photography is also recommended for comparisons between serial examinations. B-scan ultrasonography is recommended if the cornea does not allow fundus examination to rule out posterior disease. Severe optic nerve cupping may sometimes be noted on the posterior B-scan.
Pachymetry is used to measure central corneal thickness. The central cornea may be thicker due to corneal edema, and has also shown to be thinner in primary congenital glaucoma patients without corneal edema, likely due to stretching of their tissues 76. Other small studies have shown either no significant difference in central corneal thickness between normal eyes and eyes treated for primary congenital glaucoma, or the central corneal thickness was thicker in eyes treated for primary congenital glaucoma than in normal eyes 77, 78. Corneal hysteresis and corneal resistance factor have been found to be lower in eyes with primary congenital glaucoma compared to normal eyes 77, 78.
Perimetry can be attempted starting around age 7-8 years of age if the patient does not have nystagmus, cognitive impairment or severe vision loss. Quicker testing algorithms such as SITA-FAST may allow children to perform more reliably 79. Goldman perimetry can be very helpful in young children.
Standard tabletop optical coherence tomography (OCT) can be considered once a child can be examined at the regular slit lamp to evaluate the retinal nerve fiber layer and ganglion cell layer. It may be helpful especially if the child cannot perform perimetry. While devices currently do not carry normative data for children, studies have collected data on normal children 80, 81, 82, 83. Handheld and mounted spectral-domain OCT devices are emerging technologies that can be used during examination under anesthesia 84, 85.
Childhood glaucoma diagnostic criteria
Childhood glaucoma diagnostic criteria per Childhood Glaucoma Research Network definition 74:
Definition of childhood glaucoma required two or more of the main categories (1‑5)
- Intraocular pressure (IOP) >21 mm Hg (investigator discretion if examination under anaesthesia data alone)
- Optic disc cupping
- Progressive increase in cup‑disc ratio
- Cup‑disc asymmetry of ≥0.2 when disc sizes are similar
- Focal rim thinning
- Corneal findings
- Haab striae
- Diameter
- >11 mm in newborn
- >12 mm in child <1 year of age
- >13 mm any age
- Progressive myopia /myopic shift coupled with an increase in ocular dimensions out of keeping with normal growth
- Reproducible visual field defect that is consistent with glaucomatous optic neuropathy with no other observable reason for the visual field defect.
Axenfeld-Rieger syndrome differential diagnosis
- Iridocorneal Endothelial (ICE) Syndrome. Iridocorneal endothelial (ICE) syndrome is a rare eye disorder characterized by the abnormal proliferation and migration of corneal endothelial cells (CECs) onto the iris and into the iridocorneal angle. This abnormal cell growth leads to several complications, including corneal edema, iris atrophy, secondary angle-closure glaucoma, and potential vision loss. Iridocorneal endothelial (ICE) syndrome is typically unilateral and nonhereditary, is more common in women and manifests in early adulthood. Iridocorneal endothelial (ICE) syndrome is encompassed by three variations (1) Chandler’s Syndrome, (2) essential iris atrophy, (3) Cogan-Reese (iris nevus). Chandler’s syndrome is produced when the pathologic changes are confined to the inner corneal surface with dysfunction of the endothelial pump resulting in corneal edema. Essential iris atrophy is produced when the abnormal endothelium proliferates onto the iris surface with subsequent contractile membranes resulting in pupil corectopia, iris atrophy, polycoria. If the malformed and dysfunctional endothelium spreads across the anterior chamber angle, peripheral anterior synechiae develop and result in glaucoma. Cogan-Reese (iris nevus) is manifested by multiple pigmented iris nodules caused by contraction of the endothelial membranes on the surface of the iris. The unilateral nature, corneal endothelial changes, manifestation in middle age, female predominance, and lack of systemic abnormalities differentiate iridocorneal endothelial (ICE) from Axenfeld-Rieger syndrome 24, 86.
- Peters’ Anomaly. Peters anomaly is a rare congenital disorder that causes a cloudy, opaque central area in the cornea, the clear front part of the eye, associated with an absence of Descemet’s membrane and endothelial layers often with adhesions between the cornea and the iris from the border of the corneal opacity 87. Peters anomaly is a type of anterior segment dysgenesis, meaning the front part of the eye doesn’t develop properly during fetal development. This can lead to vision problems, glaucoma, and other eye-related issues 87. Around 60% of cases are bilateral and can be associated with congenital glaucoma, aniridia, and microcornea. Peters’ anomaly can also be associated with systemic abnormalities including developmental delay, heart defects, hearing loss, central nervous system (CNS) defects, gastrointestinal and genitourinary defects 87. Peters’ anomaly is usually sporadic, but it can be inherited in an autosomal dominant or recessive pattern caused by PAX6, PITX2, CYP1B1, or FOXC1 genes. Although there are many similarities, the significant corneal changes differentiate Peters’ anomaly from Axenfeld-Rieger syndrome 88.
- Aniridia (Iris Hypoplasia). Aniridia also called iris hypoplasia is a rare genetic eye condition characterized by a partial or complete absence of the iris, the colored part of the eye 89. It can affect vision and may lead to other eye complications 89. While the majority of cases are congenital, aniridia can also be acquired, such as after trauma or surgery 89. Aniridia is also characterized by pannus extending onto the central cornea from stem-cell deficiency, cataract, foveal hypoplasia, decreased vision, or nystagmus 89. Inheritance of aniridia is typically autosomal dominant, but 30% can be sporadic mutation in PAX6 89. Aniridia is associated with a 50-75% risk of glaucoma, commonly resulting from rotation of the rudimentary iris into the anterior chamber angle obstructing aqueous outflow and may not occur until the second decade of life 89. One must consider WAGR (Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation), an autosomal dominant form seen in 13% of patients with aniridia. Gillespie syndrome, an autosomal recessive form of aniridia, is associated with cerebellar ataxia and mental retardation seen in 2% of patients with aniridia. The conreal pannus, foveal hypoplasia, and iris hypoplasia differentiate aniridia from Axenfeld-Rieger syndrome 90.
- Congenital Ectropion Uveae. Congenital Ectropion Uveae is a rare genetic, non-syndromic developmental defect of the eye with unilateral anterior chamber dysgenesis that commonly leads to unilateral secondary glaucoma in the mid-teenage years 91. Congenital displacement or dragging of the posterior pigmented epithelium of the pupil caused by contraction of myofibroblasts over the papillary margin 92. Possibly a component of Axenfeld-Rieger syndrome, iridocorneal endothelial (ICE) syndrome, neurofibromatosis, facial hemihypertophy, Prader-Willi syndrome, but can be seen as an isolated finding. Congenital ectropion uveae can also be seen with rubeosis iridis. Unilateral congenital iris ectropion; smooth, cryptless iris surface; high iris insertion; dysgenesis of the anterior chamber angle; glaucoma is consistent with the diagnosis of congenital iris ectropion syndrome 93, 94.
- Ectopia Lentis et Pupillae. Ectopia lentis et pupillae is a rare, inherited eye disorder characterized by displacement of the pupil, typically inferotemporally, associated with subluxation of the lens, usually in the opposite direction 95. Ectopia lentis et pupillae often affects both eyes (bilateral) and may involve other ocular abnormalities. Ectopia lentis et pupillae is also characterized by microsperophakia, miosis, and poor papillary dilation. Pathologically thought to be secondary to a defect in neuroectodermally derived tissues including the zonules, as well as the dilator muscle and pigmented layer of the pupil. Glaucoma is typically not a feature of ectopia lentis et pupillae 96. Ectopia lentis et pupillae is typically an autosomal recessive condition, meaning individuals must inherit two copies of the affected gene to be affected 95.
- Oculodentodigital Dysplasia. Oculodentodigital dysplasia (ODDD) is a rare genetic disorder that affects many parts of the body, primarily the eyes (oculo-), teeth (dento-), and fingers (digital) 97. People with oculodentodigital dysplasia often have eye abnormalities that can lead to vision loss. These eye problems can include underdeveloped and small eyes (microphthalmia) and clouding of the lenses of the eyes (cataracts). People with oculodentodigital dysplasia may also have a condition called microcornea, in which the clear front covering of the eye (cornea) is small and abnormally curved. The tooth abnormalities seen in people with oculodentodigital dysplasia often include missing or small teeth, weak enamel, and early tooth loss. Oculodentodigital dysplasia (ODDD) can also affect the fingers and toes. Individuals with oculodentodigital dysplasia may have permanently bent fingers (camptodactyly), fingers that are unusually curved (clinodactyly), webbing of the skin (syndactyly) between the fourth and fifth fingers, and syndactyly of the toes. Individuals who have oculodentodigital dysplasia may also have abnormalities of the head and face (craniofacial anomalies), which can include a narrow nose, a small head size (microcephaly), and an opening in the roof of the mouth (cleft palate). Less common features of oculodentodigital dysplasia include sparse hair growth (hypotrichosis); brittle nails; and a skin condition called palmoplantar keratoderma that causes the skin on the palms of the hands and the soles of the feet to become thick, scaly, and calloused. Approximately 30 percent of people with oculodentodigital dysplasia experience neurological problems such as a lack of bladder or bowel control (incontinence), difficulty coordinating movements (ataxia), abnormal muscle stiffness (spasticity), and impaired speech (dysarthria). Some affected individuals develop abnormalities of the brain’s white matter that can be detected with medical imaging (leukodystrophy). Hearing loss and heart problems have also been reported in people with oculodentodigital dysplasia. The signs and symptoms of oculodentodigital dysplasia (ODDD) vary widely among affected individuals. Some features of oculodentodigital dysplasia are evident at birth, while others become apparent with age. The lack of angle changes differentiates this entity from Axenfeld-Rieger syndrome 98.
Axenfeld-Rieger syndrome treatment
Axenfeld-Rieger syndrome treatment depends on where your child has symptoms and what (if any) issues their symptoms cause. Talk to your child’s doctor or your child’s eye specialist (ophthalmologist) about which treatments your child will need and how often they need follow-up exams to monitor changes in their eyes and body.
Treating glaucoma caused by Axenfeld-Rieger syndrome
Because it’s so common for kids with Axenfeld-Rieger syndrome to develop childhood glaucoma, your child will probably need treatment for it at some point. Glaucoma is a general term doctors use to describe a group of eye disorders that damage the optic nerve. Without prompt treatment, glaucoma can cause permanent blindness. In most cases, fluid builds up in the front part of the eye. The extra fluid puts pressure on the eye, gradually damaging the optic nerve. Eye doctors refer to this pressure as intraocular pressure (IOP), or eye pressure.
The most common treatments for childhood glaucoma include:
- Medicated eye drops.
- Laser treatments to drain fluid from your child’s eyes.
- Surgery to relieve eye pressure.
Both medications and surgery have been successfully used to treat childhood glaucoma. However, medicated eye drops of congenital and childhood glaucoma is generally used as an adjunct (add-on) to surgical interventions. As in congenital glaucoma, surgery to relieve eye pressure is more efficacious than medicated eye drops in Axenfeld-Rieger syndrome 99. However, achieving long term surgical success in congenital and developmental glaucoma is also difficult and complications are common 100, 101, 102, 103, 104, 105, 106.
Treating glaucoma can slow down additional vision loss. However, it can’t restore lost vision. It’s important to see your child’s eye care specialist right away if your child has glaucoma symptoms, including:
- Eye pain.
- Severe headaches.
- Vision problems.
Medications
Medications for congenital glaucoma is typically used as an adjunct (add-on) to surgery. Most medications in the United States have not been approved for children, however many studies have been performed that inform doctors on their safety and efficacy in children. Timolol (a non-selective beta blocker) is the first choice in childhood glaucoma 107, 108. In cases with insufficient reduction of the intraocular pressure (intraocular pressure (IOP)), the combination of timolol once a day and a carbonic anhydrase inhibitor dorzolamide twice a day brings about a good control of the intraocular pressure (IOP) 109. Both medications are effective and well tolerated. Prostaglandin analogues may be used to lower intraocular pressure (IOP) 110, 111. The alpha2-agonists, especially brimonidine, have more and potentially serious adverse effects in children with potentially serious apnea, bradycardia, hypotension, hypotonia, and central nervous system (CNS) depression and are contraindicated for children younger than 2 years of age 112. Apraclonidine should be used with caution for the same considerations but seems to be safer than brimonidine 113. Latanoprost tends to be less effective in lowering intraocular pressure (IOP) in children than in adults 114.
- Beta-blockers (beta-adrenergic antagonists): Topical beta-blockers play a large role in primary congenital glaucoma treatment and include timolol (non-selective beta-1 and beta-2 blocker, concentrations of 0.1% available in some countries, 0.25% and 0.5% solutions, and 0.25% and 0.5% gel-forming solution), and betaxolol (selective beta-1-blocker, concentrations of 0.25% and 0.5% solutions). Given potentially high plasma levels of the medication from topical instillation in small children, the lowest concentration available should be initiated first. The solution drops are approved for BID dosing though may be just as effective dosed once in the morning. The gel-forming solutions are approved for once daily dosing. Beta-blockers typically reduce intraocular pressure (IOP) by 20-30%. Side effects are mainly systemic and include respiratory distress, caused by apnea or bronchospasm (which may present as coughing instead of wheezing), and bradycardia. Beta-blockers should be avoided in patients with bradycardia, second- or third-degree atrioventricular block, and active asthma or “reactive airways.” Betaxolol may be less likely to cause pulmonary distress (e.g. asthma attacks) and cardiac side effects 115.
- Carbonic anhydrase inhibitors: Oral carbonic anhydrase inhibitors include acetazolamide (Diamox, dose 10-20 mg/kg/day divided into 3 or 4 doses) and methazolamide (Neptazane, dose < 2 mg/kg/day, divided into 2 doses) 116, 74. Acetazolamide can be prepared in a flavored syrup (have the pharmacist crush the tablets and suspend the powder in syrup) with a concentration of 50 mg/ml for ease of use. Children can also take the tablet crushed in applesauce or something similar. It reduces the intraocular pressure (IOP) about 20-35%. Side effects occur in >40% of patients and include lethargy, decreased appetite, weight loss, gastrointestinal discomfort, diarrhea and metabolic acidosis. Topical carbonic anhydrase inhibitors include dorzolamide 2% (Trusopt) and brinzolamide 1% (Azopt) drops twice a day (BID) or three times a day (TID). These medications may produce less reduction in intraocular pressure (IOP) (about 25%) than oral carbonic anhydrase inhibitors, but also appear to have fewer systemic side effects. Rarely, side effects can occur, particularly in premature infants, such as metabolic acidosis 117. Topical carbonic anhydrase inhibitors ideally should be avoided or used as a later option in the setting of compromised corneas, especially of a corneal transplant 74.
- Combination beta-blocker/carbonic anhydrase inhibitor: Timolol 0.5%-dorzolamide 2% (Cosopt) drop twice a day (BID) has been shown to be effective in reducing intraocular pressure (IOP) in children requiring more than one topical medication. It is approved for twice a day (BID) dosing, but cautious use in young children is warranted due to the higher concentration of timolol.
- Adrenergic agonists (avoid or use with caution in children younger than age 6 years or weight less than 20 kg): Apraclonidine 0.5% (Iopidine) and brimonidine (Alphagan, Alphagan P, 0.1%, 0.15%, 0.2%) are alpha-2 selective agonists and are dosed twice a day (BID) to three times a day (TID). Their effectiveness has not been studied specifically for primary congenital glaucoma. The side effects in children limit their use. Due to being highly lipophilic, brimonidine passes through the blood-brain barrier potentially causing severe sleepiness, respiratory depression, apnea and coma, especially in neonates and infants, thus it is strictly contraindicated in patients 2 years old or younger. It may also cause bradycardia, hypotension, hypotonia, and hypothermia. Apraclonidine is more hydrophilic which reduces its blood-brain barrier penetration and thus has fewer central nervous system side effects than brimonidine. It must still be used with caution and is best used for short- or intermediate-term intraocular pressure (IOP) lowering. Tachyphylaxis and ocular allergy limit its effectiveness long-term.
- Combination beta-blocker/alpha-2 adrenergic agonists: Timolol 0.5%-brimonidine 0.2% (Combigan) must not be prescribed to children if there is a contraindication to the individual components.
- Prostaglandin analogs: Latanoprost 0.005% (Xalatan), travoprost 0.004% (Travatan), bimatoprost 0.01% (Lumigan), and tafluprost (Zioptan, preservative-free) are dosed nightly. Latanoprost reduces the intraocular pressure (IOP) in primary congenital glaucoma 15-20% 118. While the FDA has not approved prostaglandin analogs in children, Europe has approved latanoprost for children. Side effects mainly include lash growth, conjunctival injection, and less commonly iris pigmentation alteration, allergy, uveitis and periocular hyperpigmentation. Side effects seem more prominent with use of travoprost and bimatoprost and less with latanoprost. Long-term side effects are still unknown in children. Prostaglandin-related periorbitopathy has been described in children 119. This class of medication is relatively contraindicated when active inflammation or uveitis is present.
- Combination beta-blocker/prostaglandin analog: Available in countries outside the United States.
- Miotic agents: These do not play much of a role in primary congenital glaucoma likely due to their immature angle anatomy and high ciliary muscle insertion. They include echothiophate, phospholine iodide (irreversible cholinesterase and pseudocholinesterase inhibitors) and pilocarpine (direct parasympathomimetic). Miotic are useful perioperatively for angle surgery. Pilocarpine (0.5-6%, most common 1-2%) is dosed once to four times a day, usually 2-3 times a day after angle surgery. Side effects include miosis, decreased heart rate, apnea, sweating, and hypersalivation, and theoretically may induce cataract and retinal detachment.
- Modified prostaglandin analogs and rho-associated protein kinase inhibitors: Latanoprostene bunod (Vyzulta) and netarsudil (Rhopressa) have not been studied in patients younger than 18 years of age.
Doctors can start with a either a carbonic anhydrase inhibitor, beta-blocker or prostaglandin analog, or a combination, and progressively add another medication class, keeping in mind medications are generally a temporizing measure prior to surgery. If prescribed before initial surgery, medications should not be used without fairly frequent follow-up, and ideally surgery performed within 2 weeks of primary congenital glaucoma diagnosis. Early discussion preparing family and caregivers for surgery is necessary. Medications should be continued until surgery, and may help maximize corneal clearing by reducing the intraocular pressure (IOP). After surgery, medications may still be needed as an adjunct and family and caregivers should be made aware of this. Compliance may be an issue when the medication regimen becomes complex and should be addressed. primary congenital glaucoma requires lifelong serial measurements of intraocular pressure (IOP), corneal diameter, axial length, refractive error, and optic nerve cupping. If an adequate assessment is not possible in the outpatient clinic, an examination under anesthesia should be performed.
Eye Surgery
Surgical procedures used to treat childhood glaucoma include the following:
- Trabeculotomy and goniotomy.
- Trabeculotomy is a surgical procedure, primarily used in the treatment of childhood glaucoma, that creates a new drainage opening in the eye’s trabecular meshwork, improving the outflow of aqueous humor and reducing the intraocular pressure (IOP).
- Goniotomy is a microinvasive glaucoma surgery (MIGS) technique that improves fluid flow in the eye to lower intraocular pressure (IOP). A goniotomy involves making a small incision within the trabecular meshwork, the eye’s natural drainage system, to create a more efficient pathway for fluid outflow. This procedure can be used to treat conditions like childhood glaucoma.
- Trabeculectomy. Trabeculectomy is a surgical procedure that involves the removal of part of the trabecular meshwork drainage system, allowing the fluid to drain from the eye. Trabeculectomy works by creating a new drainage pathway for the fluid (aqueous humor) within the eye, allowing it to drain into a space beneath the outer layer of the eye (conjunctiva). This new pathway, called a bleb, helps reduce eye pressure and can slow or prevent further vision loss.
- Iridotomy. Iridotomy is a surgical procedure to treat or prevent angle-closure glaucoma, a condition where the iris (colored part of the eye) blocks the drainage angle, leading to increased eye pressure. The eye surgeon may use a laser to create this hole. Laser iridotomy involves using a laser to create a small hole in the iris, allowing fluid to flow freely and preventing or relieving pressure build-up.
- Cyclophotocoagulation. Cyclophotocoagulation is a laser procedure that uses a laser beam to freeze selected areas of the ciliary body – the part of the eye that produces aqueous humor – to reduce the production of fluid and reduce intraocular pressure (IOP). Cyclophotocoagulation is a type of cyclodestruction procedure, meaning it aims to reduce intraocular pressure (IOP) by damaging the ciliary body, a key part of the eye that produces aqueous humor. This type of surgery may be performed with severe cases of childhood glaucoma.
The primary treatment of congenital glaucoma is drainage angle surgery, either goniotomy or trabeculotomy, to lower intraocular pressure (IOP) by improving aqueous outflow. In a retrospective review of pediatric glaucoma by Bussieres et al 100, 40% of patients with Axenfeld-Rieger syndrome had goniotomy, 30% had trabeculotomy, and 2% required glaucoma drainage devices. Most of those patients required 1.5 surgeries per eye 100. In general, goniotomy and trabeculotomy are less successful interventions in congenital glaucoma than other childhood glaucomas, presumably because of the angle dysgenesis and other developmental abnormalities associated with this group of glaucomas 120, 121.
If angle surgery is not successful, trabeculectomy enhanced with mitomycin C or glaucoma implant surgery with a Molteno, Baerveldt, or Ahmed implant can be performed. Trabeculectomy with mitomycin C is associated with successful intraocular pressure (IOP) lowering effect in 82-95% of cases and long-term success around 59% at 2-year follow-up 101, 102. However, trabeculectomy enhanced with mitomycin C is associated with risk of late post-operative endophthalmitis in 7-8% of cases 102, 103.
Glaucoma drainage devices have been reported to have success rates of 70-90% with long term success reported to be 58-63% at 2-year follow-up 103, 104, 105, 106, 122. The rate of endophthalmitis is low with this procedure, 2.9% 106, but surgical revision may have to be performed for other associated complications such as dislocation, tube-cornea touch, or erosion 103, 104, 105. In addition, concomitant medical therapy is often necessary to augment IOP control with glaucoma drainage devices, and re-operation may be necessary 104, 105, 99.
In surgically refractory childhood glaucomas, specifically developmental glaucomas, glaucoma drainage devices are likely to be successful where trabeculectomy has a relatively poor chance of success 101, 105.
Cyclodestructive procedures are usually reserved for refractory glaucomas after other options have been exhausted because of low reported success rates, frequent need for re-treatment, and high complication rates 99. Cycloablation can be performed using an Nd:YAG laser, diode laser, or cryotherapy, with diode laser being the most widely used device. Medications, either topically or orally, is typically used as a temporizing measure prior to surgery and to help decrease corneal clouding to facilitate goniotomy, and to supplement intraocular pressure (IOP) control after surgery.
Surgical Complications
Surgical complications include:
- Hyphema
- Shallow anterior chamber 123
- Peripheral anterior synechiae
- Iridodialysis
- Cyclodialysis (a condition where the longitudinal ciliary muscle fibers separates from the scleral spur, the area where the muscle attaches to the eye’s wall) 124
- Cataract 125, 126
- Epithelial ingrowth
- Choroidal detachment
- Retinal detachment
- Phthisis bulbi
Filtering Procedure-Related Complications:
- Over or under-filtration
- Blebitis
- Vitreous loss
- Scleral collapse
- Scleral flap leak
- Tube lens touch
- Endothelial decompensation from tube cornea touch
- Tube erosion
- Implant migration
- Diplopia from implant-related restrictions
- Endophthalmitis
Cyclodestructive Procedure-Related Complications:
- Hypotony
- Retinal detachment 127
- Phthisis
Anesthesia-Related Complications:
- Oculocardiac reflex
- Anaphylaxis
- Malignant hyperthermia
- Cardiovascular collapse
- Hepatic porphyria
- Hypoxic brain injury.
Postoperative and Rehabilitation Care
The children undergoing surgery should be started on topical steroids, either prednisolone 1% or dexamethasone 0.1% for 1 week each using an 8/7/6/5/4/3/2/1 tapering dosage. In addition, a topical antibiotic in the form of tobramycin 0.3% or 0.3% moxifloxacin or gatifloxacin 4 times per day for 20 days should be supplemented to prevent secondary infection. These patients will need close follow-ups postsurgery to look for signs of hypotony, inflammation, or infection. Moreover, the IOP needs to be recorded every 3 to 4 months for at least 2 years postsurgery.
Cycloplegic refraction will be needed every 6 months for these patients. Moreover, lifelong regular follow-up every 6 months is needed for intraocular pressure (IOP) monitoring and early detection of any surgery-related complications. Cases of failing angle or filtration surgery should be counseled for the need for glaucoma drainage device (GDD) and the risk of subsequent failure, amblyopia, blindness, and phthisis bulbi (a shrunken, disfigured, and non-functional eye that has undergone significant damage).
Surgical follow up
In the short term, patients require frequent follow up to follow response to treatment and monitor for hypotony, infection, and excessive inflammation. For young patients, or patients with less than 2 years of intraocular pressure (IOP) control, follow-up is recommended at least every 3-4 months. Regular life-long follow-up is needed (at least every 6 months) because even if long-term intraocular pressure (IOP) control from a surgical intervention is achieved, asymptomatic relapse can occur at any time and will need to be managed with medications or further surgery. Additionally, vision-threatening complications may occur at any time, especially after filtering surgeries.
Glaucoma drainage device (GDD) patency can be assessed with B-scan ultrasonography in the clinic 128.
Painted or tinted contact lenses
As Axenfeld-Rieger syndrome patients age, they may experience significant and debilitating glare or photophobia resulting from the sometimes progressive iris atrophy, polycoria, and corectopia. In these circumstances painted or tinted contact lenses may be beneficial in reducing these symptoms.
Axenfeld-Rieger syndrome prognosis
Every case of Axenfeld-Rieger syndrome is different and prognosis varies individually as patients with Axenfeld-Rieger syndrome have varied clinical presentations and different degrees of comorbidities 9. While some patients may only present anterior eye chamber abnormalities, others may also have cardiac defects of varying severity, which may worsen their prognosis 6. The eye prognosis is good in the absence of glaucoma 9. However, glaucoma may be challenging to treat and not respond to medical therapy alone 9. Patients with Axenfeld-Rieger syndrome who require glaucoma surgery typically undergo multiple surgeries, with an average of approximately 2.2 surgeries performed on each eye 129. Late diagnosis and delayed management of glaucoma can cause blindness, phthisis bulbi (a condition where the eye is irreversibly damaged and shrunken, leading to permanent loss of vision and function) or painful blind eye 130. Talk to your child’s doctor or eye care specialist about what to watch for as your child grows and develops. The sooner you get any new symptoms examined, the better.
- Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clin Ophthalmol. 2023 Mar 10;17:819-828. doi: 10.2147/OPTH.S379853[↩]
- Khandwala NS, Ramappa M, Edward DP, Mocan MC. Axenfeld-Rieger syndrome in the pediatric population: A review. Taiwan J Ophthalmol. 2023 Nov 24;13(4):417-424. doi: 10.4103/tjo.TJO-D-23-00089[↩][↩][↩]
- Tümer, Z., Bach-Holm, D. Axenfeld–Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet 17, 1527–1539 (2009). https://doi.org/10.1038/ejhg.2009.93[↩][↩]
- Axenfeld-Rieger syndrome. https://rarediseases.org/mondo-disease/axenfeld-rieger-syndrome[↩]
- Axenfeld-Rieger syndrome. https://medlineplus.gov/genetics/condition/axenfeld-rieger-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Alward WL. Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol. 2000 Jul;130(1):107-15. doi: 10.1016/s0002-9394(00)00525-0[↩][↩][↩][↩][↩][↩][↩][↩]
- Shields MB. Axenfeld-Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc. 1983;81:736-84. https://pmc.ncbi.nlm.nih.gov/articles/instance/1312467/pdf/taos00018-0760.pdf[↩][↩][↩][↩][↩][↩][↩][↩]
- Hjalt TA, Semina EV. Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med. 2005 Nov 8;7(25):1-17. doi: 10.1017/S1462399405010082[↩][↩]
- Zamora EA, Tripathy K, Salini B. Axenfeld-Rieger Syndrome. [Updated 2024 Feb 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538504[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Sowden JC. Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond). 2007 Oct;21(10):1310-8. doi: 10.1038/sj.eye.6702852[↩]
- Badnaware S, Srivastava VK, Chandel M, Gupta P, Fulzele P. Dental and Craniofacial Manifestation of Axenfeld-Rieger Syndrome: A Case Report. Cureus. 2022 Jun 29;14(6):e26442. doi: 10.7759/cureus.26442[↩]
- Seifi M, Walter MA. Axenfeld-Rieger syndrome. Clin Genet. 2018 Jun;93(6):1123-1130. doi: 10.1111/cge.13148[↩][↩]
- Axenfeld-Rieger syndrome. https://www.orpha.net/en/disease/detail/782[↩][↩][↩][↩]
- Agarwal P, Jain K, Sandesh S, Chopra S. Axenfeld-Rieger Syndrome: Rare Case Presentation and Overview. J Maxillofac Oral Surg. 2020;19(3):364-369. doi:10.1007/s12663-019-01307-9[↩]
- Axenfeld-Rieger syndrome type 1. https://rarediseases.org/mondo-disease/axenfeld-rieger-syndrome-type-1/[↩]
- Axenfeld Rieger Syndrome. https://eyewiki.org/Axenfeld_Rieger_Syndrome[↩][↩][↩][↩]
- Akazawa, K., Yamane, S., Shiota, H., Naito, E. A case of retinoblastoma associated with Rieger’s anomaly and 13q deletion. Jpn. J. Ophthal. 25: 321-325, 1981[↩]
- Phillips JC, del Bono EA, Haines JL, Pralea AM, Cohen JS, Greff LJ, Wiggs JL. A second locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet. 1996 Sep;59(3):613-9. https://pmc.ncbi.nlm.nih.gov/articles/PMC1914897[↩]
- AXENFELD-RIEGER SYNDROME, TYPE 2; RIEG2. https://www.omim.org/entry/601499[↩][↩][↩]
- Phillips JC, del Bono EA, Haines JL, Pralea AM, Cohen JS, Greff LJ, Wiggs JL. A second locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet. 1996 Sep;59(3):613-9. https://pmc.ncbi.nlm.nih.gov/articles/instance/1914897/pdf/ajhg00022-0128.pdf[↩][↩][↩]
- Axenfeld-Rieger syndrome type 3. https://rarediseases.org/mondo-disease/axenfeld-rieger-syndrome-type-3/[↩]
- AXENFELD-RIEGER SYNDROME, TYPE 3; RIEG3. https://www.omim.org/entry/602482[↩]
- Weisschuh N, Wolf C, Wissinger B, Gramer E. A novel mutation in the FOXC1 gene in a family with Axenfeld-Rieger syndrome and Peters’ anomaly. Clin Genet. 2008 Nov;74(5):476-80. doi: 10.1111/j.1399-0004.2008.01025.x[↩]
- Shields MB. Axenfeld-Rieger and iridocorneal endothelial syndromes: two spectra of disease with striking similarities and differences. J Glaucoma. 2001 Oct;10(5 Suppl 1):S36-8. doi: 10.1097/00061198-200110001-00014[↩][↩][↩]
- George, Anjith. (2017). IMAGE BASED EYE GAZE TRACKING AND ITS APPLICATIONS. 10.13140/RG.2.2.33992.88329[↩]
- Waring GO 3rd, Rodrigues MM, Laibson PR. Anterior chamber cleavage syndrome. A stepladder classification. Surv Ophthalmol. 1975 Jul-Aug;20(1):3-27. doi: 10.1016/0039-6257(75)90034-x[↩][↩]
- Ho DK, Levin AV, Anninger WV, Piccoli DA, Eagle RC Jr. Anterior Chamber Pathology in Alagille Syndrome. Ocul Oncol Pathol. 2016;2(4):270-275. doi:10.1159/000446804[↩][↩]
- BURIAN HM, RICE MH, ALLEN L. External visibility of the region of Schlemm’s canal; report on a family with developmental anomalies of cornea, iris, and chamber angle. AMA Arch Ophthalmol. 1957 May;57(5):651-8.[↩][↩]
- Rennie, C., Chowdhury, S., Khan, J. et al. The prevalence and associated features of posterior embryotoxon in the general ophthalmic clinic. Eye 19, 396–399 (2005). https://doi.org/10.1038/sj.eye.6701508[↩][↩][↩][↩]
- Axenfeld-Rieger syndrome. https://www.eyerounds.org/atlas/pages/Axenfeld-Rieger.htm[↩]
- Axenfeld-Rieger syndrome. https://eyerounds.org/atlas/pages/Axenfeld-Rieger/index.htm#gsc.tab=0[↩]
- Trabecular Meshwork Research. https://ethier.gatech.edu/anterior-segmenttrabecular-meshwork-biomechanics[↩][↩]
- J. Buffault, A. Labbé, P. Hamard, F. Brignole-Baudouin, C. Baudouin. The trabecular meshwork: Structure, function and clinical implications. A review of the literature. Journal Français
d’Ophtalmologie, 2020, 43 (7), pp.e217-e230. https://hal.sorbonne-universite.fr/hal-02962228v1/document[↩] - Chapter 3. The Glaucoma Eye. https://eyerounds.org/books/glaucoma_guide/chapter3.html#gsc.tab=0[↩]
- Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT. A review of anterior segment dysgeneses. Surv Ophthalmol. 2006;51(3):213–231. doi: 10.1016/j.survophthal.2006.02.006[↩]
- Tanwar M, Dada T, Dada R. Axenfeld-Rieger Syndrome Associated with Congenital Glaucoma and Cytochrome P4501B1 Gene Mutations. Case Rep Med. 2010;2010:1–6. doi: 10.1155/2010/212656[↩]
- Reis LM, Semina EV. Conserved genetic pathways associated with microphthalmia, anophthalmia, and coloboma. Birth Defects Res C Embryo Today. 2015;105:96–113. doi: 10.1002/bdrc.21097[↩]
- Ma A, Yousoof S, Grigg JR, et al. Revealing hidden genetic diagnoses in the ocular anterior segment disorders. Genet Med. 2020;22(10):1623–1632. doi: 10.1038/s41436-020-0854-x[↩]
- Smith RS, Zabaleta A, Kume T, Savinova OV, Kidson SH, Martin JE, Nishimura DY, Alward WL, Hogan BL, John SW. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum Mol Genet. 2000 Apr 12;9(7):1021-32. doi: 10.1093/hmg/9.7.1021[↩]
- Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, Kanis AB, Gastier JM, Stone EM, Sheffield VC. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998 Jun;19(2):140-7. doi: 10.1038/493[↩][↩]
- Glaser T, Walton DS, Maas RL. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992 Nov;2(3):232-9. doi: 10.1038/ng1192-232[↩]
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996 Dec;14(4):392-9. doi: 10.1038/ng1296-392[↩]
- Tanwar M, Dada T, Dada R. Axenfeld-Rieger Syndrome Associated with Congenital Glaucoma and Cytochrome P4501B1 Gene Mutations. Case Rep Med. 2010;2010:212656. doi: 10.1155/2010/212656[↩]
- Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, Ritch R, Koop B, Kuo WL, Collins C, Marshall J, Gould DB, Pearce W, Carlsson P, Enerbäck S, Morissette J, Bhattacharya S, Hogan B, Raymond V, Walter MA. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet. 1998 Nov;63(5):1316-28. doi: 10.1086/302109[↩]
- Reis LM, Maheshwari M, Capasso J, Atilla H, Dudakova L, Thompson S, et al. Axenfeld-Rieger syndrome: More than meets the eye. J Med Genet. 2023;60:368–79. doi: 10.1136/jmg-2022-108646[↩][↩][↩][↩][↩]
- Strungaru MH, Dinu I, Walter MA. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007;48:228–37. doi: 10.1167/iovs.06-0472[↩][↩]
- Tümer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet. 2009;17:1527–39. doi: 10.1038/ejhg.2009.93[↩]
- Huang L, Meng Y, Guo X. Novel PITX2 Mutations including a mutation causing an unusual ophthalmic phenotype of Axenfeld-Rieger syndrome. J Ophthalmol 2019. 2019:e5642126. doi: 10.1155/2019/5642126[↩]
- Michels K, Bohnsack BL. Ophthalmological manifestations of Axenfeld-Rieger syndrome: Current perspectives. Clin Ophthalmol. 2023;17:819–28. doi: 10.2147/OPTH.S379853[↩]
- Seifi M, Walter MA. Axenfeld-Rieger syndrome. Clin Genet. 2018;93:1123–30. doi: 10.1111/cge.13148[↩]
- Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT. A review of anterior segment dysgeneses. Surv Ophthalmol. 2006 May-Jun;51(3):213-31. doi: 10.1016/j.survophthal.2006.02.006[↩][↩][↩][↩]
- Shields MB, Buckley E, Klintworth GK, Thresher R. Axenfeld-Rieger syndrome. A spectrum of developmental disorders. Surv Ophthalmol. 1985 May-Jun;29(6):387-409. doi: 10.1016/0039-6257(85)90205-x[↩][↩][↩][↩]
- Singh P, Gupta A, Tripathy K. Iridocorneal Dysgenesis. [Updated 2023 Aug 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK585064[↩]
- Churchill A, Booth A. Genetics of aniridia and anterior segment dysgenesis. Br J Ophthalmol. 1996 Jul;80(7):669-73. https://pmc.ncbi.nlm.nih.gov/articles/instance/505566/pdf/brjopthal00007-0089.pdf[↩]
- lkemade, P. Dysgenesis Mesodermalis of the Iris and the Cornea. Van Gorcum, Assen, The Netherlands. 1969.[↩]
- Tümer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet. 2009 Dec;17(12):1527-39. doi: 10.1038/ejhg.2009.93[↩]
- Tsai JC, Grajewski AL. Cardiac valvular disease and Axenfeld-Rieger syndrome. Am J Ophthalmol. 1994 Aug 15;118(2):255-6. doi: 10.1016/s0002-9394(14)72910-1[↩]
- Fitch N, Kaback M. The Axenfeld syndrome and the Rieger syndrome. J Med Genet. 1978 Feb;15(1):30-4. https://pmc.ncbi.nlm.nih.gov/articles/instance/1012820/pdf/jmedgene00296-0036.pdf[↩]
- Shields MB, Buckley E, Klintworth GK, et al. Axenfeld–Rieger syndrome. A spectrum of developmental disorders. Surv Ophthalmol. 1985;29:387–409. doi: 10.1016/0039-6257(85)90205-X[↩]
- Reis LM, Maheshwari M, Capasso J, et al. Axenfeld-Rieger syndrome: more than meets the eye [published online ahead of print, 2022 Jul 26]. J Med Genet. 2022;jmedgenet-2022-108646. doi:10.1136/jmg-2022-108646[↩][↩]
- Mandal AK. Acute Corneal Hydrops in Children with Primary Infantile Glaucoma: A Report of 31 Cases over 23 Years at the LVPEI. PLoS One. 2016 Jun 1;11(6):e0156108. doi: 10.1371/journal.pone.0156108[↩]
- Drechsler J, Lee A, Maripudi S, Kueny L, Levin MR, Saeedi OJ, Bazemore M, Karwoski B, Birdsong R, Martinez C, Jaafar MS, Yousaf S, Ahmed ZM, Madigan WP, Alexander JL. Corneal Structural Changes in Congenital Glaucoma. Eye Contact Lens. 2022 Jan 1;48(1):27-32. doi: 10.1097/ICL.0000000000000844[↩]
- Gupta S, Mahalingam K, Singh A, Selvan H, Somarajan BI, Gupta V. Posterior corneal morphological changes in primary congenital glaucoma. Indian J Ophthalmol. 2022 Jul;70(7):2571-2577. doi: 10.4103/ijo.IJO_317_22[↩][↩]
- Sihota R, Mahalingam K, Maurya AK, Sharma A, Bukke AN, Dada T. Primary congenital glaucoma: An iridotrabeculodysgenesis? Indian J Ophthalmol. 2024 Mar 1;72(3):328-334. doi: 10.4103/IJO.IJO_370_23[↩]
- Walton DS. Chronic newborn primary congenital glaucoma with secondary lens subluxation. J Pediatr Ophthalmol Strabismus. 2009 Jul-Aug;46(4):200, 231. doi: 10.3928/01913913-20090706-02[↩]
- Snehi S, Singh AK, Kaushik S. Acquired Lens Zonular Loss with Bean Pot Optic Disc Cupping in Congenital Glaucoma. Ophthalmol Glaucoma. 2023 Mar-Apr;6(2):159. doi: 10.1016/j.ogla.2023.01.001[↩]
- Gupta V, James MK, Singh A, Kumar S, Gupta S, Sharma A, Sihota R, Kennedy DJ. Differences in Optic Disc Characteristics of Primary Congenital Glaucoma, Juvenile, and Adult Onset Open Angle Glaucoma Patients. J Glaucoma. 2016 Mar;25(3):239-43. doi: 10.1097/IJG.0000000000000154[↩]
- Sihota R, Sidhu T, Agarwal R, Sharma A, Gupta A, Sethi A, Dada T, Pandey V. Evaluating target intraocular pressures in primary congenital glaucoma. Indian J Ophthalmol. 2021 Aug;69(8):2082-2087. doi: 10.4103/ijo.IJO_3473_20[↩]
- Fayed MA, Chen TC. Pediatric intraocular pressure measurements: Tonometers, central corneal thickness, and anesthesia. Surv Ophthalmol. 2019 Nov-Dec;64(6):810-825. doi: 10.1016/j.survophthal.2019.05.003[↩]
- Eisenberg DL, Sherman BG, McKeown CA, Schuman JS. Tonometry in adults and children. A manometric evaluation of pneumatonometry, applanation, and TonoPen in vitro and in vivo. Ophthalmology. 1998 Jul;105(7):1173-81. doi: 10.1016/S0161-6420(98)97016-6[↩]
- Grigorian F, Grigorian AP, Olitsky SE. The use of the iCare tonometer reduced the need for anesthesia to measure intraocular pressure in children. J AAPOS. 2012 Dec;16(6):508-10. doi: 10.1016/j.jaapos.2012.07.004[↩]
- Flemmons MS, Hsiao YC, Dzau J, Asrani S, Jones S, Freedman SF. Icare rebound tonometry in children with known and suspected glaucoma. J AAPOS. 2011 Apr;15(2):153-7. doi: 10.1016/j.jaapos.2010.11.022[↩]
- McKee EC, Ely AL, Duncan JE, Dosunmu EO, Freedman SF. A comparison of Icare PRO and Tono-Pen XL tonometers in anesthetized children. J AAPOS. 2015 Aug;19(4):332-7. doi: 10.1016/j.jaapos.2015.04.004[↩]
- Weinreb R.N., Grajewski A.L., Papadopoulos M., Grigg J.,Freedman S.F. Childhood glaucoma: the 9th consensus report of the World Glaucoma Association. Netherlands: Kugler Publications, 2013.[↩][↩][↩][↩][↩]
- Sampaolesi R, Caruso R. Ocular echometry in the diagnosis of congenital glaucoma. Arch Ophthalmol. 1982 Apr;100(4):574-7. doi: 10.1001/archopht.1982.01030030576003[↩]
- Henriques MJ, Vessani RM, Reis FA, de Almeida GV, Betinjane AJ, Susanna R Jr. Corneal thickness in congenital glaucoma. J Glaucoma. 2004 Jun;13(3):185-8. doi: 10.1097/00061198-200406000-00002[↩]
- Doozandeh A, Yazdani S, Ansari S, Pakravan M, Motevasseli T, Hosseini B, Yasseri M. Corneal profile in primary congenital glaucoma. Acta Ophthalmol. 2017 Nov;95(7):e575-e581. doi: 10.1111/aos.13357[↩][↩]
- Zareei A, Razeghinejad MR, Salouti R. Corneal Biomechanical Properties and Thickness in Primary Congenital Glaucoma and Normal Eyes: A Comparative Study. Med Hypothesis Discov Innov Ophthalmol. 2018 Summer;7(2):68-72. https://pmc.ncbi.nlm.nih.gov/articles/PMC6146241[↩][↩]
- Donahue SP, Porter A. SITA visual field testing in children. J AAPOS. 2001 Apr;5(2):114-7. doi: 10.1067/mpa.2001.113840[↩]
- Salchow DJ, Oleynikov YS, Chiang MF, Kennedy-Salchow SE, Langton K, Tsai JC, Al-Aswad LA. Retinal nerve fiber layer thickness in normal children measured with optical coherence tomography. Ophthalmology. 2006 May;113(5):786-91. doi: 10.1016/j.ophtha.2006.01.036[↩]
- Ahn HC, Son HW, Kim JS, Lee JH. Quantitative analysis of retinal nerve fiber layer thickness of normal children and adolescents. Korean J Ophthalmol. 2005 Sep;19(3):195-200. doi: 10.3341/kjo.2005.19.3.195[↩]
- Hess DB, Asrani SG, Bhide MG, Enyedi LB, Stinnett SS, Freedman SF. Macular and retinal nerve fiber layer analysis of normal and glaucomatous eyes in children using optical coherence tomography. Am J Ophthalmol. 2005 Mar;139(3):509-17. doi: 10.1016/j.ajo.2004.10.047[↩]
- Rao A, Sahoo B, Kumar M, Varshney G, Kumar R. Retinal nerve fiber layer thickness in children <18 years by spectral-domain optical coherence tomography. Semin Ophthalmol. 2013 Mar;28(2):97-102. doi: 10.3109/08820538.2012.760626[↩]
- Rotruck JC, House RJ, Freedman SF, Kelly MP, Enyedi LB, Prakalapakorn SG, Lim ME, El-Dairi MA. Optical Coherence Tomography Normative Peripapillary Retinal Nerve Fiber Layer and Macular Data in Children 0-5 Years of Age. Am J Ophthalmol. 2019 Dec;208:323-330. doi: 10.1016/j.ajo.2019.06.025[↩]
- Hsu ST, Chen X, Ngo HT, House RJ, Kelly MP, Enyedi LB, Materin MA, El-Dairi MA, Freedman SF, Toth CA, Vajzovic L. Imaging Infant Retinal Vasculature with OCT Angiography. Ophthalmol Retina. 2019 Jan;3(1):95-96. doi: 10.1016/j.oret.2018.06.017[↩]
- External Disease and Cornea, Section 8. Basic and Clinical Science Course, AAO. Pg. 300-301;2009-2010.[↩]
- Jat NS, Tripathy K. Peters Anomaly. [Updated 2023 Aug 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK580540[↩][↩][↩]
- External Disease and Cornea, Section 8. Basic and Clinical Science Course, AAO. Pg. 292-294;2009-2010.[↩]
- Tripathy K, Salini B. Aniridia. [Updated 2023 Aug 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538133[↩][↩][↩][↩][↩][↩]
- Glaucoma, Section 10. Basic and Clinical Science Course, AAO. Pg. 162;2009-2010.[↩]
- Congenital Ectropion Uveae. https://eyewiki.org/Congenital_Ectropion_Uveae[↩]
- Sridhar U, Tripathy K. Iris Ectropion Syndrome. [Updated 2023 Aug 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK580493[↩]
- Pediatric Ophthalmology and Strabismus, Section 6. Basic and Clinical Science Course, AAO. Pg. 169-170;2009-2010.[↩]
- Ophthalmic Pathology and Intraocular Tumors, Section 4. Basic and Clinical Science Course, AAO. Pg. 186;2009-2010.[↩]
- Byles DB, Nischal KK, Cheng H. Ectopia lentis et pupillae. A hypothesis revisited. Ophthalmology. 1998 Jul;105(7):1331-6. doi: 10.1016/S0161-6420(98)97043-9[↩][↩]
- Pediatric Ophthalmology and Strabismus, Section 6. Basic and Clinical Science Course, AAO. Pg. 305-306;2009-2010.[↩]
- Oculodentodigital dysplasia. https://medlineplus.gov/genetics/condition/oculodentodigital-dysplasia[↩]
- Retina and Vitreous, Section 12. Basic and Clinical Science Course, AAO. Pg. 256;2009-2010.[↩]
- Beck AD. Diagnosis and management of pediatric glaucoma. Ophthalmol Clin North Am. 2001 Sep;14(3):501-12. doi: 10.1016/s0896-1549(05)70248-0[↩][↩][↩]
- Bussières JF, Therrien R, Hamel P, Barret P, Prot-Labarthe S. Retrospective cohort study of 163 pediatric glaucoma patients. Can J Ophthalmol. 2009 Jun;44(3):323-7. doi: 10.3129/i09-065[↩][↩][↩]
- Beck AD, Wilson WR, Lynch MG, Lynn MJ, Noe R. Trabeculectomy with adjunctive mitomycin C in pediatric glaucoma. Am J Ophthalmol. 1998 Nov;126(5):648-57. doi: 10.1016/s0002-9394(98)00227-x[↩][↩][↩]
- Sidoti PA, Belmonte SJ, Liebmann JM, Ritch R. Trabeculectomy with mitomycin-C in the treatment of pediatric glaucomas. Ophthalmology. 2000 Mar;107(3):422-9. doi: 10.1016/s0161-6420(99)00130-x[↩][↩][↩]
- Coleman AL, Smyth RJ, Wilson MR, Tam M. Initial clinical experience with the Ahmed Glaucoma Valve implant in pediatric patients. Arch Ophthalmol. 1997 Feb;115(2):186-91. doi: 10.1001/archopht.1997.01100150188007[↩][↩][↩][↩]
- Eid TE, Katz LJ, Spaeth GL, Augsburger JJ. Long-term effects of tube-shunt procedures on management of refractory childhood glaucoma. Ophthalmology. 1997 Jun;104(6):1011-6. doi: 10.1016/s0161-6420(97)30193-6[↩][↩][↩][↩]
- Englert JA, Freedman SF, Cox TA. The Ahmed valve in refractory pediatric glaucoma. Am J Ophthalmol. 1999 Jan;127(1):34-42. doi: 10.1016/s0002-9394(98)00292-x[↩][↩][↩][↩][↩]
- Djodeyre MR, Peralta Calvo J, Abelairas Gomez J. Clinical evaluation and risk factors of time to failure of Ahmed Glaucoma Valve implant in pediatric patients. Ophthalmology. 2001 Mar;108(3):614-20. doi: 10.1016/s0161-6420(00)00603-5[↩][↩][↩]
- Boger WP 3rd, Walton DS. Timolol in uncontrolled childhood glaucomas. Ophthalmology. 1981 Mar;88(3):253-8. doi: 10.1016/s0161-6420(81)35055-6[↩]
- Hoskins HD Jr, Hetherington J Jr, Magee SD, Naykhin R, Migliazzo CV. Clinical experience with timolol in childhood glaucoma. Arch Ophthalmol. 1985 Aug;103(8):1163-5. doi: 10.1001/archopht.1985.01050080075024[↩]
- Portellos M, Buckley EG, Freedman SF. Topical versus oral carbonic anhydrase inhibitor therapy for pediatric glaucoma. J AAPOS. 1998 Feb;2(1):43-7. doi: 10.1016/s1091-8531(98)90109-4[↩]
- Enyedi LB, Freedman SF, Buckley EG. The effectiveness of latanoprost for the treatment of pediatric glaucoma. J AAPOS. 1999 Feb;3(1):33-9. doi: 10.1016/s1091-8531(99)70092-3[↩]
- Black AC, Jones S, Yanovitch TL, Enyedi LB, Stinnett SS, Freedman SF. Latanoprost in pediatric glaucoma–pediatric exposure over a decade. J AAPOS. 2009 Dec;13(6):558-62. doi: 10.1016/j.jaapos.2009.10.003[↩]
- Carlsen JO, Zabriskie NA, Kwon YH, Barbe ME, Scott WE. Apparent central nervous system depression in infants after the use of topical brimonidine. Am J Ophthalmol. 1999 Aug;128(2):255-6. doi: 10.1016/s0002-9394(99)00083-5[↩]
- Wright TM, Freedman SF. Exposure to topical apraclonidine in children with glaucoma. J Glaucoma. 2009 Jun-Jul;18(5):395-8. doi: 10.1097/IJG.0b013e31818624e5[↩]
- Coppens G, Stalmans I, Zeyen T, Casteels I. The safety and efficacy of glaucoma medication in the pediatric population. J Pediatr Ophthalmol Strabismus. 2009 Jan-Feb;46(1):12-8. doi: 10.3928/01913913-20090101-05[↩]
- Buckley MM, Goa KL, Clissold SP. Ocular betaxolol. A review of its pharmacological properties, and therapeutic efficacy in glaucoma and ocular hypertension. Drugs. 1990 Jul;40(1):75-90. doi: 10.2165/00003495-199040010-00005[↩]
- Maren TH, Haywood JR, Chapman SK, Zimmerman TJ. The pharmacology of methazolamide in relation to the treatment of glaucoma. Invest Ophthalmol Vis Sci. 1977 Aug;16(8):730-42.[↩]
- Capino AC, Dannaway DC, Miller JL. Metabolic Acidosis with Ophthalmic Dorzolamide in a Neonate. J Pediatr Pharmacol Ther. 2016 May-Jun;21(3):256-9. doi: 10.5863/1551-6776-21.3.256[↩]
- Maeda-Chubachi T, Chi-Burris K, Simons BD, Freedman SF, Khaw PT, Wirostko B, Yan E; A6111137 Study Group. Comparison of latanoprost and timolol in pediatric glaucoma: a phase 3, 12-week, randomized, double-masked multicenter study. Ophthalmology. 2011 Oct;118(10):2014-21. doi: 10.1016/j.ophtha.2011.03.010[↩]
- Kim JS, Blizzard S, Woodward JA, Leyngold IM, Liss J, Freedman SF. Prostaglandin-Associated Periorbitopathy in Children and Young Adults with Glaucoma. Ophthalmol Glaucoma. 2020 Jul-Aug;3(4):288-294. doi: 10.1016/j.ogla.2020.03.009[↩]
- Luntz MH. Congenital, infantile, and juvenile glaucoma. Ophthalmology. 1979 May;86(5):793-802. doi: 10.1016/s0161-6420(79)35451-3[↩]
- Taylor RH, Ainsworth JR, Evans AR, Levin AV. The epidemiology of pediatric glaucoma: the Toronto experience. J AAPOS. 1999 Oct;3(5):308-15. doi: 10.1016/s1091-8531(99)70028-5[↩]
- Chen TC, Bhatia LS, Walton DS. Ahmed valve surgery for refractory pediatric glaucoma: a report of 52 eyes. J Pediatr Ophthalmol Strabismus. 2005 Sep-Oct;42(5):274-83; quiz 304-5. doi: 10.3928/0191-3913-20050901-09[↩]
- Christy J, Jain N, Gurnani B, Kaur K. Twinkling Eye -A Rare Presentation in Neovascular Glaucoma. J Glaucoma. 2019 May 23. doi: 10.1097/IJG.0000000000001287[↩]
- Gurnani B, Kaur K, Sekaran S. First case of coloboma, lens neovascularization, traumatic cataract, and retinal detachment in a young Asian female. Clin Case Rep. 2021 Aug 30;9(9):e04743. doi: 10.1002/ccr3.4743[↩]
- Gurnani B, Kaur K, Gireesh P. A rare presentation of anterior dislocation of calcified capsular bag in a spontaneously absorbed cataractous eye. Oman J Ophthalmol. 2021 Jun 28;14(2):120-121. doi: 10.4103/ojo.OJO_65_2019[↩]
- Gurnani B, Kaur K, Gireesh P. Rare Coexistence of Bilateral Congenital Sutural and Cortical Blue Dot Cataracts. J Pediatr Ophthalmol Strabismus. 2020 Jan 1;57(1):68. doi: 10.3928/01913913-20191011-01[↩]
- Kaur K, Gurnani B, Kannusamy V, Yadalla D. A tale of orbital cellulitis and retinopathy of prematurity in an infant: First case report. Eur J Ophthalmol. 2022 Nov;32(6):NP20-NP23. doi: 10.1177/11206721211026098[↩]
- Baig NB, Lin AA, Freedman SF. Ultrasound evaluation of glaucoma drainage devices in children. J AAPOS. 2015 Jun;19(3):281-4. doi: 10.1016/j.jaapos.2015.02.001[↩]
- Zepeda EM, Branham K, Moroi SE, Bohnsack BL. Surgical outcomes of Glaucoma associated with Axenfeld-Rieger syndrome. BMC Ophthalmol. 2020 May 1;20(1):172. doi: 10.1186/s12886-020-01417-w[↩]
- Tripathy K, Chawla R, Temkar S, Sagar P, Kashyap S, Pushker N, Sharma YR. Phthisis Bulbi-a Clinicopathological Perspective. Semin Ophthalmol. 2018;33(6):788-803. doi: 10.1080/08820538.2018.1477966[↩]




{kind=link}